
Abstract
The single-crystal structure of anagliptin, N-[2-({2-[(2S)-2-cyanopyrrolidin-1-yl]-2-oxoethyl}amino)-2-methylpropyl]-2-methylpyrazolo[1,5-a]pyrimidine-6-carboxamide, was determined. Two independent molecules were held together by intermolecular hydrogen bonds, and the absolute configuration of the 2-cyanopyrrolidine ring delivered from l-prolinamide was confirmed to be S. The interactions of anagliptin with DPP-4 were clarified by the co-crystal structure solved at 2.85 Å resolution. Based on the structure determined by X-ray crystallography, the potency and selectivity of anagliptin were discussed, and an SAR study using anagliptin derivatives was performed.
Introduction
In 1966, dipeptidyl peptidase IV (DPP-4, EC 3.4.14.5) was discovered as a new aminopeptidase that hydrolyses glycyl-dl-prolyl-β-naphthylamideCitation1. In 1993, one of the substrates of this enzyme was reported to be glucagon-like peptide-1 (GLP-1)Citation2. GLP-1 is a peptide stimulating insulin secretion depending on the blood glucose concentration level, and is inactivated by cleavage of two N-terminal amino-acid residues by DPP-4. Accordingly, DPP-4 inhibitors can function as diabetes medicines by minimizing the reduction in GLP-1 concentration. The effects of DPP-4 inhibition via the inhibitor val-pyrrolidine have been investigated in anesthetized pigsCitation3, and a perspective on the use of DPP-4 inhibitors for the treatment of type 2 diabetes was reported in 1998Citation4. DPP-4 consists of 766 amino acids and belongs to the prolyloligopeptidase family of serine proteases; its crystal structure was revealed in 2003Citation5–9.
The DPP-4 crystal structure forms a homodimer, suggesting that it is an in vivo form. Each subunit consists of two domains, an α/β-hydrolase domain and an eight-bladed β-propeller domain. The active site was revealed by observation of the tetrahedral intermediate of a peptide inhibitor, diprotin A (Ile-Pro-Ile)Citation7. The catalytic triad including catalytic residue Ser630 is located at the interface of the two domains, and the S1 subsite is formed by the hydrophobic residues. The N-terminal amino group of the substrate is recognized in the S2 subsite that consists of the acidic residues Glu205 and Glu206.
In a subsequent report, Engel et al. focused on whether the substrates approached the active site by opening a side or a propeller region. They found that a substrate approaching via the side opening would be in the proper orientation for docking to the active siteCitation10.
Until recently, various inhibitors of DPP-4 were developed by chemical design and synthesis, as well as by biological evaluation with consideration for hypoglycaemic activity and – as a safety concern – with consideration for selectivity against DPP-4-related proteases such as DPP8, DPP9 and fibroblast activation protein (FAP). These efforts resulted in numerous inhibitors; the eight inhibitors shown in are currently available for useCitation11. Sitagliptin was the first of these inhibitors to be approved, gaining authorization in 2006 in the US and in 2007 in the EU. Vildagliptin was approved next, in 2008 in the EU. Subsequently, saxagliptin (in 2009), linagliptin (in 2011) and alogliptin (in 2013) were approved in both the US and EU, respectively. Gemigliptin was approved in Korea in 2012, and teneligliptin and anagliptin were approved in Japan in 2012. With the exceptions of gemigliptin and anagliptin, Nabeno et al. categorised all of these inhibitors into three classes according to the binding subsites of DPP-4, which tend to be characteristic of their chemical structuresCitation12. Specifically, the inhibitors binding to only the S1 and S2 subsites were categorised into Class 1, those binding to the S1′ and/or S2′ along with the S1 and S2 subsites were categorised into Class 2, and those binding to the S2 extensive subsite along with the S1 and S2 subsites were categorised into Class 3. The S2 extensive subsite was defined as the site beyond the S2 subsite by Yoshida et alCitation13; natural substrates do not interact with the subsite.
Figure 1. Launched DPP-4 inhibitors.
