
Abstract
This study is based on our attempts to further explore the structure–activity relationship (SAR) of VX-148 (3) in an attempt to identify inosine 5′-mono-phosphate dehydrogenase (IMPDH) inhibitors superior to mycophenolic acid. A five-point pharmacophore developed using structurally diverse, known IMPDH inhibitors guided further design of novel analogs of 3. Several conventional as well as novel medicinal chemistry strategies were tried. The combined structure- and ligand-based approaches culminated in a few analogs with either retained or slightly higher potency. The compounds which retained the potency were also checked for their ability to inhibit human peripheral blood mononuclear cells proliferation. This study illuminates the stringent structural requirements and strict SAR for IMPDH II inhibition.
Introduction
Inosine 5′-monophosphate dehydrogenase (IMPDH, EC 1.1.1.205) catalyzes a crucial step in the de novo synthesis of guanine nucleotides. It oxidizes inosine 5′-monophosphate (IMP) to xanthosine 5′-monophosphate (XMP) in a nicotinamide adenine dinucleotide (NAD+)-dependent mannerCitation1,Citation2. Owing to its critical position in the purine biosynthesis pathway, the inhibition of IMPDH (leading to reduced cellular levels of guanine nucleotides) is an important strategy for antiproliferative, antiviral and antiparasitic effectsCitation3. It is also a useful strategy for producing immunosuppression as IMPDH-dependent de novo synthesis of guanine nucleotides is responsible for maintaining the growth and differentiation of human B and T lymphocytesCitation4.
Two isoforms of human IMPDH, I and II (514 amino acids each, 84% sequence identity) have been identifiedCitation5. IMPDH I is constitutively expressed and the majority of this isoform is found in normal lymphocytes, whereas type II is inducible and is closely linked to cell differentiation and neoplastic transformationCitation4. There is some ambiguity about the importance of one isoform over the other in terms of therapeutic utilityCitation6. Mycophenolic acid (MPA) (1, ), a natural product, is a reversible, potent and uncompetitive inhibitor of IMPDH. Mycophenolate mofetil (MMF, CellCept®), a prodrug of MPA, has been approved for the treatment of acute allograft rejection following kidney transplantCitation7. MPA and its related forms [MMF or MPA sodium (Myfortic®)] cause dose-limiting gastrointestinal (GI) toxicity in addition to their faster metabolic liability. Thus, high doses (CellCept®, 1 g b.i.d. and Myfortic®, 720 mg b.i.d.) are required to maintain the therapeutic plasma levelsCitation8. The competitive IMPDH inhibitors such as Mizoribine (Bredinin®) and Ribavirin (Virazole®) (after intracellular activation by phosphorylation) are nucleoside analogs and have unfavorable tolerability profiles tooCitation3. Thus, there is a need for newer, safer, potent and orally bioavailable IMPDH inhibitors (selective or nonselective). The outcome of this search may further help to deduce the importance of one isoform over the other in a particular disease state.
Figure 1. Chemical structures of select IMPDH inhibitors used for the pharmacophore model developmentCitation12.
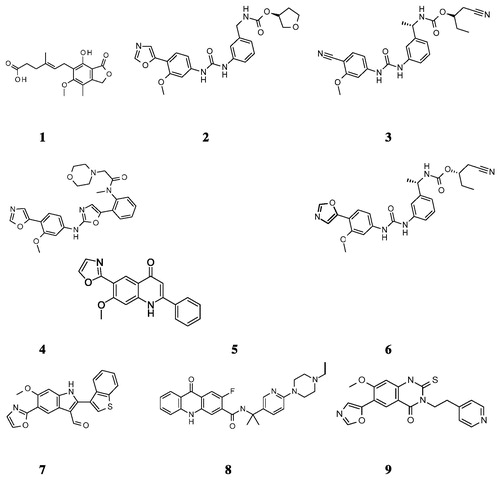
Nearly a decade ago, a new series of biaryl ureas exemplified by VX-497 (2, ) appeared in the literatureCitation9. This was followed by another round of structural modifications of 2 which led to VX-148 (3, )Citation10. Bioisosteric replacement of urea moiety in 3 led to a new class, N-aryl-2-aminooxazoles [e.g. BMS-337197 (4, )], of IMPDH inhibitorsCitation11. There are several reports of novel and potent IMPDH inhibitors with varied pharmacokinetic and pharmacodynamic profilesCitation12,Citation13. Despite the availability of several potent IMPDH inhibitors (), the quest for new inhibitors continues owing to the involvement of the target in several disease states.
This study focuses on our in-house efforts to develop novel IMPDH inhibitors, which will be devoid of the side effect profiles of MPA and related drugs. These novel inhibitors can be potentially useful in several diseases/disorders (e.g. arthritis, inflammatory bowel disease, psoriasis, dermatitis, prevention of transplant rejection, etc.) in which immunosuppression is desired.
