
Abstract
Recent advances in microbial genomics, synthetic organic chemistry and X-ray crystallography provided opportunities to identify novel antibacterial targets for the development of new classes of antibiotics and to design more potent antimicrobial compounds derived from existing antibiotics in clinical use for decades. The antimetabolites, sulfa drugs and trimethoprim (TMP)-like agents, are inhibitors of three families of enzymes. One family belongs to the carbonic anhydrases, which catalyze a simple but physiologically relevant reaction in all life kingdoms, carbon dioxide hydration to bicarbonate and protons. The other two enzyme families are involved in the synthesis of tetrahydrofolate (THF), i.e. dihydropteroate synthase (DHPS) and dihydrofolate reductase. The antibacterial agents belonging to the THF and DHPS inhibitors were developed decades ago and present significant bacterial resistance problems. However, the molecular mechanisms of drug resistance both to sulfa drugs and TMP-like inhibitors were understood in detail only recently, when several X-ray crystal structures of such enzymes in complex with their inhibitors were reported. Here, we revue the state of the art in the field of antibacterials based on inhibitors of these three enzyme families.
Introduction
Carbon biological cycle
Living organisms play an important role in the movement of carbon between land, ocean and atmosphere through the processes of photosynthesis and respirationCitation1. Carbon dioxide is the final product of the respiration in all domains of life, from microorganisms to mammalsCitation2,Citation3. Virtually all life on earth depends on the production of sugars from sunlight and carbon dioxide (photosynthesis) and the metabolic breakdown (respiration) of those sugars to produce the energy needed for movement, growth and reproduction. Carbon dioxide (CO2) and its hydration product bicarbonate () are substrates and products of various metabolic reactions in the cells of different living organismsCitation4,Citation5. The interconversion of CO2 and
is spontaneously balanced to maintain the equilibrium between dissolved inorganic carbon dioxide, carbonic acid and carbonate of which
is physiologically the most important one as it is a substrate for several carboxylating enzymes involved in biosynthetic pathways such as biosynthesis of fatty acids, amino acids and nucleotidesCitation6,Citation7. The uncatalyzed hydration–dehydration of CO2-
is slow at physiological pH and thus, in biological systems, the reaction is accelerated by an enzymatic catalyst, called carbonic anhydrase (CA). CAs (EC 4.2.1.1) catalyze a simple but physiologically relevant reaction in all life kingdoms, carbon dioxide hydration to bicarbonate and protonsCitation8,Citation9:
These enzymes are thus involved in many physiologic processes in microorganisms, such as photosynthesis, respiration, CO2 transport, as well as metabolism of xenobiotics (e.g. cyanate in Escherichia coli)Citation10–13.
Folic acid synthesis pathway
Prokaryotes and unicellular eukaryotes such as yeasts or Plasmodium are able to synthesize folate de novo via the folic acid synthesis pathway (). The steps of this pathway include: (i) condensation of dihydropteridine pyrophosphate (DHPP) 1 with p-amino benzoic acid (pABA) 2 by dihydropteroate synthase (DHPS, EC 2.5.1.15) to form dihydropteroate (DHP) 3; (ii) addition of glutamate to DHP by dihydrofolate synthase (DHFS, EC 6.3.2.12) to form dihydrofolate (DHF) 4 and (iii) reduction of DHF to form tetrahydrofolate (THF) 5, catalyzed by dihydrofolate reductase (DHFR, EC 1.5.1.3)Citation14.
Figure 1. Biosynthesis of THF 5 from dihydropteridine pyrophosphate 1 (see text for details).
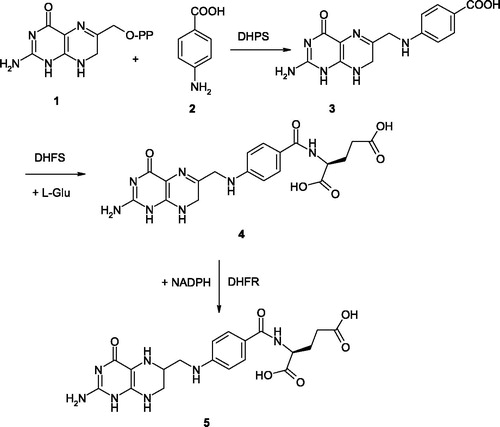
DHPS is thus an essential enzyme responsible for the de novo synthesis of the folate molecule. THF produced by this pathway is required for one carbon transfer reactions in the biosynthesis of a range of biomolecules, such as nucleotides and amino acidsCitation15. DHFR is the enzyme responsible for the maintenance of folate pools in their physiologic, reduced states. DHFR, in fact, is needed for the intracellular conversion of synthetic folic acid into the THF forms that can participate in the folate/homocysteine metabolism. Reduction of DHFR enzymatic activity diminishes the THF pool inside the cells, affecting the level of folate coenzymes and thus purine and pyrimidine biosynthesisCitation16. This may as well influence homocysteine levels and methylation processes, since methyl-THF is needed for the re-methylation of homocysteine to form methionine, thereby ensuring the provision of S-adenosylmethionine (SAM) necessary for most biological methylation processesCitation17.
In contrast to prokaryotes and lower eukaryotes, higher eukaryotic cells cannot synthesize folates de novo, being totally dependent on exogenous folic acid. Thus, the folic acid synthesis pathway represents a convenient target for obtaining drugs for the treatment of infectious diseases, caused by bacteria and protozoan parasites. Indeed, at least two steps in this pathway are targeted by antibacterial drugs in clinical use for decades: DHPS is the target of the sulfa drugs, whereas DHFR is targeted by trimethoprim (TMP) (and its congeners)Citation5–7. These enzymes have been validated as antimicrobial therapeutic targets by chemical and genetic means at the beginning of research in the field of antibacterialsCitation18–20.
