
Abstract
We describe the characterisation of a series of 4,4′-biphenylsulfonamides as selective inhibitors of matrix metalloproteases MMP-2 and -13, two enzymes involved in cell invasion and angiogenesis. Double-inhibitor studies in the presence of acetohydroxamic acid show that these molecules do not bind the catalytic zinc. Moreover, two of the characterised inhibitors (11 and 19) act as non-competitive inhibitors, whereas the para-methyl ester derivative 13 behaves as a competitive inhibitor. This finding suggests that this class of molecules binds to a catalytic subsite, possibly the S1′-pocket. Moreover, since these compounds also act as inhibitors of carbonic anhydrases (CAs), another family of enzymes involved in cell invasion, they could be potentially useful as CA/MMP dual target inhibitors with increased efficacy as anticancer agents.
Introduction
Matrix metalloproteases (MMPs) are a class of zinc-dependent endopeptidases which catalyse the cleavage of extracellular matrix macromolecules such as the interstitial and basement membrane collagens, proteoglycans and accessory proteins such as fibronectinCitation1. The general structure of MMPs consists of a propeptide with a cysteine-switch motif, a catalytic domain and, with few exceptions (MMP-7, -23 and -26), a C-terminal hemopexin-like domain that is linked to the catalytic domain via a flexible hinge regionCitation1. The early reports on MMPs as tumour-associated and tumour-promoting enzymesCitation2 boosted the development of synthetic MMP inhibitors (MMPIs) as promising anti-metastatic drugs. The first generation of MMPIs consisted of peptidomimetic compounds that chelated the active site zinc through a zinc-binding group (ZBG) such as acetohydroxamic acid (AHA)Citation3. These broad-spectrum inhibitors (e.g. as marimastat and ilomastat, ) failed in clinical trials due to lack of therapeutic efficacy, poor outcome or unacceptable side effects such as musculoskeletal syndromeCitation4,Citation5. Nowadays, it is widely accepted that the lack of efficacy was generally due to administration to patients with advanced diseases, whereas the side effects were due to cross-inhibition of anti-target proteasesCitation6.
Figure 1. Structures of representative MMPIs. (A) Broad-spectrum MMPIs and AHA. (B) Non-zinc binding MMPIs.
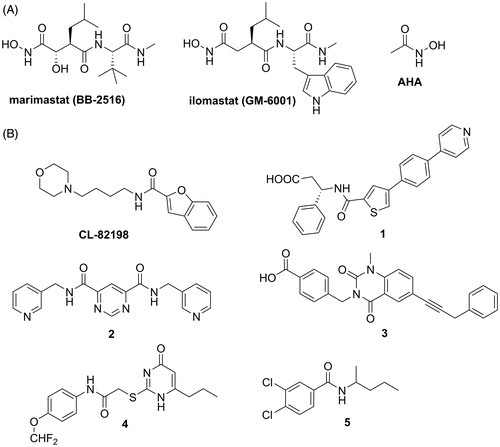
A way to develop more selective MMPIs is removing the ZBG and targeting directly specific pocket(s) within the catalytic site cleft. In 2000, Chen et al.Citation7 reported a compound (CL-82198), devoid of any obvious ZBG, which was a weak inhibitor of MMP-13 (). CL-82198 interacted specifically with the deep S1′-pocket of MMP-13. Pfizer identified a biphenyl(thienyl) derivative (compound 1, ) which inhibited potently MMP-12 and -13 with modest selectivity over MMP-2, -3, -8 and -9Citation8,Citation9. Although it contains another potential ZBG, a carboxylic group, the crystal structure of 1 with MMP-12 revealed that this inhibitor partially occupies the deep S1′-pocket of MMP-12, without chelating the catalytic zincCitation8. In 2005, a group from Sanofi-Aventis reported that a pyrimidine dicarboxamide (compound 2, ) exhibited micromolar activity against MMP-13 without inhibiting other MMPsCitation10. Crystal structure analysis of 2 with MMP-13 revealed that this compound, devoid of any ZBG, binds to a unique side pocket of S1′ cavity of MMP-13, referred to as S1′*-pocketCitation11. Pfizer reported a 1,4-dihydroquinazolinone highly selective for MMP-13 (compound 3, ) for osteoarthritis treatment that effectively reduced cartilage damage in vivo and did not induce joint fibroplasia in a rat model of musculoskeletal syndromeCitation12,Citation13. This inhibitor is a non-zinc binding ligand which confers an ordered structure to the S1′-specificity loop of MMP-13 that is otherwise flexible. Platinum complexes with the ability to inhibit MMPs through a non-competitive mechanism have also been reportedCitation14.