Design strategy
Recently, a chemical feature-based pharmacophore model of IMPDH inhibitors consisting of six features (one H-bond donor, one H-bond acceptor, one aromatic ring, one hydrophobic and two excluded volumes) appeared in the literatureCitation14. Further, the pharmacophore features were confirmed by molecular docking analyses. In the present study, the molecular docking studies were initially performed using IMPDH II crystal structure (PDB ID 1JR1)Citation6 to generate the “binding poses”, which were subsequently used as an input for developing the pharmacophore model. A total of nine potent and structurally diverse IMPDH inhibitors belonging to classes biarylurea (2, 3 and 5, Charts 1 and 2), oxazolamines (4, ), quinolone (6, ), indole (7, ), acridone (8, ) and quinazoline thione (9, ) in addition to 1 were used in the process. The figures depicting the binding modes of few of these inhibitors (2 and 3) can be found in the Supplementary material section. A five-point pharmacophore model was selected based on: (a) the mutual agreement between the observed features and the corresponding interactions with IMPDH II residues and (b) pharmacophore fitness score and related statistics. The model AADHR (A – Acceptor; D – Donor; H – Hydrophobic; R – Ring features) along with the interfeature distances is shown in .
Figure 2. The five-point pharmacophore model – AADHR – for IMPDH inhibitors with VX-148 (3, ) and the design strategy based on 3 (Parts A, B and C) utilized for systematic modifications. A: Acceptor (light pink); D: Donor (cyan); H: Hydrophobic (green) and R: Ring (orange) features.
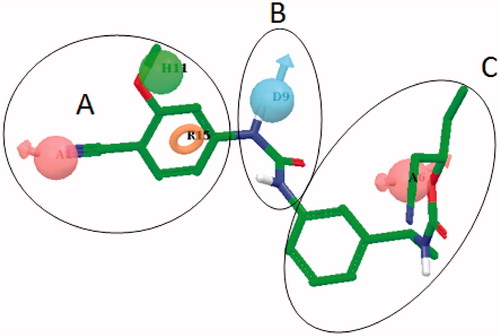
Compound 3 was selected for further investigations based on the in-house early absorption, distribution, metabolism and excretion (eADME) evaluations (data not shown). In fact, the oxazole-containing compounds were excluded because they were found to inhibit CYP 3A4 and CYP 2D6 significantly (data not shown). Compound 3 was divided into three parts – proximal (phenyl ring with 4–CN and 3–OMe substitutions), middle (urea) and the distal (remaining part) (). Given the importance of the H-bond acceptor feature (), several Part A modifications were tried to probe the binding cavity features () with the retention of at least one H-bond acceptor feature. The distances between the A and R features () spanned a greater range (data not shown) compared to the corresponding distance in the pharmacophore model (5.08 Å). Upon obtaining the results from Part A modifications, significant synthetic efforts were invested in finding a suitable urea replacement (Part B modifications). The combined results of Part A and B modifications along with the clues from the pharmacophore model were utilized to design suitable Part C modifications. Herein, our attempts to develop novel and potent IMPDH inhibitors based on compound 3 with the help of ligand-based and partly structure-based design strategies are summarized.
Table 1. In vitro inhibitory and cellular potencies of Part A modifications.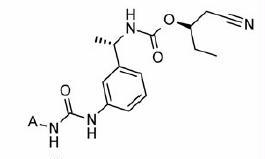
Materials and methods
General
All the chemicals (intermediates and reagents) and solvents (including dry solvents) were procured from commercial suppliers such as Sigma-Aldrich Chemical Co. (St. Louis, MO), Thermo Fisher Scientific Inc. (Waltham, MA), VWR International (Radnor, PA), etc., and were used as received unless otherwise indicated. All reactions were performed under an inert atmosphere (argon or N2) unless otherwise noted. Analytical silica gel 60 F254-coated thin-layer chromatography (TLC) plates were purchased from Merck Chemicals (Whitehouse Station, NJ), and were visualized with UV light or by treatment with TLC reagents such as ninhydrin, Dragandorff’s or phosphomolybdic acid (PMA). Flash-column chromatography was carried out on Combiflash Rf _silica gel (230–400 mesh). 1H-NMR spectra were routinely recorded on Bruker 300 MHz FT NMR, with tetramethylsilane (TMS) as an internal standard. Reversed-phase high-performance liquid chromatography (RP-HPLC) analyses were performed with a Waters Acquity UPLC instrument (Waters, Milfort, MA) equipped with Waters PDA absorption detector. Analytical ultra-performance liquid chromatography (UPLC) analyses were carried out with a Waters BED C18 column (100 × 50 mm × 5 µm) using acetonitrile:water solvent system (flow rate 1 mL/min, injection volume 10 µL). The gradient used was – water 80:acetonitrile 20 (0 min) to water 20:acetonitrile 80 (20 min). The detection was done using UV/Vis detector (λmax 254 nm). For semipreparative analyses, a Waters Xtera BED C18 column (100 × 50 mm × 5 µm) was used. The optical purity of chiral amine was determined using a Thermo GC instrument (Thermo Electron Corporation, San Jose, CA). Analytical gas chromatography (GC) analyses were carried out with a SUPELCO 30 M × 0.25 mm × 0.25 µm column (Sigma-Aldrich Chemical Co., St. Louis, MO). Optical rotations were recorded on a PerkinElmer 241 polarimeter (PerkinElmer, Waltham, MA). Melting points were recorded using a MEL-TEMP II capillary melting point apparatus (Laboratory Devices Inc., Holliston, MA), and are uncorrected.
cDNA coding for IMPDH II (accession number BC0155) (UniProt P12268 IMDH2_HUMAN) was obtained from Open Biosystems and cloned into pET21 Eschericia coli expression vector. Escherichia coli DH5α, E. coli DE3 (BL21) and plasmid pET-21b were purchased from Novagen (Billerica, MA). Restriction enzymes, Pfu DNA Polymerase, DNA marker were purchased from NEB. Plasmid DNA purification kit was purchased from Qiagen (Hilden, Germany). Oligonucleotides, Protease inhibitor cocktail, IMP, NAD, guanine monophosphate (GMP), MPA and routine chemicals were obtained from Sigma-Aldrich Chemical Co. The 96-well plate for UV assay was purchased from Costar.