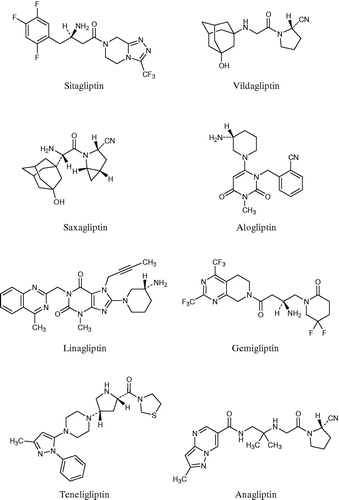
AnagliptinCitation14 is one of the DPP-4 inhibitors launched in Japan. In this paper we report its crystal structure and its interactions with DPP-4 as determined by X-ray crystallography, and discuss its potency and selectivity.
Materials and methods
Compounds
AnagliptinCitation14, sitagliptinCitation15, vildagliptinCitation16 and alogliptinCitation17 were prepared by known methods, respectively.
Crystallographic study of anagliptin
A colourless single crystal (0.3 × 0.3 × 0.08 mm3) was obtained by standing at room temperature after heating to dissolve in ethyl acetate. The data were collected by the ω scan method with 0.3° in scan width and 10 s in exposure time at room temperature, using a SMART 1000 two-dimensional X-ray diffractometer (Bruker; Mo Kα, 50 kV, 40 mA) with a 0.5-mmϕ collimator. The structure was solved by the direct method using the SHELXS-97 program (Göttingen, Germany), and refined by the full-matrix least-squares method using the SHELXL-97 program. The crystal structure data has been deposited to the Cambridge Crystallographic Data Centre (CCDC 1003533).
Expression and purification of DPP-4
Human DPP-4 (hDPP-4) was expressed in the Silkworm–Baculovirus System produced by ProCube (Sysmex Corporation; http://procube.sysmex.co.jp/eng/). The hDPP-4 gene encoding Ser39-Pro766 was amplified by PCR from the synthesised full-length hDPP-4 gene template with optimised codon usage for Bombyx mori. The PCR was run using a forward primer containing a BglII site and a cleavage site for HRV 3C protease (5′-GGAAGATCTCTGGAAGTTCTGTTCCAGGGGCCCTCAAGGAAGACTTATACATT-3′) and a reverse primer containing an EcoRV site (5′-GGCGATATCTTATGGTAAACTGAAACATTG-3′). The PCR product was inserted between BglII and EcoRV in the multiple cloning site of the pM24 vector (Sysmex Corporation) based on the vector pUC19 for B. mori nucleopolyhedrovirus (BmNPV). The vector was designed to express a fusion protein containing a DDDDK-tag and HRV3C protease recognition site at the N-terminal fragment of hDPP-4. This transfer vector was co-transfected with BmNPV DNA into a B. mori-derived cell line (BmN). After 7 days of incubation, the recombinant baculovirus was injected into the body of silkworm pupae. The pupae were harvested 6 days after infection, frozen and homogenised with PBS buffer (pH 7.4) containing a protease inhibitor cocktail. The homogenised solution was centrifuged to separate the supernatant and precipitate using an ultracentrifuge (100 000 g) for 1 h at 4 °C. The supernatant containing DDDDK-tag fusion hDPP-4 was applied to DDDDK-tagged Protein PURIFICATION GEL (MBL Co., Ltd.), and the fusion protein was eluted with DDDDK peptide containing PBS buffer (pH 7.4). The eluted sample was treated with HRV 3C protease (Wako Pure Chemical Industries, Ltd.) for 16 h at 4 °C to remove the N-terminal DDDDK-tag from the recombinant fusion protein and then applied to a Glutathione Sepharose 4B column (GE Healthcare) to remove the HRV 3C protease. The flow-through fraction was collected and rapidly purified by TOYOSCREEN SuperQ (TOSOH Corporation), which is an ion exchange column with a linear gradient of 0–300 mM NaCl in Tris–HCl buffer (pH 8.0). Finally, hDDP4 was further purified by gel filtration chromatography using a Superdex 200 column (GE Healthcare) with 20 mM Tris–HCl (pH 8.0) buffer containing 100 mM NaCl. The purified hDPP-4 was concentrated using an Apollo spin concentrator (Orbital Biosciences, LLC) to 10 mg/mL for the crystallisation sample.