An alternative approach has been to target potential regulatory sites (i.e. allosteric sites) that have been predicted to be specific for each MMPCitation15. Dufour et al.Citation16 reported a pyrimidinone derivative which specifically binds to the hemopexin domain of MMP-9 (compound 4, ). The interaction between 4 and the hemopexin domain resulted in the abrogation of MMP-9 homodimerisation and subsequent blockage of a downstream signalling pathway required for MMP-9-mediated cell migrationCitation17. An MT1-MMP inhibitor which significantly inhibited tumour growth in an animal model has been reported by Remacle et al.Citation18 as an example of selective allosteric MMPI (compound 5, ). This inhibitor exerted its action by preventing MT1-MMP dimerisationCitation18.
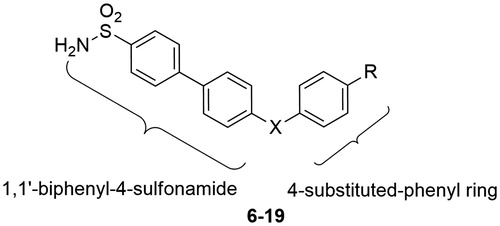
In the present study, we investigated the ability of a series of 4,4′-biphenylsulfonamides (compounds 6–19, ), devoid of an obvious ZBG, to inhibit the catalytic activity of two MMP members, MMP-2 and -13, both of them implicated in cancer angiogenesis and tumour growthCitation19–21. These compounds were originally developed as competitive inhibitors of carbonic anhydrasesCitation22 (CAs), a class of zinc-dependent enzymes unrelated to MMPs.
Figure 2. General structure of 4,4′-biphenylsulfonamides 6–19.
Methods
Reagents
The fluorogenic peptides FS6 [Mca-Lys-Pro-Leu-Gly-Leu-Dap(Dnp)-Ala-Arg-NH2] and M-9545 [Mca-Arg-Pro-Lys-Pro-Val-Glu-Nva-Trp-Arg-Lys(Dnp)-NH2] were purchased from Bachem AG (Bubendorf, Switzerland). Recombinant human pro-MMP-1, pro-MMP-2, pro-MMP-3, pro-MMP-13 and MMP-7 were purchased from Calbiochem (Merck, Nottingham, UK). Pro-MMP-8 was a kind gift from Prof. Gillian Murphy, University of Cambridge, Cambridge, UK. APMA (p-aminophenylmercuric acetate), DMSO (dimethyl sulfoxide) and Tris base [tris(hydroxymethyl)-aminomethane] were purchased from Sigma-Aldrich (Milano, Italy). Microtitre plates with 96 wells were from Corning (Sigma-Aldrich, Milano, Italy). The synthesis of the various inhibitors is described elsewhereCitation22.
Determination of IC50
All enzyme assays were conducted in Fluorometric Assay Buffer (FAB: 50 mM Tris base, 10 mM CaCl2, 150 mM NaCl, 1% DMSO, 0.05% Brij-35, pH 7.5) at 37 °C, using a SPECTRAmax Gemini XS Microplate Spectrofluorometer (Molecular Devices, Sunnyvale, CA). MMP-1, -2, -8 and -13 pro-enzymes were activated immediately prior to use with APMA (final concentration: 2 mM for 2 h at 37 °C for MMP-1, 2 mM for 1 h at 37 °C for MMP-2 and -8 and 1 mM for 35 min at 37 °C for MMP-13). Pro-MMP-3 was activated with trypsin (4.4 μg/ml) for 30 min at 37 °C followed by the addition of soybean trypsin inhibitor (61.5 μg/ml). MMP-7 was catalytically active.