Chemistry
The syntheses of the final compounds are outlined in . Synthetic details of the representative compounds 13, 30 and 45 are shown for reference. Similar details for all other compounds are found in the Supplementary material section.
Scheme 1. Syntheses of compounds 13–27. Reagents and conditions: (a) NaBH4, EtOH, RT; (b) DPPA, DBU, toluene; (c) PPh3, THF:Water; (d) L-(+)-tartaric acid, MeOH; (e) NaOH, EtOAc; (f) H2, Pd(OH)2/C, EtOH; (g) KCN, DMSO; (h) 10, CDI, EtOAc, RT; (i) phenyl chloroformate, DCM, RT; (j) DIPEA, EtOAc, RT; (k) DCM, RT, 6 h.
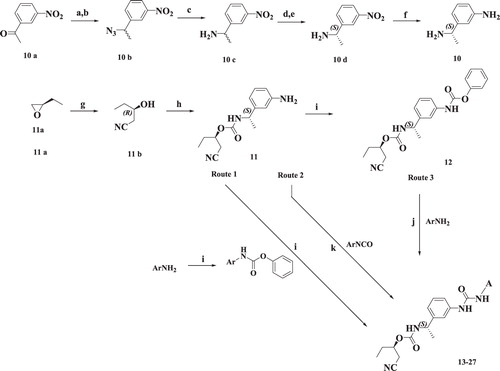
Scheme 2. Syntheses of compounds 30–35. Reagents and conditions: (a) DCM, RT; (b) (i) CuSO4:SiO2; (ii) methanolic NH3, THF; (c) MeI, K2CO3, acetone; (d) NaNHCN, 2-propanol, 80 °C, 1 h; (e)1,1′-thiocarbonyldi-2(1H)-pyridone, DCM, RT; (f) (i) HCl:1,4-dioxane, RT; (ii) 11b, CDI, EtOAc, RT; (g) MeI, DMF, RT; (h) EtOH, reflux.
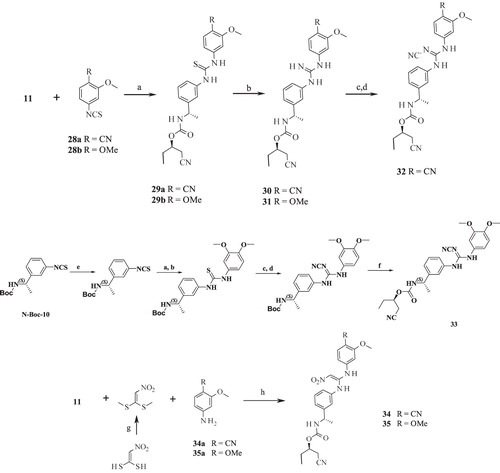
Scheme 3. Syntheses of compounds 36–39. Reagents and conditions: (a) KI (cat.), AcCN, RT; (b) HATU, DIPEA, THF, RT.
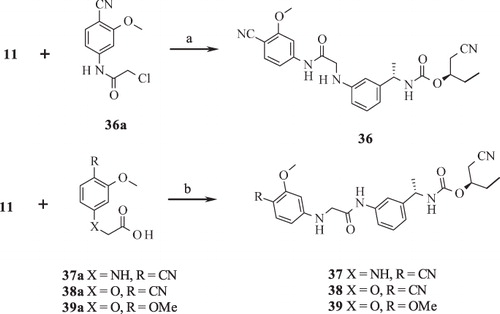
Scheme 4. Syntheses of compounds 40–44. Reagents and conditions: (a) NaH, THF, reflux; (b) DIPEA, THF, RT; (c) 1,1′-carbonothionyldipyri-din-2(1H)-one, DCM, RT; (d) silver trifluoroacetate, TEA, AcCN, reflux; (e) HATU, DIPEA, THF, RT; (f) (i) DCM, RT; (ii) HgO, S, EtOH, reflux.
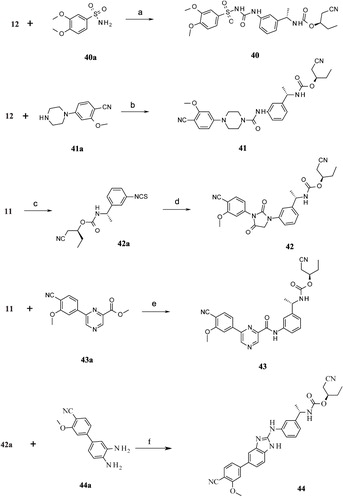
Scheme 5. Syntheses of compounds 45–53. Reagents and conditions: (a) (i) DCM, RT; (ii) MeI, K2CO3, RT; (b) NaNHCN, 2-propanol, reflux; (c) LiOH, THF:H2O (2:1), RT.