Crystallographic study of the DPP-4 complex with anagliptin
The complex was prepared by adding a 10-fold molar excess of anagliptin in a buffer (20 mM Tris–HCl, 0.1 M NaCl, pH 8.0) to the enzyme solution (20 mM Tris–HCl, 0.1 M NaCl, pH 8.0, concentration 9.6 mg/mL). The crystals of the complex were grown from a solution containing 18% PEG400, 15% PEG 1000 and 0.15 M Na/K phosphate (pH 6.5) at 293 K by the sitting-drop vapour-diffusion method. These crystallisation trials were conducted using the LIGHT method (laser irradiated growth technique)Citation18 to produce high-quality crystals. Before data collection, the crystal was cryo-cooled using a mixture of 25% glycerol, 18% PEG400, 15% PEG1000 and 0.15 M Na/K phosphate (pH 6.5). X-ray diffraction data were collected on BL41XU at SPring-8 (Harima, Japan) at 100 K using a MX225HE (Rayonix, Evanston, IL) detector.
The structure of the DPP-4–anagliptin complex was solved by the molecular replacement method with the program MOLREPCitation19 using the structure of DPP-4–saxagliptin complexCitation20 refined to 2.85 Å resolution (PDB code 3BJM) as a model structure. The rigid body refinement was performed with the program REFMACCitation21. The refinements for the range of 47.7–2.85 Å resolution were performed with the program PrimeX, version 2.2 (Schrödinger, LLC, New York, NY). The coordinates of the structure were deposited in the Protein Data Base under code 3WQH.
Measurements of in vitro activities
Inhibition of DPP-4 activity was determined by measuring the rate of hydrolysis of a surrogate substrate, Gly-Pro-4-methylcoumarin-7-amide (Gly-Pro-MCA; Peptide Institute, Osaka, Japan). Ten microliters of various concentrations of the test compound solutions and 45 μL of human recombinant DPP-4 (R&D Systems, Minneapolis, MN) with an assay buffer (25 mM HEPES, 140 mM NaC1, 0.1 mg/mL BSA, pH 7.8) were added to a black 96-well microplate and mixed. After a 30-min preincubation at room temperature, the enzymatic reaction was initiated by the addition of 45 μL of 0.4 mM Gly-Pro-MCA and allowed to react for 20 min. The reaction was stopped by the addition of 100 μL of 25% acetic acid solution, and the fluorescence intensity was measured at an excitation wavelength of 390 nm and a fluorescence wavelength of 460 nm. As standards, the fluorescence intensity of 7-amino-4-methylcoumarin (AMC) solutions plus 25% acetic acid solution was measured. The percent inhibition relative to the control without inhibitor was calculated and the IC50 values were determined by a nonlinear regression model.
The levels of inhibition of the DPP8 and DPP9 activities were determined by the degradation of the fluorescent substrate Gly-Pro-MCA. Human DPP8 and DPP9 were expressed in baculovirus-infected Sf9 insect cells and purified using his-tagged protein purification resins. Inhibition of the DPP8 and DPP9 activities was determined as described above. Ten microliters of the test compounds and 50 μL of 1.0 mM Gly-Pro-MCA in buffer solution (50 mM HEPES, 0.1 mg/mL BSA, pH 8.0) were added to black 96-well microplates, and the reaction was initiated by the addition of 40 μL of the diluted enzyme solution for 20 min at room temperature. The reaction was stopped by the addition of 25% acetic acid solution and the fluorescence intensity was measured.
Inhibition of FAP activity was determined by the degradation of the fluorescent substrate Ala-Pro-7-amino-3-trifluoromethylcoumarine (Ala-Pro-AFC; Bachem AG, Switzerland). Human FAP was expressed in baculovirus-infected Sf9 insect cells and purified using his-tagged protein purification resins. The inhibition of FAP activities was measured as described above. Ten microliters of the test compound solutions and 50 μL of 1.0 mM Ala-Pro-AFC in buffer solution (50 mM Tris hydrochloride–150 mM sodium chloride, 0.1 mg/mL BSA, pH 8.0) were added to black 96-well microplates. The reaction was initiated by the addition of 40 μL of the diluted enzyme solution for 60 min at room temperature. The fluorescence intensity was measured at an excitation wavelength of 400 nm and a fluorescence wavelength of 505 nm. To establish the standards, various concentrations of AFC solutions were added to a 96-well black microplate, and the fluorescence intensities were measured.
Docking simulation
The initial ligand structure was prepared from the single-crystal structure of anagliptin determined in this paper. All ligands for docking simulation were provided from anagliptin analogues using the LigPrep software package, version 2.8 (Schrödinger, LLC, New York, NY, 2013). These ligand-calculation conditions adopted conventional values. The molecular-docking simulation of compounds against the DPP-4 crystal structure, PDB code 3QWH, was performed by GlideCitation22.