Assays were performed in a total volume of 200 μl per well in 96-well microtitre plates. Stock solutions (10 mM) of the inhibitors in DMSO were further diluted in FAB (solubility > 500 μM) for 4 h at 25 °C in the presence of the activated enzyme (final concentration: 1 nM for MMP-1, 1.12 nM for MMP-2, 5 nM for MMP-3, 4 nM for MMP-7, 1.5 nM for MMP-8 and 0.66 nM for MMP-13). The hydrolysis of FS6 (final concentration: 2 μM) was monitored every 3 s for 15 min at 37 °C at the excitation and emission wavelengths of 325 and 395 nm, respectively. For MMP-3, the substrate used was M-9545 at a final concentration of 2 μM. In this case, the excitation and emission wavelengths were 328 and 393 nm, respectively. Enzymatic rates were corrected for spontaneous hydrolysis of the substrate. IC50 values were determined using the formula vi/v0 = 1/{1 + ([I]/IC50}, where vi is the initial velocity of substrate cleavage in the presence of the inhibitor at concentration [I] and v0 is the initial velocity in the absence of the inhibitor. Results were analysed using SoftMax Pro software and Prism Software version 5.0 (GraphPad Software, Inc., La Jolla, CA).
Double-inhibitor studies
Double-inhibitor experiments were conducted as described by Yonetani and TheorellCitation23. Inhibitors were incubated with MMP-2 or -13 in the presence and the absence of a fixed concentration (10 or 15 mM, respectively) of AHA. IC50 values were determined in the absence () and in the presence of AHA (
). In the latter condition, the activity measured in the presence of AHA and enzyme alone was taken as 100%. To compensate for the addition of AHA, an equal volume of DMSO was added to the reactions containing enzyme alone so that total DMSO concentration was kept at 3% (v/v).
Determination of inhibition mode
Inhibitors were incubated with MMP-13 in the presence of increasing concentrations of substrate (0.5–3 μM). Data were arranged in a Dixon Plot (1/v versus [I])Citation24 and fitted to a linear polynomial equation using GraphPad 5.0 (GraphPad Software, Inc.) and the Ki values were determined on the basis of the point of intersection of the curves.
Results
Inhibitory activity of 4,4′-biphenylsulfonamides on MMPs
A series of sulfonamides (6–19, ) initially designed as CA inhibitorsCitation22 was screened against MMP-2 and -13 by a fluorometric assay (). All these compounds had a bended 4-substituted phenyl ring linked by a 1C bridge to the 4′-position of a 1,1′-biphenyl-4-sulfonamide scaffold and generally resembled the structure of some previously reported MMP-13 non-zinc binding inhibitors (). In fact, in the scaffolds of these known inhibitors there is a linear aromatic or heterocyclic central core properly decorated with a bended aromatic or hetero-aromatic side chain (see compounds 1–5, ). Here, the simplest structure among the sulfonamides 6–19 was represented by compound 6, which was a weak inhibitor (IC50 = 300 µM) of both tested enzymes. The introduction of various R substituents in para position of the phenyl ring led to a moderate increase of inhibitory activity. The best results were obtained for compound 10, bearing a carboxylic acid in R, which showed an improved activity against MMP-2 (IC50 = 40 µM). The introduction of a hydroxyl group (X = CHOH) on the bridging chain of 6, led to inhibitor 11 which exhibited an improved activity against both enzymes with respect to its simpler starting compound. The presence of both this hydroxymethyl substituent in 4′ and a para substituent R on the bended phenyl ring led to a further improvement of activity. Surprisingly, the best results were given by the para substituted methyl ester derivative 13 (IC50 = 40 µM on MMP-2 and 50 µM on MMP-13) rather than its carboxylic analogue 14. Finally, a series of rigid carbonyl analogues (X = CO) of sulfonamides 6–14 was evaluated. The R unsubstituted derivative 15 showed a very weak inhibitory activity, just like the starting derivative 6. Also in this series of compounds, the introduction of substituents R in para position on the phenyl ring led to an increase of activity. In particular, the R carboxylic acid derivative 19 showed the best results on both MMP-2 and -13 with IC50 values in the low-micromolar range. Selectivity of the most active compounds (11, 13 and 19) was then tested on a plethora of MMPs (). All three compounds resulted in 10-fold less active against MMP-1 and -7, two MMPs which are characterised by shallow S1′-pockets, and 2–40-fold less active against MMP-3 and -8, which are characterised by deep S1′-pocketsCitation3.
Table 1. In vitroa inhibitory activity (IC50 μM values) of sulfonamides 6–19 and hydroxamate 20.
Table 2. Selectivity profile of compounds 11, 13 and 19.