Enzymatic targets for sulfa- and TMP-like drugs
Carbonic anhydrases
CAs (EC 4.2.1.1) catalyze a simple but physiologically relevant reaction in all life kingdoms, carbon dioxide hydration to bicarbonate and protonsCitation8. Five genetically distinct CA families are known to date, the α-, β-, γ-, δ- and ζ-CAs, and all of them are metalloenzymes, using Zn(II), Cd(II) or Fe(II) at their active sitesCitation8,Citation12,Citation21. Bacteria encode for enzymes belonging to the α-, β- and γ-classes. shows the phylogenetic relation between the α-, β- and γ-classes from different species. The phylogenetic analysis also shows three different clusters denoting the existence of these three evolutionary classes of CAs, as mentioned above (we did not include δ- and ζ-CAs in our phylogenetic analysis as these enzymes are present only in diatoms)Citation10. CAs thus represent an excellent example of Darwinian convergent evolution of catalytic function. Intriguingly, the phylogenetic analysis produced two clusters, one of which includes the α- and γ-CAs () suggesting that these classes of enzymes might have originated from an ancestral gene different from the gene, which led to the β-CAs.
Figure 2. Phylogenetic tree of the α, β and γ CAs from selected eukaryotic and prokaryotic species. The tree was constructed using the program PhyML 3.0. Branch support values are reported at branch points. Organisms, accession numbers and cryptonyms of the sequences used for the phylogenetic analysis have been indicated in .
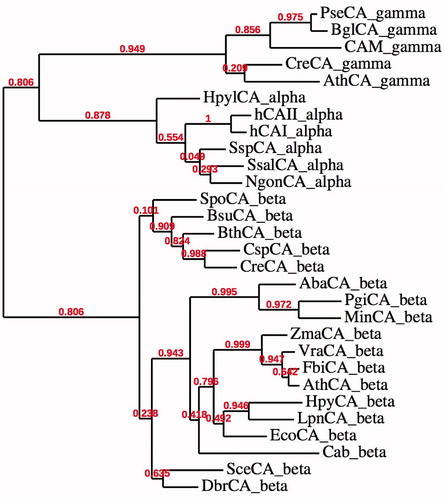
Table 1. CA class, organisms, accession numbers and cryptonyms of the sequences used in the phylogenetic analysis.
Most of these enzymes contain zinc ion (Zn2+) in their active site, coordinated by three histidine residues and a water molecule/hydroxide ion (in the α- and γ-classes) or by two Cys and one His residues (in the β-CA classes), with the fourth ligand being a water molecule/hydroxide ionCitation8,Citation12,Citation22,Citation23. Some of the catalytically active α-CAs can also catalyze the hydrolysis of esters, for example, 4-nitrophenyl acetate (4-NpA) and other hydrolytic reactions. Recently, CA enzymes were investigated in detail in pathogenic (as well as nonpathogenic) bacteria such as Brucella spp., Mycobacterium tuberculosis, Streptococcus spp., Helicobacter pylori, Salmonella enterica and Sulfurihydrogenibium spp.in the search for antibiotics with a novel mechanism of action, since it has been demonstrated in many of these organisms. CAs are essential for their life cycleCitation8,Citation21,Citation24. For some of them, X-ray crystal structures of the encoded CAs were also determined, and in vitro and in vivo inhibition studies with various classes of inhibitors, such as anions, sulfonamides and sulfamates have been reportedCitation8,Citation9,Citation24–28. Although the bacterial CA inhibition studies are in their infancy at this moment, cloning of more bacterial genomes may lead to the discovery of genes and proteins, which may have interesting applications both in the biomedical and biotechnological fieldsCitation8,Citation9,Citation21,Citation24–26.
DHP synthase
DHPS activity was identified in E. coli by BrownCitation29 and later in crude cell extracts of several organisms by Shiota et al.Citation30,Citation31. Humans lack an equivalent to DHPS, which is an advantage from the drug design viewpoint, since no toxicity to the host should emerge by using compounds targeting the bacterial/protozoan enzyme. DHPSs are inhibited by sulfonamide, which function as competitive inhibitor of one of the DHPS substrates, pABA 2. The DHPS encoded by the folP gene in Bacillus anthracis is also inhibited by sulfonamidesCitation32. The protein was shown to be a homodimer of two 30 kDa subunits. The chromosomal gene that codes for DHPS in Streptococcus pneumoniae was cloned, sequenced, and shown to code for a protein of 34 kDaCitation33. A similar sequence was also identified in a Bacillus subtilis folic acid biosynthetic operonCitation34. Two other genes (sulI and sulII) that code for plasmid-borne sulfonamide-resistant DHPSs have also been sequencedCitation35,Citation36. DHPS possesses a classic (β/α) TIM barrel structure, and the pABA binding site is at the edge of the barrel, being comprised of loop regions where the mutations that confer sulfonamide resistance are foundCitation37. In contrast, the other substrate of DHPS dihydropterin pyrophosphate (DHPP) 1, binds in a deep, structured pocket within the DHPS β-barrel. This pocket is highly conserved among various bacterial speciesCitation37.