Results and discussion
Crystal structure of anagliptin
The crystal structure of anagliptin was solved and refined by single-crystal X-ray analysis. Anagliptin has a single asymmetric carbon on its pyrrolidine moiety. Since the pyrrolidine moiety is prepared from l-prolinamide without racemization, the stereochemistry must be 2S, like that of l-prolinamide. The structure having a 2S configuration was therefore refined; the final R index was 0.0454 and the absolute structure parameter χCitation23 was 0.0. Consequently, it was clarified that the configuration was 2S (space group: P32). For comparison, a 2R configuration (space group: P31) was modelled and refined. In this case the final R index was 0.0459 and the χ parameter was 1.1; these parameters showed that the 2R configuration was not correct. The crystallographic data and refinement are shown in (CCDC 1003533).
Table 1. Crystallographic data and refinement of anagliptin.
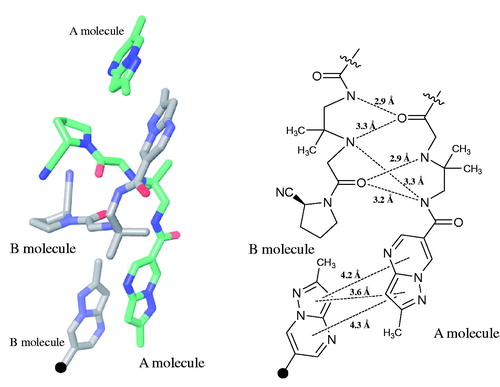
The solved structure consisted of two independent molecules which were formed by intermolecular hydrogen bonds; the ORTEP diagram is shown in . The ethylenediamine moieties and carbonyl groups linked to the pyrrolidine ring in the pair of molecules were contributed as proton donors and acceptors, respectively, and the pair of A and B molecules formed a crystalline lattice by helical π–π stackingCitation24 of the pyrazolopyrimidine rings ().
Figure 2. ORTEP diagram of the A and B molecules.
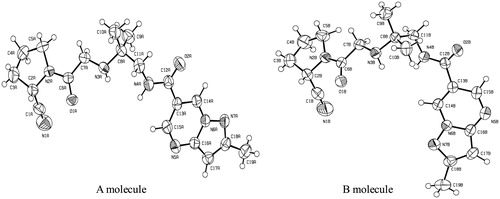
Figure 3. Interactions of the A and B molecules in single-crystals of anagliptin (left). The schematic diagram shows the distances of intermolecular hydrogen bonds and π/π stacking of A and B molecules (right).
Co-crystal structure of DPP-4 with anagliptin
The recombinant human DPP-4 expressed in the silkworm baculovirus system was purified by ion exchange column chromatography and gel filtration. The single crystals were grown from the complex with anagliptin by the sitting-drop vapour-diffusion method. The X-ray diffraction data were collected on BL41XU at SPring-8 at 100 K. The structure was solved by the molecular replacement method and refined. The crystallographic data and refinement data are shown in (PDB code 3WQH). The 2Fobs − Fcalc electron density map (σ = 1.1) around anagliptin after refinement is represented in .
Figure 4. 2Fobs − Fcalc electron density map (σ = 1.1, magenta) around anagliptin (green) after refinement.
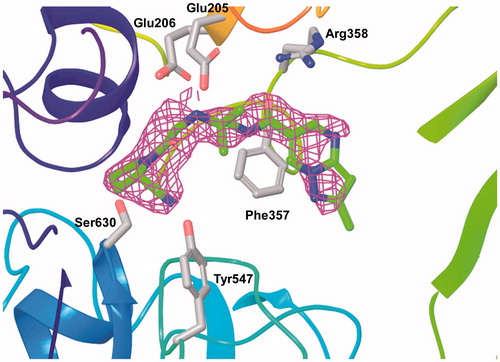
Table 2. Crystallographic data and refinement of the complex of DPP-4 with anagliptin.