Dual inhibition studies
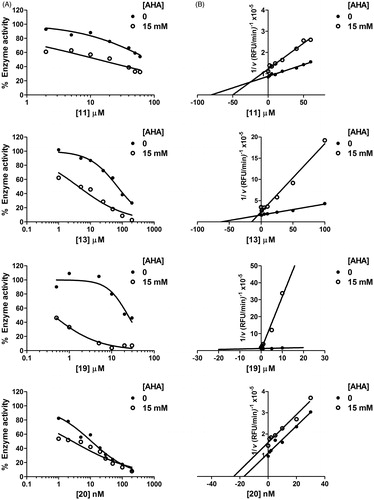
Since some of the biphenylsulfonamides described here are devoid of an obvious ZBG and still retained inhibitory activity, we hypothesised that they do not interact with the catalytic zinc ion of MMP active site. To investigate this possibility, we performed dual inhibition kinetics using AHA (), a known zinc-binder and competitive millimolar-range inhibitor of many MMPsCitation11, in combination with selected compounds 11, 13 and 19. For comparison, a previously reported zinc-chelating MMPI, 20Citation25, was used (). At the AHA concentrations used here (10 mM for MMP-2 and 15 mM for MMP-13), the percentage of enzyme inhibition is ∼35–50% (IC50 of AHA is 10 mM for MMP-2 and 30 mM for MMP-13). The inhibitory potency (IC50 values) of compounds 11, 13, 19 and 20 was measured in the presence and in the absence of AHA. In the presence of AHA, we took as 100% the residual activity of the reaction containing only enzyme and AHA. These studies would show zero interaction if the effect of the combination is that expected from the dose-response curves of each inhibitor (/
= 1); synergism, if the effect is greater than expected (
/
< 1) and antagonism, if the effect is less than expected (
/
> 1). Compounds 11 and 13 showed increased potency in the presence of AHA. The synergistic effect was considerable, with
/
ratios significantly lower than unity (). Quite surprisingly, the same effect was observed for the carboxylic acid derivative 19. The increase in inhibitory potency against MMP-2 was 70-fold for 19, 4-fold for 11 and 2.5-fold for 13. Similar results were observed in the case of MMP-13 inhibition, with increases in inhibitory potency of 27-fold for 19 and 2-fold for 11 and 13. In contrast, the hydroxamate MMPI 20, with potency in the low-nanomolar range, showed antagonism with AHA, with
/
ratios higher than unity (). Differences in IC50 values between and are probably due to the different DMSO concentration in the assay buffer (3 versus 1%), which can affect compound solubility.
Table 3. Effect of AHA on in vitro activity of compounds 11, 13, 19 and 20.
Initial velocities were then organised in a Yonetani–Theorell Plot ( and ). These studies, also known as mutual exclusivity studies, can predict the binding cooperativity between two inhibitors. In this analysis, only inhibitor concentrations able to produce between 50 and 70% inhibition were used due to high uncertainties in measuring very low enzymatic rates. In the case of compounds 11, 13 and 19, we observed lines which intersected above the x-axis ( and ). In a Yonetani–Theorell plot, intersecting lines indicate simultaneous (i.e. non-mutually exclusive) binding of both inhibitors to the enzymeCitation23. However, the hydroxamate-based compound 20 showed parallel (non-intersecting) lines in the Yonetani–Theorell plot. This indicates mutually exclusive binding between the inhibitor and AHA, i.e. competition between the two MMPIs for binding to the catalytic zinc.
Figure 3. AHA increases inhibitory potency of compounds 11, 13 and 19 on MMP-2. Dose-response (A) and Yonetani–Theorell plots (B) for MMP-2 inhibition by compounds 11, 13, 19 and 20 in the absence and presence of a fixed concentration (10 mM) of AHA. In A, residual activity refers to the uninhibited enzyme.
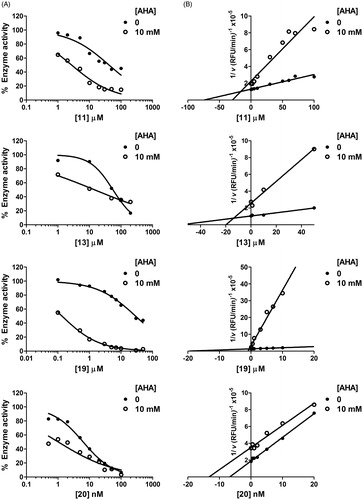
Figure 4. AHA increases inhibitory potency of compounds 11, 13 and 19 on MMP-13. Dose-response (A) and Yonetani–Theorell plots (B) for MMP-13 inhibition by compounds 11, 13, 19 and 20 in the absence and presence of a fixed concentration (15 mM) of AHA. In A, residual activity refers to the uninhibited enzyme.