Dihydrofolate reductase
DHFR is the enzyme responsible for the nicotinamide adenine dinucleotide phosphate (NADPH)-dependent reduction of 5,6-DHF to 5,6,7,8-THF, an essential cofactor used in the biosynthetic pathways of purines, thymidylate, methionine, glycine, pantothenic acid and N-formyl-methionyl tRNACitation38,Citation39. DHFR is ubiquitously expressed in all organisms, being found in all dividing cells of prokaryotes and eukaryotes. The mammalian enzymes are all highly similar in sequence, while each bacterial form is distinct. The DHFR sequence in humans is 30% similar to that of E. coli and 70% similar to other mammalian DHFRsCitation40. Human DHFR is a small enzyme with a primary structure of 186 amino acid residues and a molecular weight of about 20 kDaCitation40. Structural studies show that DHFR is a monomeric molecule with many secondary structural elements. The protein is divided into two subdomains, the adenosine-binding subdomain and the loop subdomainCitation41. The adenosine-binding subdomain is the larger of the two and binds the adenosine moiety of NADPH. The loop subdomain contains three loops. Between the two subdomains is the active site, where folate and NADPH bind. The size of the active site is regulated by the movements of the two subdomainsCitation41–43. At the transcriptional level, DHFR is characterized by a TATA-less promoter that is controlled by numerous transcription factors, including Sp1 and E2F, which are important for their regulation throughout the cell cycleCitation16,Citation44. In addition to key roles in folate metabolism, DHFR can reduce 7,8-dihydrobiopterin (BH2) to 5,6,7,8-tetrahydrobiopterin (BH4)Citation45, the natural cofactor required for the hydroxylation of aromatic amino acids, such as phenylalanine, tyrosine and tryptophan in higher animalsCitation46. Also, BH4 is an essential cofactor for nitric oxide synthesis involved in oxidative stress as a regulator of nitric oxide synthase or as a direct radical scavengerCitation47. In bacteria, there are DHFR encoded within mobile elements (e.g. plasmids), which are rapidly spread within a bacterial communityCitation48. They are grouped into two main families, A and BCitation48,Citation49. The A gene (dfrA) family encodes proteins of 152–189 amino acids with identity levels of 20–90% and some structural and sequence similarities to the chromosomal enzyme. At least 20 different dfrA sequences have been reported so farCitation50,Citation51. The B gene (dfrB) family, by contrast, encodes a unique group of enzymes referred to as DfrB, which are completely unrelated to the chromosomal DHFRs or to other DHFRs in terms of sequence and structure. The dfrB genes encode similar and much shorter proteins (78 residues) with identity levels of 75% or above, which are extremely resistant to TMPCitation52–54.
Clinically used inhibitors
Sulfa drug inhibitors
The primary sulfonamides were the first antimicrobial drugs, discovered in 1935 by Domagk, and they paved the way for the antibiotic revolution in medicineCitation55. The first sulfonamide showing effective antibacterial activity, Prontosil 6, was in fact a prodrug, with the real antibacterial agent being sulfanilamide 7, a compound isosteric/isostructural with pABA 2. Sulfanilamide is generated by in vivo reduction of prontosilCitation55,Citation56. In fact the aniline moiety is common in both compounds 2 and 7, whereas the carboxyl moiety of pABA is mimicked by the primary sulfamoyl moiety of sulfanilamide. In the following years after the discovery of sulfanilamide as a bacteriostatic agentCitation56, a range of analogs have entered into clinical use (constituting the so-called sulfa drug class of antibacterials), and many of these compounds are still widely used (mainly in combination with DHFR inhibitors)Citation57.
The clinically used sulfa drugs (including prontosil which is no longer used) are shown in , and they include Sulfamethoxazole 8, Sulfisomidine 9, Sulfacetamide 10 and Sulfathiazole 11 among othersCitation57,Citation58. All these derivatives are competitive inhibitors (with pABA) of DHPS, the first enzyme in the biosynthesis of THF. However, significant resistance developed over the years to these drugs, which drastically limit their use in the clinic (see later in the text). In fact an intermediate obtained by reaction of the sulfa drug with DHP led to a covalent product, which has been characterized by means of X-ray crystallography ()Citation57.
Figure 3. Sulfa drugs in clinical use (7–11) and prontosil 6, the lead molecule generating this class of pharmacological agents. These compounds are structurally similar to pABA 2 and compete with this compound for the biosynthesis of DHP 3Citation58. Many other analogs are knownCitation58.
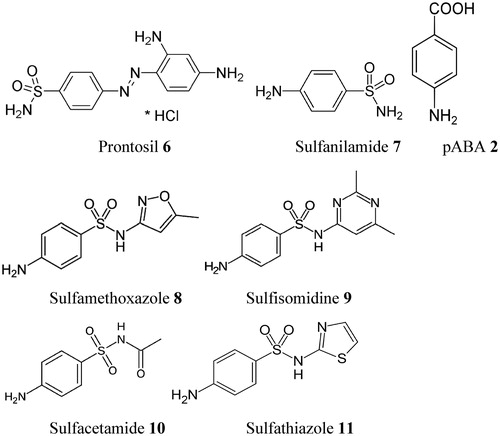
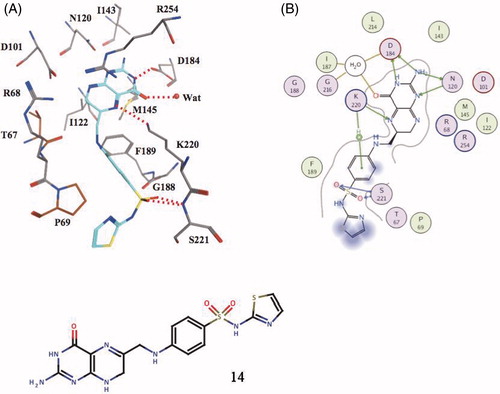
Figure 4. Covalent adduct of DHPP with the sulfa drug sulfathiazole, of type 14, as determined by X-ray crystallography (A) and the detailed interactions in which 14 participates when bound to the enzyme (B)Citation58.