General procedure 1 (Route 1)
To a solution of intermediate 12 (190 mg, 0.5 mmol) and the corresponding aromatic amine (0.5 mmol) in EtOAc (5 mL) was added N,N-diisopropylethylamine (0.16 mL, 1 mmol) dropwise. The reaction mixture was refluxed overnight, cooled to room temperature (RT) and washed with water (3 × 10 mL). Combined organic layers were separated and dried (anhydrous Na2SO4). The solvent was evaporated and the crude product was purified by flash chromatography.
General procedure 2 (Route 2)
To a solution of intermediate 11 (200 mg, 0.77 mmol) in DCM (5 mL) at 0 °C was added corresponding isocyanate (0.8 mmol) and the reaction mixture stirred at RT for 4 h. The reaction was monitored by TLC. Reaction mixture was extracted with water (3 × 10 mL). Organic layers were dried (anhydrous Na2SO4). The crude product obtained after removing the solvent was purified using flash chromatography.
General procedure 3 (Route 3)
Compound 11 (200 mg, 0.77 mmol) and the respective carbamate (0.8 mmol) were dissolved in EtOAc (5 mL). To this solution was added N,N-diisopropylethylamine (0.16 mL, 1 mmol) dropwise. The reaction mixture was refluxed overnight and cooled to RT. The reaction was terminated by adding water (10 mL). The organic layer was separated and the aqueous layer was further extracted with EtOAc (2 × 10 mL). Combined organic phase was dried (anhydrous Na2SO4) and the solvent evaporated to obtain crude product that was further purified by flash chromatography.
(2R)-1-Cyanobutan-2-yl-[(1S)-1-(3-{[(3,4-dimethoxyphenyl)carbamoyl]amino}phenyl)- ethyl] carbamate (13)
General procedure 1 was used. Off-white solid; yield of 13: 100 mg (40%); Rf = 0.25 (DCM:MeOH 9:1); purity (HPLC) = 92%; LC-MS (ESI) (M + H)+: 441.17; HRMS (ESI) (M + H)+ calcd for C23H29N4O5 441.2129, found 441.2132; 1H-NMR (300 MHz, DMSO-d6) δ ppm 0.90 (t, J = 7.4 Hz, 3H), 1.34 (d, J = 6.8 Hz, 3H), 1.56–1.70 (m, 2H), 2.84 (d, J = 4.9 Hz, 2H), 3.71 (s,3H), 3.74 (s, 3H), 4.56–4.74 (m, 2H), 6.86 (s, 2H), 6.92 (d, J = 7.6 Hz, 1H), 7.17–7.24 (m, 2H), 7.26–7.34 (m, 1H), 7.38 (s, 1H), 7.90 (d, J = 7.9 Hz, 1H), 8.48 (s, 1H), 8.54 (s, 1H).
(2R)-1-Cyanobutan-2-yl[(1S)-1-{3-[N′-(4-cyano-3-methoxyphenyl)carbamimidamino]-phenyl}ethyl] carbamate (30)
Intermediate 29a (250 mg, 0.55 mmol) (see Supplementary material), NEt3 (0.77 mL, 0.554 mmol), anhydrous CuSO4 (1 g), SiO2 (1 g) in THF (5 mL) was stirred at RT for 30 min. Methanolic NH3 (5 mL) was added and the reaction mixture was stirred at RT for another 30 min. It was filtered through celite and filtrate was concentrated in vacuo. Crude product was purified by column chromatography (DCM:MeOH, 95:5) to produce 30 as yellow solid. Yield of 30: 50 mg (23%); Rf = 0.25 (DCM:MeOH, 95:5); purity (HPLC) = 90%; LC-MS (ESI) (M + H)+: 435.25; HRMS (ESI) (M + H)+ calcd for C23H27N6O3 435.2137, found 435.2139; 1H NMR (DMSO-d6) δ ppm 0.87–0.90 (t, J = 7.4 Hz, 3H), 1.3–1.5 (m, 3H), 1.6–1.8 (m, 2H), 2.73–2.8 (d, J = 5.3 Hz, 2H), 3.82 (s, 4H), 4.586 (m, 2H), 5.46 (d, J = 6.0 Hz, 2H), 6.56 (brs, 2H), 6.86–6.88 (d, J = 6.8 Hz, 2H), 7.13–7.16 (t, J = 7.9 Hz, 2H), 7.44–7.47 (d, J = 8.3 Hz, 2H), 7.81–7.83 (d, J = 7.9 Hz, 2H).
General procedure 4
To a solution of isothiocynate (1 mmol) () in DCM (5 mL) amine (1 mmol) was added slowly. Reaction mixture was stirred at RT for 6 h and extracted with water (3 × 5 mL). Combined organic layers were dried (anhydrous Na2SO4). Crude thiourea was purified using flash chromatography. To a solution of thiourea (1 mmol) in acetone (5 mL), MeI (2.5 mmol) and K2CO3 (345 mg, 2.5 mmol) were added. The contents of the flask were stirred at RT for 4 h and the solvent evaporated in vacuo. The residue was taken up in EtOAc (10 mL) and washed with water (3 × 10 mL). Organic layer was dried (anhydrous Na2SO4) and crude product was purified using flash chromatography to yield a colorless liquid. To a solution of this S-methyl intermediate (1 mmol) in IPA (5 mL), sodium cyanamide (160 mg, 2.5 mmol) was added and the reaction mixture was refluxed for 4 h, cooled to RT and the solvent evaporated in vacuo. The residue was dissolved in EtOAc (10 mL) and washed with water (3 × 10 mL). Organic layer was dried (anhydrous Na2SO4) and the solvent evaporated in vacuo and the crude product was subjected to purification by flash chromatography.