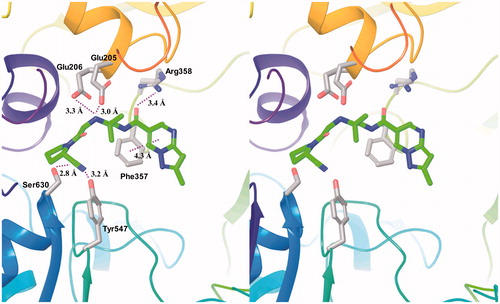
The solved DPP-4 structure complexed with anagliptin consisted of two independent protein molecules in an asymmetric unit, one anagliptin molecule bound to each protein molecule. The three interaction subsites of anagliptin with DPP-4 were revealed; shows the interactions with the B molecule of two proteins. (1) The first site was the S1 subsite, in which the cyano group had a dipole interaction. The distance between the cyano carbon atom and the Oγ of Ser630 (catalytic residue) was 2.8 Å, and the distance between the cyano nitrogen atom and the Oη of Tyr547 (proton donor) was 3.2 Å. (2) The second site was the S2 subsite, in which the secondary amine formed double salt bridges. The distances of 3.0 and 3.3 Å between the nitrogen atom of the secondary amine and each Oɛ of Glu205 and Glu206 were appropriate distances for salt-bridge formation. (3) The third site was the S2 extensive subsiteCitation13, in which the carbonyl group linked to the pyrazolopyrimidine ring formed a hydrogen bond with the Nη of Arg358, and the pyrazolopyrimidine ring underwent a π–π stacking interaction with the phenyl ring of Phe357. The distance of 3.4 Å between the oxygen atom of the carbonyl group and the Nη of Arg358 was within a hydrogen bond. The distance of 4.3 Å between the centres of each six-membered ring of the heterocycle and the phenyl ring of Phe357 was within an energetically-favoured range for π–π stackingCitation24.
Figure 5. Interactions of anagliptin in the active site of DPP-4. The distances of their interactions (dotted lines) are represented.
As described above, the interacting subsites of anagliptin with DPP-4 were the S1, S2 and S2 extensive subsites. Anagliptin should thus be included in class 3 according to the categorization by Nabeno et alCitation12. The covalent imidate intermediates with the Oγ of Ser630 in the S1 subsite are observed in the X-ray analyses of the inhibitors having a cyano group, vildagliptinCitation27 and saxagliptinCitation20.
However, the cyano group of anagliptin underwent dipole interactions in the S1 subsite. These interactions did not lead to covalent binding with the Oγ of Ser630, but could have resulted in a transition state in the formation of the covalent imidated intermediate. Based on the study of 14C-labelled anagliptin, the major metabolite of anagliptin is known to be a hydrolysis product of the cyano groupCitation28. The metabolic mechanism of anagliptin therefore likely proceeds through the S1 subsite via an imidate intermediate.
The S2 extensive subsite contributes to the affinity and selectivity as described in the characterizations of sitagliptinCitation15 and teneligliptinCitation29. The selectivity against DPP-4 homologues in particular has been reported to be important for safetyCitation11,Citation30. Rummey et al. reported the homology models of DPP8 and DPP9 based on the DPP-4 X-ray structure, and suggested that the selectivity of sitagliptin was based on the absence of key interactions in DPP8/9Citation31. In their models, the three residues of DPP-4 (Phe357, R358 and Ser209), which had the key interactions with sitagliptin, were not retained in either DPP8 or DPP9, and the corresponding residues in DPP8 and DPP9 lost the interactions with sitagliptin. Since anagliptin also exhibited interactions with Phe357 and Arg358 at the S2 extensive subsite, its selectivity can be accounted for in the same way.
Potency and selectivity of DPP-4 inhibitors
The in vitro activities of DPP-4 inhibitors against DPP-4 and its homologues DPP8, DPP9 and FAP were measured. Their selectivities were calculated from the IC50 values as shown in . The results showed that the IC50 values of vildagliptin and anagliptin were lower than those of sitagliptin and alogliptin. We considered that this difference was likely attributable to the different binding modes in the S1 subsite. Vildagliptin and anagliptin, both of which can form a covalent bond as described in the previous section, might bind more slowly than sitagliptin and alogliptin, which undergo hydrophobic interactions at the subsite. Presumably, since the incubation time was relatively long by this method, the IC50 values of vildagliptin and anagliptin were probably lowered due to their binding modes.
Table 3. In vitro IC50 values (nM) of DPP-4 inhibitors against enzymes.
With regard to the selectivity, anagliptin showed high selectivities against DPP8, DPP9 and FAP (). The high selectivities were probably attributable to the interactions with the Phe357 and Arg358 in the S2 subsite as discussed in the previous section.