DHFR inhibitors
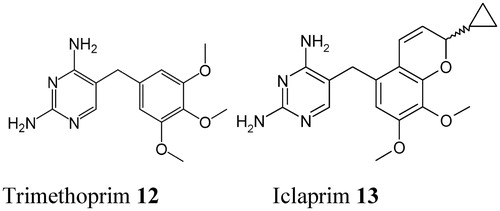
DHFR inhibitors such as TMP 12, are in clinical use since the 1960sCitation54, whereas newer analogs (e.g., brodimoprim, tetroxoprim, iclaprim) were constantly discovered and developedCitation57 (). Such compounds compete with the pteridine moiety of DHF for binding to the enzyme () and thus impair the generation of THF, a compound crucial in many biosynthetic processes (as shown in the “Introduction” section). Sulfa drugs and DHFR inhibitors are commonly used in combination as antibacterial drugs, just to avoid the drug-resistance problems typical of these pharmacologic agentsCitation57,Citation58.
Figure 5. Chemical structure of the DHFR inhibitor trimethoprim 12. Its congeners (brodimoprim, tetroxoprim, etc.) have slightly different moieties replacing the 4-methoxy group of 12Citation57. The new DHFR inhibitor iclaprim 13 is also shownCitation57.
CA inhibitors with antibacterial activity
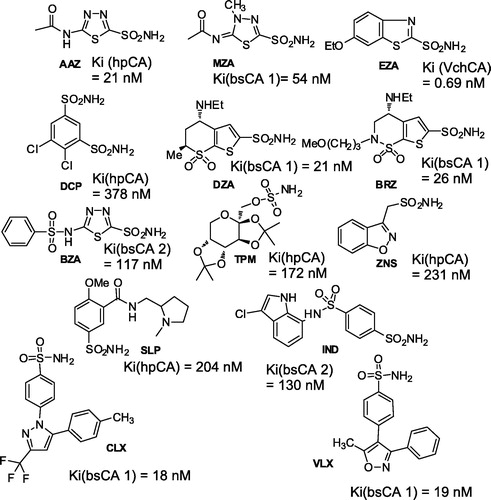
One of the best studied bacterial α-CA is the one from the gastric pathogen provoking ulcer and gastric cancer, H. pylori, hpαCACitation59,Citation60. The genome project of H. pylori identified, in fact, two different classes of CAs, with different subcellular localization: a periplasmic α-class CA (hpαCA) and a cytoplasmic β-class CA (hpβCA)Citation59,Citation60. These two CAs were shown to be catalytically efficient with almost identical activity to that of the human isoform hCA I, for the CO2 hydration reaction, and highly inhibited by many sulfonamides/sulfamates, including acetazolamide, ethoxzolamide, topiramate and sulpiride, all clinically used drugs ()Citation59,Citation60. Furthermore, certain CAIs, such as acetazolamide and methazolamide, were shown to inhibit the bacterial growth in cell culturesCitation59,Citation60. Since the efficacy of H. pylori eradication therapies currently employed has been decreasing due to drug resistance and side effects of the commonly used drugs, the dual inhibition of α- and/or β-CAs of H. pylori could be applied as an alternative therapy in patients with Helicobacter pylori infection or for the preve of gastroduodenal diseases caused by this widespread pathogenCitation59,Citation60.
Figure 6. Inhibition data of hpCA (α isoform), bsCA 1, bsCA 2 and VchCA with sulfonamides/sulfamates in clinical useCitation12,Citation13,Citation59,Citation61.
As mentioned in the introduction, the β-CA class is most widespread in bacteriaCitation8,Citation10,Citation21. The proof-of-concept study that such an enzyme may be a drug target has been published by Nishimori et al.Citation59 who cloned and purified the H. pylori enzyme (hpβCA), showing that it is highly susceptible to be inhibited by sulfonamides and sulfamates, of the same type as those mentioned above for hpαCA ().
Recently, Capasso’s groupCitation61,Citation62 reported the purification and characterization of another interesting enzyme, the α-CA from V. cholerae (denominated VchCA), a Gram-negative bacterium, which is the causative agent of cholera. This bacterium colonizes the upper small intestine where sodium bicarbonate is present at a high concentration. Sodium bicarbonate is an inducer of virulence gene expression. V. cholerae utilizes the CA system to accumulate bicarbonate into its cells, suggesting a pivotal role of this metalloenzymes in the microbial virulence. VchCA showed a low esterase activity with 4-nitrophenyl acetate as substrate, and a high activity for the hydration of CO2 to bicarbonateCitation61,Citation62. The inhibition study with sulfonamides and sulfamates led to the detection of a large number of low nanomolar inhibitors, among which are methazolamide, acetazolamide, ethoxzolamide, dorzolamide, brinzolamide, benzolamide and indisulam (KI values in the range 0.69–8.1 nM) (). As bicarbonate is a virulence factor of this bacterium and since ethoxzolamide was shown to inhibit the in vivo virulence, it was proposed that VchCA may be a target for antibiotic development, exploiting a mechanism of action rarely considered until nowCitation61,Citation62. Some glycosylsulfanilamide derivatives (in addition to the sulfonamides/sulfamates shown in ) were also shown to have potent antibacterial effects, in vivo against Brucella suis, inhibiting the growth of the pathogen in culturesCitation63. These compounds act as effective, nanomolar inhibitors of the two β-CAs present in this pathogen, bsCA 1 and bsCA 2Citation12,Citation13.