Scheme 6. Syntheses of compounds 54–60. Reagents and conditions: (a) 3-Bromoaniline, DCM, RT; (b) MeI, K2CO3, RT; (c) NaNHCN, 2-propanol, reflux; (d) substituted arylboronic acid, Cs2CO3, SPhos, Pd(OAc)2/EtOAc:toluene (1:1), 100 °C.
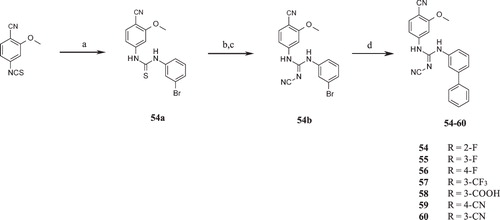
Isopropyl 2-[3-[[(E)-N′-cyano-N-(4-cyano-3-methoxyphenyl)carbamimidoyl]amino]phen-oxy]acetate (45)
Synthesized according to general procedure 4. Analytical data of 45: white solid; yield of 45: 200 mg (65%); Rf = 0.2 (hexanes:EtOAc 6.5:4.5); purity (HPLC) = 98%; LC-MS (ESI) (M + H)+: 408.25; HRMS (ESI) (M + H)+ calcd for C18H16N5O4 408.1661, found 408.1666; 1H NMR (DMSO-d6) δ ppm 1.11–1.40 (m, 6H), 1.99 (s, 1H), 3.6–3.85 (s, 3H), 4.71 (s, 2H), 4.84–5.12 (m, 1H), 6.62-6.80 (m, 1H), 6.88–6.98 (m, 2H), 7.02 (dd, J = 8.7, 1.9 Hz, 1H), 7.14 (d, J = 1.5 Hz, 1H), 7.26 (t, J = 8.3 Hz, 1H), 7.65 (d, J = 8.7 Hz, 1H), 9.88 (br. s., 1H), 9.92 (brs, 1H).
Molecular modeling
All the molecular modeling studies were performed on a Hewlett-Packard xw4600 computer workstation with (Hewlett-Packard, Palo Alto, CA) 2 GB memory and Intel Core® 2 Duo processors (Intel Corp., Santa Clara, CA) of 2.66 GHz each running under Red Hat Enterprise Linux (RHEL), version 5 (Red Hat, Inc., Raleigh, NC). The molecular modeling operations were performed using Schrödinger suite 2011Citation15.
lists the structures of IMPDH inhibitors used for the development of pharmacophore (IC50 6–30 nM). For developing the docking protocol, Chinese hamster (Cricetulus griseus) IMPDH II crystal structure (PDB ID 1JR1)Citation6 was used (details not shown). Phase, version 3.3Citation16,Citation17 was utilized for the development of the pharmacophore model with the top ligand poses from the docking runs as the starting point. The algorithm searches the common features and scores the hypotheses. The end result of the scoring process is an overall ranking of all hypotheses from which user can select best candidate hypothesis using several criteria such as the alignment of site points and vectors, selectivity, volume overlap, number of ligands matched, relative conformational energy and activity, etc.Citation16,Citation17. The top-scoring hypothesis consisting of five features (AADHR) was selected for evaluating new designs based on the template structure VX-148.
Screening, cloning, expression and purification of recombinant IMPDH II
Cloning strategy and primer designing
The sequences of the clones were verified with the Genebank sequence (accession number BC015567). Primers were designed to include restriction enzyme sites HindIII and XhoI to 5′- and 3′-ends, respectively. Primers were synthesized from Sigma genosys (Forw – GCGTCAAGCTTATGGCCGACTACCTG; Rev – AGCTGACCTCGAGGAAAAGCCGCTTCTC). PCR was performed using Phusion polymerase enzyme (Finnzyme) for 28 cycles as per the manufacturer’s instructions. Each cycle included 98 °C/10 s, 61 °C/30 s, 72 °C/90 s and final extension at 72 °C for 10 min. Amplified PCR product was gel purified and digested with HindIII and XhoI restriction enzymes. Digested PCR product was ligated into pET-21b (Novagen) vector and transformed into E. coli strain.
Selection and confirmation of clones
Clones from the antibiotic plate were selected and analyzed for presence of insert by colony PCR using the above mentioned protocol. Colony 1 resulted in expected band size of 1.6 kb and further plasmid was amplified from this clone and sequence was confirmed by automated DNA sequencing. No mutations were found after the sequence check. The verified clone was transformed into expression host BL21(DE3) PlysS for expression optimization.
Cells were grown to an OD600 of 0.3 in lysogeny broth medium supplemented with 100 µg/mL ampicillin at 37 °C. Protein expression was induced with the addition of 0.3 mM IPTG followed by incubation for 5 h at 225 rpm, 25 °C. Cells were harvested by centrifugation at 4000 rpm, corresponding to 3400g using HERAEUS Megafuge 1.0R (Kendro Laboratory Products GmbH, Hanau, Germany) for 30 min and lysed the cells by sonication at 70% amplitude, with a burst size of 5 min, 30 s on/off. The clarified cell lysate was subjected for 40% w/v ammonium sulfate fractionation and pelleted down the protein by spinning the contents at 10 000 rpm for 30 min at 4 °C. The pellet was suspended in KH2PO4 buffer and applied the sample on Sephadex G-25 column to desalt. These fractions were pooled and the total protein was estimated to be 1.3 mg/mL by Bradford assay. Bovine serum albumin (BSA) was used to generate standard curve for the Bradford assay. The activity of the enzyme with this preparation was found satisfactory with purity of the enzyme estimated to be >75%.