Determination of inhibition modality
Since AHA is known to bind to the catalytic zinc ion of MMPs, compounds 11, 13 and 19 most likely do not act by binding the active site zinc. Therefore, we examined the mode of action of these MMPIs (). The Michaelis–Menten constant, Km, of MMP-2 for FS-6 substrate is >30 μMCitation26. At these concentrations, auto-quenching of the substrate must be taken in consideration and this can complicate the kinetic analysis. Therefore, we chose to characterise the mechanism of inhibition of these inhibitors against MMP-13, whose Km value for FS-6 is in the low-micromolar rangeCitation26.
Figure 5. Compound 13 is a competitive inhibitor of MMP-13, whereas 11 and 19 show a non-competitive inhibition mode. Inhibitors were incubated with MMP-13 in the presence of increasing concentrations of substrate and data were arranged in a Dixon Plot. RFU, raw fluorescence units.
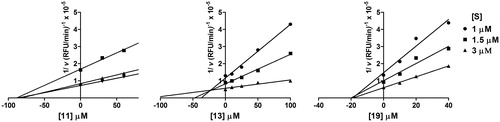
In the case of 11 and 19, the curves in the Dixon plot intersected on the x-axis indicating classical non-competitive inhibition. By contrast, in the presence of 13, the curves in the Dixon plots intersected above the x-axis indicating competitive inhibition. The Ki values estimated by the intersections of the curves on the Dixon plots () were in good agreement with the IC50 values in the case of non-competitive inhibitors 11 and 19, whereas in the case of the competitive inhibitor 13 the estimated Ki value (24 μM) is similar to the IC50 value corrected by the Cheng–Prusoff equation (33 μM) {Ki = IC50/(1 + [S]/Km)}Citation27.
Table 4. Mechanism of inhibition of MMP-13 by compounds 11, 13 and 19.
Discussion and conclusions
All the CA inhibitors in clinical use such as acetazolamide and ethoxzolamide contain a terminal primary sulfonamide (RSO2NH2; where, R is generally an aromatic/heteroaromatic moiety) as the ZBGCitation28. Since some MMP members are involved in carcinogenesis and tumour progressionCitation1,Citation19 and CA inhibitors are being tested for a therapeutic use as anti-tumour agentsCitation29, dual inhibition of both MMP and CA members is attracting pharmacological interest. A dual inhibition has been achieved by conjugation in the same molecular entity of two ZBGs such as benzenesulfonamide/1-hydroxy-2(1H)-pyrimidinone and AHA for inhibition of CA and MMP, respectivelyCitation30,Citation31. Here, we have shown that primary biphenylsulfonamides previously discovered as nanomolar CA inhibitorsCitation22, can act also as non-zinc binding MMPIs.
The majority of MMPIs are based on a ZBG and a hydrophobic portion protruding into the S1′-subsite. Since the ZBG can mimic one of the transition states occurring during the catalysis, these compounds are competitive inhibitors and therefore act by decreasing the apparent substrate-binding affinity (Km) with minimal effect on maximum activity (Vmax). A potential drawback of competitive inhibitors is that their potency can be diminished by mass-action competition with the substrate and the increase in inhibition constant (Ki) can be calculated from the Cheng–Prusoff equation {Ki = IC50/(1 + [S]/Km}Citation27. In open systems, which are characterised by a constant supply of substrates and elimination of products, competitive inhibitors do not provide long-term inhibition due to accumulation of substrate to a new steady-state levelCitation32. By inhibiting several MMP members and hence impairing alternative degradation pathways of MMP substrates, broad-spectrum MMPIs are likely to produce substrate accumulation which eventually will overcome MMP inhibition. In the case of uncompetitive inhibitors, no recovery of enzymatic activity is observed in open systems, whereas in the case of non-competitive inhibitors only partial recovery is observedCitation32. Therefore, uncompetitive or non-competitive inhibitors should be preferred over competitive inhibitors in open systems (e.g. in vivo) and, in particular, when a therapeutic application is desired.