SAR of anagliptin derivatives
Some of the anagliptin derivatives are shown in along with their inhibitory activitiesCitation14. The monosubstitution at the 7 position (R3) of the heterocycle tended to decrease the inhibitory activity (compounds 2 and 3), and disubstitution at the 5 and 7 positions (R2 and R3) significantly decreased the activity (compounds 4 and 5). In this X-ray study, the heterocycle and the linked carbonyl group of anagliptin were coplanar as shown in . These findings suggested that the substituent introductions at the ortho position of the heterocycle caused steric hindrance or distortion of the planarity, and thereby reduced the effects of the hydrogen bond of Arg358 and π–π stacking of Phe357 to decrease the IC50 values.
Table 4. In vitro IC50 values (nM) of and GlideScores of anagliptin derivatives. 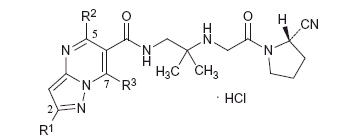
On the other hand, replacement of the methyl group by the tert-butyl group at the 2-position (R1) had no influence on the inhibitory activity, based on the comparisons between compounds 2 and 3 and between compounds 4 and 5 (). It was considered that the extra space around R1 allowed introduction of the bulky substituent.
We therefore employed the flexible ligand-docking program GlideCitation22 to assess the influences of the substituents at R1 based on the co-crystal structure of human DPP-4 with anagliptin determined in this study. The extra precision mode (XP mode) of Glide was adopted in this simulation, and the compound-inhibitory activities were estimated using the GlideScore value. The docking simulation showed that the best binding model of anagliptin in the active site of DPP-4 presented an RMSD of 1.1 Å relative to the X-ray structure determined in this study. Using these docking conditions, we obtained a moderate correlation coefficient of r = 0.801 between the log(IC50) values of the five compounds 1–5 and their GlideScores, as shown in . Virtual compounds 6–9 having polar and non-polar substituents were then evaluated under these docking conditions, as shown in . Interestingly, both virtual compounds 8 and 9 having a polar group at R1 were hydrogen-bonded to the Oη of Tyr585 as shown in ; these compounds had the better GlideScores, −8.55 and −8.59. In addition, this simulation suggested that T-shaped interactions occurred between compounds 8 and 9 and the phenyl ring of Phe357, respectively, although anagliptin showed a slipped parallel interaction with the Phe357. This simulation therefore indicated that compounds 8 and 9 simultaneously obtained their interaction energies for T-shaped interaction and hydrogen bonding. In their quantum chemical study of the benzene–dimer interaction, Tsuzuki et al. reported that the interaction energy of the slipped parallel interaction was approximately equal to that of the T-shaped interactionCitation24. Ninković et al. also recently reported that a large number of phenyl–phenyl contacts in proteins had tilt angles above 60°, corresponding to the T-shaped interaction in the aromatic–aromatic interaction study at a tilt angle range from 0° (parallel) to 90° (T-shaped) by using PDB dataCitation32. With regard to the hydrogen bonding with the Tyr585, Ahn et al. showed that a carboxyl substituent on the homopiperazine derivative appeared to interact with the Oη of Tyr585 in the co-crystal structure of DPP-4, and the acid moiety could regulate CYPs activitiesCitation33.
Figure 6. Co-crystal structure of anagliptin (red) determined by X-ray crystallography, and virtual compounds 8. (green) and 9. (blue) by the docking simulation.
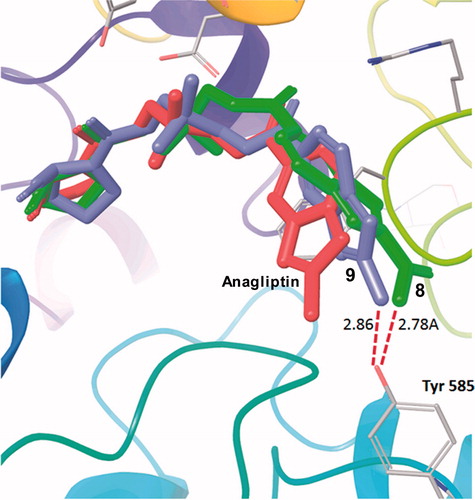
Table 5. GlideScores of virtual compounds by docking simulations*.
As a result of the SAR study, we therefore concluded that the introductions of polar substituents at R1 were likely to be an effective approach for further development of the derivatives.