Enzyme assay
The assay was performed in a 100 μL final volume in 96-well UV plates (Costar, 3635) with a reaction buffer composed of 50 mM Tris–HCl (pH 8.0), 100 mM KCl, 3 mM EDTA and 1 mM dithiothreitol, 4% v/v DMSO plus or minus inhibitory compound and 0.75 µg of purified IMPDH II enzyme per well (from 1 mg/mL stock concentration). The final volume of the enzyme stock solution per well was 0.75 μL which was insignificant to cause any change in the final assay buffer composition. The reaction was initiated by the addition of 300 μM of IMP and 500 µM of NAD+ and the assay was allowed to proceed at 37 °C for 50 min before stopping by the addition of 15 mM GMP. The NADH generated was measured by reading the absorbance at 340 nm. At this wavelength, a background of <0.1 optical density (OD) was observed with negligible crosstalk between wells. Molecular Devices Spectramax 384 plus (Molecular Devices, LLC, Sunnyvale, CA) was used for recording the absorbance, which allows for selection of up to six wavelengths at a time for absorbance detection in the UV-vis range (190–1000 nm). Initial absorbance values (before substrate addition) were subtracted from the final absorbance to rule out compound auto fluorescence, if any. Mycophenolic acid (10 µM) used as a positive control and DMSO as a vehicle control. For IC50 determinations, a total of eight concentrations in triplicates with half log dilutions were used. For majority of the compounds, concentration ranged from 10 µM to 3 nM. Percent inhibition was calculated based on the following formula for each concentration and IC50 analyzed using the curve fitting program GraphPad Prism software version 6 using nonlinear regression analysis.
O.D. − test compound inhibition; positive control − O.D. with inhibitor; negative control − O.D. without inhibitor (100% activity).
PBMC proliferation assay
Human peripheral blood mononuclear cells (PBMCs) were isolated from citrate blood by means of a Ficoll–Hypaque density-gradient centrifugation. After isolation, PBMCs were seeded into 96-well cell culture plates in 10% v/v fetal calf serum containing RPMI-10 medium (1.5 × 105 cell/100 µL/well). Test compounds were added in a concentration ranging from 10 nM to 10 µM. DMSO to a final concentration of 0.1 % v/v DMSO was serving as a vehicle control. In order to verify the specificity of a potential growth inhibition by the IMPDH inhibitors, another control containing the test compound at the highest concentration of 10 µM and 100 µM GMP was added. After a period of 30 min of pre-incubation with the compounds, Concanavalin A was added to a final concentration of 3 µg/mL. The cells were subsequently cultured for a period of 48 h. Within the last 12 h of this period, 3H-thymidine (1 µCu/mL) was added to the wells. Finally, cells were harvested on 96-well filter plates by means of a cell harvester. After several washing steps with PBS, the filter plates were dried for 3 h at 60 °C, scintillation fluid was added to each well, plates were sealed and incorporated. 3H-thymidine was counted using a TopCount (Perkin Elmer, Waltham, MA). The IC50 values were calculated using GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA).
Results and discussion
In this study, our attempts to discover potent and novel IMPDH inhibitors using 3 as a starting point [advanced chemical analog (ACA) approach] are outlined. The division of 3 in three parts () allowed us to explore the SAR systematically. For designing the analogs of 3, a five-point pharmacophore hypothesis was developed using potent and structurally diverse IMPDH inhibitors. The top ranking hypothesis – AADHR () – formed the basis of new molecular entity (NME) design. Three out of five features [acceptor (A), hydrophobic (H) and ring (R)] resided in Part A of 3, whereas Parts B and C each contained one feature, a donor (D) and an acceptor (A), respectively (). The first part of our study was to find a suitable replacement for 4-cyano-3-methoxyphenyl (Part A) substructure which would possess corresponding pharmacophoric features A, H and R. These modifications (13–27) are listed in along with their corresponding pIC50 values for IMPDH II inhibition.
Replacement of 4-CN (acceptor feature) in 3 with 4-OMe group (13) led to a slight gain in potency. Substitution of 4-CN with 4-Me (14) dropped the potency by ∼2 log units indicating the stringent requirements for H-bond interaction. Another modification wherein –CN was replaced with morpholine ring (15) disappointed us in terms of potency despite showing good pharmacophore fitness (data not shown). It was speculated that the alicyclic ring might be little bigger in terms of size and shape compared to planar, yet small –CN group. Further, substitution of morpholine in 15 with 3,6-dihydro-2H-pyran (16) gained some potency, although one log unit less than 3. Our logic that a planar substituent should behave similar to –CN group was correct to some extent, although a slightly bigger ring size was probably an impediment to binding. Compound 16 with 4-tetrahydropyran substitution also proved to be detrimental for IMPDH II inhibition despite showing good pharmacophore fitness. Having ascertained the importance of planarity and presence of H-bond acceptor feature, four new bicyclic analogs 18–20 and 24 () were designed. But to our disappointment, none of these exhibited pIC50 above five. A second aromatic ring containing at least one H-bond acceptor feature placed at a nearby position was then introduced to probe the adjacent areas in the enzyme (exemplified by 21–23 and 25–27. Compound 21 contained 1-methylbenzimidazole ring system wherein N-3 was postulated to mimic the 4-CN group, albeit placed at a distance shorter than –CN would approach. Similarly, 22 had 2-methoxyquinoxaline substructure in which 2-OMe group was slightly moved away compared to 3. In case of 23, both the methoxy groups were placed farther due to insertion of additional phenyl ring. Compound 25 was the structural isomer of 22 where the positions of the ring N and –OMe groups were interchanged. The last two designs 26 and 27 had chromone substructure wherein the –C=O would be a putative H-bond acceptor. All these modifications did not show any improvement in potency. Hence, 4-cyano-3-methoxyphenyl (present in 3) and 3,4-dimethoxyphenyl substructures were fixed as Part A to be used in the next round of structural alterations.