Compounds 11, 13 and 19 do not act through a zinc-binding mechanism. The potent synergistic effect observed in the presence of AHA () is consistent with the binding of these compounds in a site proximal to the S1′-pocket. In the case of carboxylic acid 19, this result is quite unexpected. It is possible that the presence of AHA may force the compound in the S1′ site with no chelation exerted by the putative ZBG (the carboxyl group). This behaviour is similar to that of the thiophene amido carboxylic acid 1 developed by PfizerCitation9. However, the similar inhibitory potency showed by the other derivatives such as 11 and 13 suggest that the biphenylsulfonamide moiety is responsible for most of the inhibitory activity. Moreover, the non-competitive behavior of 19 () excludes binding of the catalytic zinc ion by the carboxylic acid.
Compound 13 is a competitive inhibitor of MMP-13 () even though it shows a synergistic effect with AHA (). Since its analogue 11 is a non-competitive inhibitor, the para-methyl ester group of 13 should be responsible for the observed competition with the substrate, possibly by protruding in the groove where the peptide backbone of the substrate would bind. Similarly, a non-zinc binding inhibitor recently reported by AstrazenecaCitation33 binds with the majority of the molecule within the MMP-13 S1′-pocket, whereas the pyridyl motif lies in the substrate-binding cleft. Although the inhibitory mechanism of this compound has not been characterised, it would likely display a competitive mechanism against peptide substrates. As a consequence, the absence of a ZBG is not an obvious guarantee of a non-competitive/uncompetitive mechanism, and a detailed kinetic characterisation is required.
The fact that all three compounds spare MMP-1 and -7 (), two MMPs with shallow S1′-pocketsCitation3 strongly suggests this pocket as a binding site. Moreover, the lower inhibitory potency against MMP-3 and -8, which are instead characterised by a much larger S1′-pocketCitation3, underlies the ability of non-zinc binding MMPIs to recognise structural differences within this subsite. MMP-3 and -8 are currently regarded as anti-targets in cancer therapyCitation19, therefore sparing these enzymes will likely result in a better therapeutic index.
Assuming that the compounds described here bind in the proximity of catalytic zinc, a possible optimisation may involve linking of the sulfonamide scaffold to a strong ZBG, thus mimicking double-inhibitor experiments and consequently increasing the inhibitory potency. However, such compounds would likely suffer from the same drawbacks of other zinc-binding MMPIs. An alternative may be increasing affinity by appropriate substitutions of the biphenyl sulfonamide scaffold in order to obtain nanomolar non-competitive inhibitors. Ultimately, the binding site for the compounds described here should be further experimentally determined by X-ray crystallography, NMR or cross-linking experiments.
In summary, we described and characterised here three 4,4′-biphenylsulfonamides as non-zinc binding MMP-2 and -13 selective inhibitors. These same compounds also inhibit CAsCitation22 and could be potentially useful as CA/MMP dual target inhibitors with increased efficacy as anticancer agents.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
This study was supported by funding from the Italian Ministry of Education, University and Research (MIUR, PRIN 2007, A.R.) and from the University of Pisa (Fondi di Ateneo 2012, rating 2013 to A.R. and E.N.).
References
- Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinas: regulators of the tumor microenvironment. Cell 2010;141:52–67
- Wojtowicz-Praga SM, Dickson RB, Hawkins MJ. Matrix metalloproteinase inhibitors. Invest New Drugs 1997;15:61–75
- Whittaker M, Floyd CD, Brown P, Gearing AJ. Design and therapeutic application of matrix metalloproteinase inhibitors. Chem Rev 1999;99:2735–76
- Zucker S, Cao J, Chen WT. Critical appraisal of the use of matrix metalloproteinase inhibitors in cancer treatment. Oncogene 2000;19:6642–50
- Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science 2002;295:2387–92
- Vandenbroucke RE, Libert C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat Rev Drug Discov 2014;13:904–27
- Chen JM, Nelson FC, Levin JI, Mobilio D, Moy FJ, Nilakantan R, et al. Structure-based design of a novel, potent and selective inhibitor for MMP-13 utilizing NMR spectroscopy and computer-aided molecular design. J Am Chem Soc 2000; 122:9648–54