Molecular mechanisms of drug resistance
The molecular mechanisms of drug resistance both to sulfa drugsCitation58 and TMP-like DHFR inhibitorsCitation52,Citation57 are understood in detail due to the fact that several X-ray crystal structures of DHPS and/or DHFR (both wild-type and mutated proteins) are available, in complex with substrates and/or inhibitors.
Resistance to sulfa drugs
Crystallographic work with Bacillus anthracis (BaDHPS) and Yersinia pestis (YpDHPS) DHPS, complexed with DHPP 1, pABA 2 and the pABA analog p-hydroxybenzoic acid, afforded a deep understanding of the catalytic, inhibition and resistance mechanisms to sulfa drugs of this enzymeCitation58. First, the reaction between 1 and 2 occurs not via an SN-2 like but via an SN-1 reaction mechanism with formation of a DHP carbocation (at C-9) which then attacks the weakly nucleophilic amino group of pABA (or its analog p-hydroxybenzoic acid), with formation of DHP 3 (the adduct of 3 and BaDHPS has been isolated and characterized at atomic level). Furthermore, when BaDHPS was soaked in DHPP 1 and sulfathiazole 11, the covalent compound 14 () was observed in the X-ray crystal structure, bound within the active site of the enzyme and stabilized by the so-called loop 1–loop 2 substructure of the proteinCitation58. shows the detailed interactions between the covalent adduct 14 and the enzyme and also the amino acid residues involved in the sulfa drug resistance.
Indeed, the same crystallographic study from White’s groupCitation58 showed that in the YpDHPS – sulfa drug complexes, the sulfa drug perfectly fits the pABA binding pocket, with the SO2 oxygens matching the COOH group of pABA, and the phenylene moieties of the two type of compounds being superposable and engaging the same hydrophobic pocket in the loop 1 – loop 2 substructure of the enzymeCitation58. The resistance mutations in DHPS all cluster around this substructure, and comprise residues Phe33, Thr67 and Pro69. These amino acids form key elements for the pABA binding site. The sulfa drugs have been observed positioned (with their heterocyclic ring, which is attached to the SO2NH moiety) outside the DHPS substrate envelope, being located in such a way that mutations at Phe33 and pro69 can impede the binding of the drugCitation58. Thus, the presence of the heterocyclic ring (or the acetamido moiety in 10) in the sulfa drugs (which has no counterpart in the substrate molecule, pABA) leads to the generation of mutations within the DHPS active site and the consequent drug-resistance problems.
Resistance to TMP-like DHFR inhibitors
TMP is a synthetic drug that was introduced for clinical use in Western Europe in the early 1960sCitation54. Although its structural similarity to DHF makes it a competitive inhibitor of the ubiquitous chromosomal DHFR in bacteria, fungi and protozoa, mammalian DHFRs are resistant to TMPCitation54. Therefore, the specificity and selectivity of this antifolate drug have led to its widespread use in the treatment of human infections. However, bacteria have developed resistance mechanisms including the acquisition of plasmid-derived versions of DHFR, leading to high-level resistance or the point mutations of the chromosomal DHFR, which have been demonstrated to confer intermediate resistanceCitation52.
Just a single amino acid substitution, Phe98 to Tyr98, in DHFR was shown to be the molecular origin of TMP 12 resistance (in Staphylococcus aureusCitation52). This active site amino acid substitution was found in all S. aureus TMP-resistant clinical isolates tested so far. Ternary complexes of the chromosomal S. aureus DHFR (SaDHFR) with methotrexate and TMP, in the presence of NADPH as well as that of mutant Phe98Tyr DHFR SaDHFR (F98Y) ternary folate-NADPH complex have been obtained and their structure determined by high-resolution X-ray crystallographyCitation52. Critical evidence concerning the resistance mechanism has also been provided by NMR spectral analyses of Citation15N-labelled TMP in the ternary complexes of both wild-type and mutant enzyme. These studies show that the mutation results in loss of a hydrogen bond between the 4-amino group of TMP and the carbonyl oxygen of Leu5. This mechanism of resistance is predominant in both transferable plasmid-encoded and non-transferable chromosomally encoded resistanceCitation52. Knowledge of the resistance mechanism at a molecular level could help in the design of antibacterials against multi-resistant Staphylococcus aureus (MRSA), one of today’s most serious problems in clinical infectology, which is, in fact, not yet resolved.
Resistance to CAIs
Resistance to the sulfonamide or sulfamate CAIs has not been reported up until now.
Clinical impact of resistance to sulfa drugs and DHFR inhibitors
The wide use (and misuse) of antibacterial treatment led to the selection for bacterial strains with physiologically or genetically enhanced capacity to survive high doses of antibacterials of several pharmacological classes including the DHFR inhibitors and the sulfa drugsCitation52,Citation58,Citation64,Citation65. The biochemical mechanisms by which these processes occur have been outlined earlier.