Several of the compounds with Part A modifications were further investigated for their ability to inhibit Concanavalin A (ConA)-induced proliferation of PBMCs. The proliferation of human PBMCs, which majorly consist of human T- and B-cells, is highly dependent on the generation of GMP by reduction of IMP by IMPDH. GMP acts as a precursor for cGMP, which is required for DNA synthesis, as a prerequisite of proliferation. In accordance with the enzymatic potency, compound 13 turned out to be the most potent inhibitor of PBMC proliferation in this series of modifications with a pIC50 of 6.25. This inhibition was almost completely reversed by the addition of GMP, the end product of the metabolic pathway IMPDH is part of. This indicates that the effect of the compound is target specific. As also indicated in , examples of compounds were identified (e.g. compounds 20, 21, 22, 23, 25, 26), which showed no inhibition of IMPDH but of PBMC proliferation, which was not reversed by 100 µM GMP. Thus, those compounds are supposed to have a further mode of action affecting PBMC proliferation.
Part B modifications were aimed at finding a suitable replacement for the linker urea. The donor (D) feature of AADHR five-point pharmacophore, represented by NCitation1 of urea (), was found to form a critical H-bond with Asp274 of IMPDH II (see Supplementary material section). Most of the Part B designs were based on this observation so as to contain at least one H-bond donor. The first two designs were based on the replacement of urea 3 with guanidine (30 and 31, ). The presence of =NH instead of =O (as in 3) was not tolerated as both these compounds failed to exhibit any inhibition at concentrations tested. Our next strategy was to replace urea with N-cyanoguanidine. Compounds 32 and 33 exhibited some retention of IMPDH II inhibitory activity an order of magnitude less than 3. Excited by these results, compounds 34 and 35 were designed in which =NCN was replaced with =C-NO2. To our disappointment, both these compounds were inactive due to their poor permeability characteristics (data not shown). Further modifications to insert a link in the urea nitrogens and the carbonyl O were attempted. These designs “glycinamide (36)” and the “reverse glycinamide (37)” also failed to show any enzyme inhibition. Similar results were obtained when essential NCitation1 of urea in 3 was replaced with O and a methylene linker was introduced between O and –C=O (38 and 39, ). Replacing urea with sulfonylurea also did not help in terms of potency gain. Insertion of piperazine (41) wherein the donor feature was removed turned out to be inactive. Other modifications such as conformationally restricting urea in the form of imidazolidine-2,4-dione (42), insertion of either monocyclic (43) or bicyclic heteroaromatic ring (44) led to a similar drop in the potency compared to 3. All these urea replacements (Part B modifications) turned out to be futile except N-cyanoguanidine where some retention of inhibitory activity was observed. Hence, Part B was fixed as –NHC(=NCN)NH– to be used in combination with Part A in subsequent modifications in Part C.
Table 2. In vitro inhibitory and cellular potencies of Part B modifications.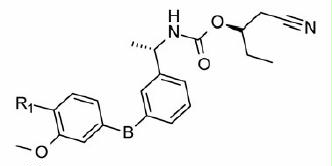
PBMC proliferation inhibition experiments using compounds with Part B modifications indicated that compound 32 most potently inhibited proliferation in a manner that is reversible by adding GMP. However, also in this series, two compounds were identified, which inhibited PBMC proliferation unspecifically and which had no activity on IMPDH at all (compounds 43 and 44).
Our attention was then focused on finding a substructure that would be a replacement for the benzyl carbamate moiety present on the distal phenyl ring. Our initial efforts were directed at removing the two chiral centers present in Part C for the ease of synthesis. The acceptor (A) feature (represented by carbamate –C=O, ) was part of five-point pharmacophore. Compounds 45 (ester) and 46 (acid) () contain this feature at a similar distance as found in 3. Both these compounds failed to exhibit any activity. Substitution by N-methyltetrazole group also led to an inactive compound (47, ). Insertion of a methylene linker between the tetrazole and the distal Ph ring to introduce some conformational flexibility did not exhibit any activity. Further attempts (50–53, ) to vary the distance of the acceptor feature with or without conformational restriction (51) also led to a complete loss of inhibitory potency. As a last resort, an additional Ph ring was introduced with an acceptor feature placed at different position to tap the features of the binding cavity. The groups which would behave as acceptor included –F, –CF3,–COOH and –CN. Thus, any Part C modification we tried led to loss of IMPDH II inhibitory activity. Accordingly, all these compounds also turned out to be inactive with respect to PBMC proliferation inhibition. In all, Parts A, B and C modifications demonstrated a steep SAR for IMPDH II inhibition. It was disappointing to see that most of these compounds did not turn out to be the potent inhibitors of IMPDH. Further investigations related to the involvement of other targets pertaining to the observations of the present investigation may be helpful to the researchers working in the IMPDH field.