- Morales R, Perrier S, Florent JM, et al. Crystal structures of novel non-peptidic, non-zinc chelating inhibitors bound to MMP-12. J Mol Biol 2004;341:1063–76
- Dublanchet AC, Ducrot P, Andrianjara C, et al. Structure based design and synthesis of novel non-zinc chelating MMP-12 inhibitors. Bioorg Med Chem Lett 2005;15:3787–90
- Engel CK, Pirard B, Schimanski S, et al. Structural basis for the highly selective inhibition of MMP-13. Chem Biol 2005;12:181–9
- Gooljarsingh LT, Lakdawala A, Coppo F, et al. Characterization of an exosite binding inhibitor of matrix metalloprotease 13. Protein Sci 2008;17:66–71
- Johnson AR, Pavlovsky AG, Ortwine DF, et al. Discovery and characterization of a novel inhibitor of matrix metalloprotease-13 that reduces cartilage damage in vivo without joint fibroplasia side effects. J Biol Chem 2007;282:27781–91
- Li JJ, Nahra J, Johnson AR, et al. Quinazolinones and pyrido-[3,4-d]pyrimidin-4-ones as orally active and specific matrix metalloprotease-13 inhibitors for the treatment of osteoarthritis. J Med Chem 2008;51:835–41
- Sasanelli R, Boccarelli A, Giordano D, et al. Platinum complexes can inhibit matrix metalloproteinase activity: platinum-diethyl[(methylsulfinyl) methyl]phosphonate complexes as inhibitors of matrix metalloproteinases 2, 3, 9, and 12. J Med Chem 2007;50:3434–41
- Udi Y, Fragai M, Grossman M, et al. Unraveling hidden regulatory sites in structurally homologous metalloproteases. J Mol Biol 2013;425:2330–46
- Dufour A, Zucker S, Sampson NS, et al. Role of matrix metalloproteinase-9 dimers in cell migration: design of inhibitory peptides. J Biol Chem 2010;285:35944–56
- Dufour A, Sampson NS, Li J, et al. Small-molecule anticancer compounds selectively target the hemopexin domain of matrix metalloproteinase-9. Cancer Res 2011;71:4977–88
- Remacle AG, Golubkov VS, Shiryaev SA, et al. Novel MT1-MMP small-molecule inhibitors based on insights into hemopexin domain function in tumor growth. Cancer Res 2012;72:2339–49
- Overall CM, Kleifeld O. Tumour microenvironment – opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat Rev Cancer 2006;6:227–39
- Kudo Y, Iizuka S, Yoshida M, et al. Matrix metalloproteinase-13 (MMP-13) directly and indirectly promotes tumor angiogenesis. J Biol Chem 2012;287:38716–28
- Shah M, Huang D, Blick T, et al. An MMP13-selective inhibitor delays primary tumor growth and the onset of tumor-associated osteolytic lesions in experimental models of breast cancer. PLoS One 2012;7:e29615
- La Regina G, Coluccia A, Famiglini V, et al. Synthesis and biological evaluation of 4,4′-biphenylylsulfonamides as a new class of potent and selective carbonic anhydrase XIV inhibitors. ACS Med Chem Lett
- Yonetani T, Theorell H. Studies on liver alcohol hydrogenase complexes. 3. Multiple inhibition kinetics in the presence of two competitive inhibitors. Arch Biochem Biophys 1964;106:243–51
- Dixon M, Webb EC, Thorne CJR, Tipton KF. Enzymes. 3rd ed. London: Longmans, Green & Co; 1979
- Nuti E, Panelli L, Casalini F, et al. Design, synthesis, biological evaluation, and NMR studies of a new series of arylsulfones as selective and potent matrix metalloprotease-12 inhibitors. J Med Chem 2009;52:6347–61
- Neumann U, Kubota H, Frei K, et al. Characterization of Mca-Lys-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2, a fluorogenic substrate with increased specificity constants for collagenases and tumor necrosis factor converting enzyme. Anal Biochem 2004;328:166–73
- Cheng YC, Prusoff, WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (IC50) or an enzymatic reaction. Biochem Pharm 1973;22:3099–108
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77
- Marques SM, Nuti E, Rossello A, et al. Dual inhibitors of matrix metalloproteinases and carbonic anhydrases: iminodiacetyl-based hydroxamate-benzenesulfonamide conjugates. J Med Chem 2008;51:7968–79
- Esteves MA, Ortet O, Capelo A, et al. New hydroxypyrimidinone-containing sulfonamides as carbonic anhydrase inhibitors also acting as MMP inhibitors. Bioorg Med Chem Lett 2010;20:3623–7
- Westley AM, Westley J. Enzyme inhibition in open systems. Superiority of uncompetitive agents. J Biol Chem 1996;271:5347–52
- De Savi C, Morley AD, Ting A, et al. Selective non-zinc binding inhibitors of MMP13. Bioorg Med Chem Lett 2011;21:4215–9