Co-trimoxazole, a combination of TMP 12 and sulfamethoxazole 8 (in the ratio of 1:5) is still widely clinically used, although significant resistance problems to both drugs emergedCitation64,Citation65. The synergistic effects of the two drugs are due to the fact they inhibit successive steps in folate synthesis ().
Presently, sulfa drugs/DHFR inhibitors are primarily used for the treatment of uncomplicated urinary tract infections (cystitis) (mainly TMP-sulfa-methoxazole), this regimen being preferred to the quinolones (even though approximately 20% of E. coli are resistant to co-trimoxazole)Citation66. Skin infections are sometimes treated with sulfacetamide or a sulfacetamide-silver(I) saltCitation67. With its greater efficacy against a limited number of bacteria, co-trimoxazole remains indicated for some infections; for example, it is used as prophylaxis in patients at risk for Pneumocystis jirovecii pneumonia and as therapy in Whipple's diseaseCitation64,Citation65,Citation68. Gram-positive bacteria are generally moderately susceptible to this combination therapyCitation68. However, these drugs were not combined, as far as we know, with CAIs which specifically and potently inhibit bacterial CAs, as those discussed here. It would be of interest to investigate whether such a combination of three different enzyme inhibitors has a more potent antibacterial effect compared to the separate compounds used alone. We hypothesize that a synergistic effect between these different classes of pharmacological agents may have a beneficial and strong antibacterial effect against some pathogens.
Conclusions
Since the introduction of the sulfa drugs into clinical use in the 1940s, large numbers of antibiotics have been developed, possessing a wide range of mechanisms of action. However, their extensive use and misuse raised a serious public health problem due to the emergence of multi-antibiotic resistant bacterial pathogens worldwide. There is thus a stringent need to develop new antibiotics to keep pace with the bacterial resistance. Recent advances in microbial genomics, synthetic organic chemistry and X-ray crystallography provided opportunities to identify novel antibacterial targets for the development of new classes of antibiotics and to design more potent antimicrobial compounds derived from existing antibiotics in clinical use for decades, such as the sulfa drugs or the TMP-like DHFR inhibitors. Although resistance to these pharmacological agents constitutes a serious problem, sulfa drugs and DHFR inhibitors still have a firm place in the antibacterials’ armamentarium and may lead to new agents in which the resistance problem may be overcome by structured-based drug design. Furthermore, the recent cloning and characterization of many CAs in pathogenic bacteria, and the proof-of-concept studies showing that these targets are druggable and can lead to the inhibition of growth of these bacteria, offer interesting new alternatives that were not yet exploited clinically. Indeed, low nanomolar CAIs targeting the α- and/or β-CAs from many pathogenic bacteria are available, and further studies are necessary to see whether they may be synergistic with other antibacterials, such as the sulfa drugs or the DHFR inhibitors. Undoubtedly, these are fascinating research fields that can lead to interesting developments in the antibacterial drug research.
Declaration of interest
The authors declare no conflict of interest.
This research was financed by a grant of the “Accordo di Programma CNR-MSE” and by two EU projects (Metoxia and Dynano).
Acknowledgements
The authors are grateful to Dr Atilla Akdemir, Bezliaalem University, Istanbul, Turkey, for generating for us.
References
- Kronholm S, Capel P. Concentrations, loads, and yields of organic carbon in streams of agricultural watersheds. J Environ Qual 2012;41:1874–83
- Wingate L, Ogee J, Cuntz M, et al. The impact of soil microorganisms on the global budget of delta18O in atmospheric CO2. Proc Natl Acad Sci USA 2009;106:22411–15
- Freeman C, Kim SY, Lee SH, et al. Effects of elevated atmospheric CO2 concentrations on soil microorganisms. J Microbiol 2004;42:267–77
- Johnson X, Alric J. Interaction between starch breakdown, acetate assimilation, and photosynthetic cyclic electron flow in Chlamydomonas reinhardtii. J Biol Chem 2012;287:26445–52
- Tcherkez G, Boex-Fontvieille E, Mahe A, et al. Respiratory carbon fluxes in leaves. Curr Opin Plant Biol 2012;15:308–14
- Casey JR. Why bicarbonate? Biochem Cell Biol 2006;84:930–9
- Kozliak EI, Fuchs JA, Guilloton MB, et al. Role of bicarbonate/CO2 in the inhibition of Escherichia coli growth by cyanate. J Bacteriol 1995;177:3213–19
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discover 2008;7:168–81
- Supuran CT, Casini A, Scozzafava A. Protease inhibitors of the sulfonamide type: anticancer, antiinflammatory, and antiviral agents. Med Res Rev 2003;23:535–58
- Smith KS, Ferry JG. Prokaryotic carbonic anhydrases. FEMS Microbiol Rev 2000;24:335–66