Table 3. In vitro inhibitory and cellular potencies of Part C modifications.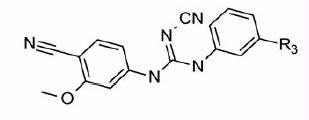
Conclusions
In this study, systematic investigations of the SAR of 3 in order to discover potent and novel IMPDH II inhibitors were carried out. A five-point pharmacophore model based on structurally diverse and potent inhibitors was used to guide the design of new molecules. The information about the key binding interactions of these inhibitors obtained from docking analyses was utilized as well in the design process. Initially, Part A was fixed as 4-cyano-3-methoxyphenyl or 3,4-dimethoxyphenyl substructures which were in agreement with the proposed pharmacophore. In our meticulous attempts to replace the urea (Part B), N-cyanoguanidine was identified as a suitable alternative from among the various substructures tried. Having fixed Parts A and B, further investigations were centered on doing suitable Part C modifications. Of the several Part C designs that were tried, none exhibited promising enzyme inhibitory activity. Hence, it was concluded that the structural requirements for IMPDH II inhibition were strict and the binding space was very restrictive in terms of tolerating any chemical modifications.
Supplementary information
The syntheses and the associated spectral data for compounds 14–27, 31–44, 46–60 are given. The dose–response curves for IMPDH II inhibition and PBMC proliferation inhibition by reference compounds 1, 2 and 3 are also included. The binding poses of few reference compounds with IMPDH II (PDB ID 1JR1) are shown.
Declaration of interest
There is no conflict of interest.
Acknowledgements
The authors are thankful to Jürgen Volz and colleagues for providing the HRMS data, Katharina Rossbach and colleagues for providing HTS samples and the Analytical Department, Global Discovery, Mumbai, for their help in recording the required analytical data.
References
- Crabtree GW, Henderson JF. Rate-limiting steps in the interconversion of purine ribonucleotides in Ehrlich ascites tumor cells in vitro. Cancer Res 1971;31:985–91
- Jackson RC, Weber G, Morris HP. IMP dehydrogenase, an enzyme linked with proliferation and malignancy. Nature 1975;256:331–3
- Franchetti P, Grifantini M. Nucleoside and non-nucleoside IMP dehydrogenase inhibitors as antitumor and antiviral agents. Curr Med Chem 1999;6:599–614
- Wu JC. Mycophenolate mofetil: molecular mechanism of action. Perspec Drug Discov Des 1994;2:185–204
- Natsumeda Y, Carr SF. Human type I and II IMP dehydrogenases as drug targets. Ann NY Acad Sci 1993;696:88–93
- Sintchak MD, Fleming MA, Futer O, et al. Structure and mechanism of inosine monophosphate dehydrogenase in complex with the immunosuppressant mycophenolic acid. Cell 1996;85:921–30
- Sollinger HW. Mycophenolate mofetil for the prevention of acute rejection in primary cadaveric renal allograft recipients (US Renal Transplant Mycophenolate Mofetil Study Group). Transplantation 1995;60:225–32
- Sievers TM, Rossi SJ, Ghobrial RM, et al. Mycophenolate mofetil. Pharmacotherapy 1997;17:1178–97
- Jain J, Almquist SJ, Shlyakhter D, Harding MW. VX-497: a novel, selective IMPDH inhibitor and immunosuppressive agent. J Pharm Sci 2000;90:625–37
- Jain J, Almquist SJ, Heiser AD, et al. Characterization of pharmacological efficacy of VX-148, a new, potent immunosuppressive inosine 5′-monophosphate dehydrogenase inhibitor. J Pharmcol Exper Ther 2002;302:1272–7
- Dhar TG, Shen Z, Guo J, et al. Discovery of N-[2-[2-[[3-methoxy-4-(5-oxazolyl)phenyl]amino]-5-oxazolyl]phenyl]-N-methyl-4-morpholineacetamide as a novel and potent inhibitor of inosine monophosphate dehydrogenase with excellent in vivo activity. J Med Chem 2002;45:2127–30
- Dhar TG, Shen Z, Gu HH, et al. 3-Cyanoindole-based inhibitors of inosine monophosphate dehydrogenase: synthesis and initial structure-activity relationships. Bioorg Med Chem Lett 2003;13:3557–60
- Stamos D, Trudeau M, Bethiel S. Inhibitors of IMPDH enzyme Patent WO0056331 (Vertex Pharma, USA)
- Yang N, Wang J, Li J, et al. A three-dimensional pharmacophore model for IMPDH inhibitors. Chem Biol Drug Des 2011;78:175–82
- Schrödinger Suite 2011 was available from Schrödinger, LLC, New York, NY, 2011
- Phase, version 3.3, Schrödinger, LLC, New York, NY, 2011
- Dixon SL, Smondyrev AM, Knoll EH, et al. PHASE: a new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening. 1. Methodology and preliminary results. J Comput Aided Mol Des 2006;20:647–71