- Maeda S, Price GD, Badger MR, et al. Bicarbonate binding activity of the CmpA protein of the cyanobacterium Synechococcus sp. strain PCC 7942 involved in active transport of bicarbonate. J Biol Chem 2000;275:20551–5
- Joseph P, Ouahrani-Bettache S, Montero JL, et al. A new beta-carbonic anhydrase from Brucella suis, its cloning, characterization, and inhibition with sulfonamides and sulfamates, leading to impaired pathogen growth. Bioorg Med Chem 2011;19:1172–8
- Joseph P, Turtaut F, Ouahrani-Bettache S, et al. Cloning, characterization, and inhibition studies of a beta-carbonic anhydrase from Brucella suis. J Med Chem 2010;53:2277–85
- Djapa LY, Zelikson R, Delahodde A, et al. Plasmodium vivax dihydrofolate reductase as a target of sulpha drugs. FEMS Microbiol Lett 2006;256:105–11
- Hartman PG. Molecular aspects and mechanism of action of dihydrofolate reductase inhibitors. J Chemother 1993;5:369–76
- Chen MJ, Shimada T, Moulton AD, et al. The functional human dihydrofolate reductase gene. J Biol Chem 1984;259:3933–43
- Sohn KJ, Jang H, Campan M, et al. The methylenetetrahydrofolate reductase C677T mutation induces cell-specific changes in genomic DNA methylation and uracil misincorporation: a possible molecular basis for the site-specific cancer risk modification. Int J Cancer 2009;124:1999–2005
- Khalil I, Ronn AM, Alifrangis M, et al. Dihydrofolate reductase and dihydropteroate synthase genotypes associated with in vitro resistance of Plasmodium falciparum to pyrimethamine, trimethoprim, sulfadoxine, and sulfamethoxazole. Am J Trop Med Hyg 2003;68:586–9
- Mockenhaupt FP, Teun Bousema J, Eggelte TA, et al. Plasmodium falciparum DHFR but not DHPS mutations associated with sulphadoxine-pyrimethamine treatment failure and gametocyte carriage in northern Ghana. Trop Med Int Health 2005;10:901–8
- Benkovic SJ, Fierke CA, Naylor AM. Insights into enzyme function from studies on mutants of dihydrofolate reductase. Science 1988;239:1105–10
- Supuran CT. Bacterial carbonic anhydrases as drug targets: toward novel antibiotics? Front Pharmacol 2011;2:1--6
- Krungkrai J, Supuran CT. The alpha-carbonic anhydrase from the malaria parasite and its inhibition. Curr Pharm Des 2008;14:631–40
- Reungprapavut S, Krungkrai SR, Krungkrai J. Plasmodium falciparum carbonic anhydrase is a possible target for malaria chemotherapy. J Enzyme Inhib Med Chem 2004;19:249–56
- Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2010;20:3467–74
- Supuran CT. Indisulam: an anticancer sulfonamide in clinical development. Expert Opin Investig Drugs 2003;12:283–7
- Supuran CT. Carbonic anhydrases as drug targets—an overview. Curr Top Med Chem 2007;7:825–33
- Supuran CT. Antiepileptic activity of zonisamide on hippocampal CA3 neurons does not depend on carbonic anhydrase inhibition. Epilepsy Res 2008;82:109, author reply 10
- Supuran CT. Carbonic anhydrase inhibition/activation: trip of a scientist around the world in the search of novel chemotypes and drug targets. Curr Pharm Des 2010;16:3233–45
- Brown GM. The biosynthesis of folic acid. II. Inhibition by sulfonamides. J Biol Chem 1962;237:536–40
- Shiota T, Disraely MN, McCann MP. The enzymatic synthesis of folate-like compounds from hydroxymethyldihydropteridine pyrophosphate. J Biol Chem 1964;239:2259–66
- Shiota T, Baugh CM, Jackson R, et al. The enzymatic synthesis of hydroxymethyldihydropteridine pyrophosphate and dihydrofolate. Biochemistry 1969;8:5022–8
- Valderas MW, Andi B, Barrow WW, et al. Examination of intrinsic sulfonamide resistance in Bacillus anthracis: a novel assay for dihydropteroate synthase. Biochim Biophys Acta 2008;1780:848–53
- Lopez P, Espinosa M, Greenberg B, et al. Sulfonamide resistance in Streptococcus pneumoniae: DNA sequence of the gene encoding dihydropteroate synthase and characterization of the enzyme. J Bacteriol 1987;169:4320–6
- Slock J, Stahly DP, Han CY, et al. An apparent Bacillus subtilis folic acid biosynthetic operon containing pab, an amphibolic trpG gene, a third gene required for synthesis of para-aminobenzoic acid, and the dihydropteroate synthase gene. J Bacteriol 1990;172:7211–26
- Radstrom P, Swedberg G. RSF1010 and a conjugative plasmid contain sulII, one of two known genes for plasmid-borne sulfonamide resistance dihydropteroate synthase. Antimicrob Agents Chemother 1988;32:1684–92
- Sundstrom L, Radstrom P, Swedberg G, et al. Site-specific recombination promotes linkage between trimethoprim- and sulfonamide resistance genes. Sequence characterization of dhfrV and sulI and a recombination active locus of Tn21. Mol Gen Genet 1988;213:191–201
- Morgan RE, Batot GO, Dement JM, et al. Crystal structures of Burkholderia cenocepacia dihydropteroate synthase in the apo-form and complexed with the product 7,8-dihydropteroate. BMC Struct Biol 2011;11:1--9
- Blakley RL. Eukaryotic dihydrofolate reductase. Adv Enzymol Relat Areas Mol Biol 1995;70:23–102
- Heaslet H, Harris M, Fahnoe K, et al. Structural comparison of chromosomal and exogenous dihydrofolate reductase from Staphylococcus aureus in complex with the potent inhibitor trimethoprim. Proteins 2009;76:706–17
- Pan YC, Domin BA, Li SS, et al. Studies of amino-acid sequence in dihydrofolate reductase from a human methotrexate-resistant cell line KB/6b. Structural and kinetic comparison with mouse L1210 enzyme. Eur J Biochem 1983;132:351–9
- Kovalevskaya NV, Smurnyy YD, Polshakov VI, et al. Solution structure of human dihydrofolate reductase in its complex with trimethoprim and NADPH. J Biomol NMR 2005;33:69–72
- Cody V, Luft JR, Ciszak E, et al. Crystal structure determination at 2.3 A of recombinant human dihydrofolate reductase ternary complex with NADPH and methotrexate-gamma-tetrazole. Anticancer Drug Des 1992;7:483–91
- Cody V, Wojtczak A, Kalman TI, et al. Conformational analysis of human dihydrofolate reductase inhibitor complexes: crystal structure determination of wild type and F31 mutant binary and ternary inhibitor complexes. Adv Exp Med Biol 1993;338:481–6
- Jensen DE, Black AR, Swick AG, et al. Distinct roles for Sp1 and E2F sites in the growth/cell cycle regulation of the DHFR promoter. J Cell Biochem 1997;67:24–31
- Curtius HC, Heintel D, Ghisla S, et al. Tetrahydrobiopterin biosynthesis. Studies with specifically labeled (2H)NAD(P)H and 2H2O and of the enzymes involved. Eur J Biochem 1985;148:413–19
- Kim HL, Choi YK, Kim do H, et al. Tetrahydropteridine deficiency impairs mitochondrial function in Dictyostelium discoideum Ax2. FEBS Lett 2007;581:5430–4
- Channon KM. Tetrahydrobiopterin: regulator of endothelial nitric oxide synthase in vascular disease. Trends Cardiovasc Med 2004;14:323–7
- Huovinen P, Sundstrom L, Swedberg G, et al. Trimethoprim and sulfonamide resistance. Antimicrob Agents Chemother 1995;39:279–89
- Huovinen RL, Alanen KA, Collan YU. Cell proliferation in dimethylbenz(A)anthracene(DMBA)-induced rat mammary carcinoma treated with antiestrogen toremifene. Acta Oncol 1995;34:479–85
- Kehrenberg C, Schwarz S. dfrA20, A novel trimethoprim resistance gene from Pasteurella multocida. Antimicrob Agents Chemother 2005;49:414–17
- Alonso H, Gready JE. Integron-sequestered dihydrofolate reductase: a recently redeployed enzyme. Trends Microbiol 2006;14:236–42
- Dale GE, Broger C, D'Arcy A, et al. A single amino acid substitution in Staphylococcus aureus dihydrofolate reductase determines trimethoprim resistance. J Mol Biol 1997;266:23–30
- Howell EE. Searching sequence space: two different approaches to dihydrofolate reductase catalysis. Chem Biochem 2005;6:590–600
- Skold O. Resistance to trimethoprim and sulfonamides. Vet Res 2001;32:261–73
- Domagk G. [Experimental bases in chemotherapy of bacterial infections with sulfonamides and related substances with special reference to its application in surgery]. Langenbecks Archiv fur klinische Chirurgie vereinigt mit Deutsche Zeitschrift fur Chirurgie 1950;264:102–23
- Tréfouël JT, Nitti F, Bovet D. Activité du p.aminophénylsulfamide sur l’infection streptococcique expérimentale de la souris et du lapin. C R Soc Biol 1935;120:756–61
- Hawser S, Lociuro S, Islam K. Dihydrofolate reductase inhibitors as antibacterial agents. Biochem Pharmacol 2006;71:941–8
- Yun MK, Wu Y, Li Z, et al. Catalysis and sulfa drug resistance in dihydropteroate synthase. Science 2012;335:1110–14
- Nishimori I, Minakuchi T, Morimoto K, et al. Carbonic anhydrase inhibitors: DNA cloning and inhibition studies of the alpha-carbonic anhydrase from Helicobacter pylori, a new target for developing sulfonamide and sulfamate gastric drugs. J Med Chem 2006;49:2117–26
- Chirica LC, Petersson C, Hurtig M, et al. Expression and localization of alpha- and beta-carbonic anhydrase in Helicobacter pylori. Biochim Biophys Acta 2002;1601:192–9
- Del Prete S, Isik S, Vullo D, et al. DNA cloning, characterization, and inhibition studies of an alpha-carbonic anhydrase from the pathogenic bacterium Vibrio cholerae. J Med Chem 2012;55:10742–8
- Vullo D, Isik S, Del Prete S, et al. Anion inhibition studies of the alpha-carbonic anhydrase from the pathogenic bacterium Vibrio cholerae. Bioorg Med Chem Lett 2013;23:1636–8
- Vullo D, Nishimori I, Scozzafava A, et al. Inhibition studies of a beta-carbonic anhydrase from Brucella suis with a series of water soluble glycosyl sulfanilamides. Bioorg Med Chem Lett 2010;20:2178–82
- Dini L, du Plessis M, Frean J, et al. High prevalence of dihydropteroate synthase mutations in Pneumocystis jirovecii isolated from patients with Pneumocystis pneumonia in South Africa. J Clin Microbiol 2010;48:2016–21
- Tadesse DA, Zhao S, Tong E, et al. Antimicrobial drug resistance in Escherichia coli from humans and food animals, United States, 1950–2002. Emerg Infect Dis 2012;18:741–9
- Tauber MG, Muhlemann K. Antibiotic therapy in the outpatient setting: update 2009. Praxis 2009;98:877–83
- Mastrolorenzo A, Supuran CT. Antifungal activity of Ag(I) and Zn(II) complexes of sulfacetamide derivatives. Met Based Drugs 2000;7:49–54
- Yoneyama H, Katsumata R. Antibiotic resistance in bacteria and its future for novel antibiotic development. Biosci Biotechnol Biochem 2006;70:1060–75