Conclusion
The single-crystal X-ray analysis of anagliptin was performed. Anagliptin is not hygroscopic, but is very soluble in water. This favourable property for a pharmaceutical ingredient is probably attributable to the unique crystal structure. The intermolecular hydrogen bonds of the hydrophilic moieties and stackings of hydrophobic bicyclic-hetero rings could prevent contact with moisture, accounting for the non-hygroscopicity of the crystal, while the hydrophilic moieties could promote the solvation with water once the lattice breaks up due to contact with excess water.
The three interaction subsites of anagliptin with DPP-4 were revealed by the co-crystal structure analysis. Interestingly, a dipole interaction of the cyano group was observed in the S1 subsite in contrast to the covalent bonds observed in the analyses of vildagliptinCitation27 and saxagliptinCitation20. This novel non-covalent bond interaction of the cyano group was considered to be a transition state before formation of a covalent imidate, which was supported by the facts that both anagliptin and vildagliptin were highly potent in this analysis, and a major metabolite of anagliptin was a hydrolysed compound of the cyano group.
We concluded that the high selectivity of anagliptin was attributable to the utilization of the S2 extensive site as in the cases of sitagliptinCitation31 and teneligliptinCitation29.
Further, the SAR study of anagliptin derivatives reasonably accounted for the influence of the substituent on the heterocycle, which interacted at the S2 extensive site. The docking simulation results suggested a direction for further optimization.
Thus, the chemical structure of anagliptin was characterised and shown to have favourable properties for medicinal application. Continued study of the DPP-4 inhibitors, including anagliptin, is expected to underscore their efficacy for the treatment of type 2 diabetes.
Acknowledgements
We are grateful to Dr. Shigeru Sugiyama (Associate Professor, Osaka University, Japan) for useful suggestions and discussion.
Declaration of interest
The authors report no declarations of interest.
Related Research Data
References
- Hopsu-Havu VK, Glenner GG. A new dipeptide naphthylamidase hydrolyzing glycyl-prolyl-β-naphthylamide. Histochemie 1966;7:197–201
- Mentlein R, Gallwitz B, Schmidt WE. Dipeptidyl-peptidase IV hydrolyses gastric inhibitory polypeptide, glucagon-like peptide-1(7-36)amide, peptide histidine methionine and is responsible for their degradation in human serum. Eur J Biochem 1993;214:829–35
- Deacon CF, Hughes TE, Holst JJ. Dipeptidyl peptidase IV inhibition potentiates the insulinotropic effect of glucagon-like peptide-1 in the anesthetized pig. Diabetes 1998;47:764–9
- Holst JJ, Deacon CF. Inhibition of the activity of dipeptidyl-peptidase IV as a treatment for type 2 diabetes. Diabetes 1998;47:1663–70
- Rasmussen HB, Branner S, Wiberg FC, Wagtmann N. Crystal structure of human dipeptidyl peptidase IV/CD26 in complex with a substrate analog. Nat Struct Biol 2003;10:19–25
- Hiramatsu H, Kyono K, Higashiyama Y, et al. The structure and function of human dipeptidyl peptidase IV, possessing a unique eight-bladed β-propeller fold. Biochem Biophys Res Commun 2003;302:849–54
- Thoma R, Loffler B, Stihle M, et al. Structure basis of proline-specific exopeptidase activity as observed in human dipeptidyl peptidase-IV. Structure 2003;11:947–59
- Engel M, Hoffmann T, Wagner L, et al. The crystal structure of dipeptidyl peptidase IV (CD26) reveals its functional regulation and enzymatic mechanism. Proc Natl Acad Sci USA 2003;100:5063–8
- Oefner C, Arcy AD, Sweeney AM, et al. High-resolution structure of human apo dipeptidyl peptidase IV/CD26 and its complex with 1-[({2-[5-iodopyridin-2-yl)amino]-ethyl}amino)-acetyl]-2-cyano-(S)-pyrrolidine. Acta Crystallogr 2003;D59:1206–12
- Engel M, Hoffmann T, Manhart S, et al. Rigidity and flexibility of dipeptidyl peptidase IV: crystal structures of and docking experiments with DPIV. J Mol Biol 2006;355:768–83
- Juillerat-Jeanneret L. Dipeptidyl peptidase IV and its inhibitor: therapeutics for type 2 diabetes and what else? J Med Chem 2014;57:2197–212
- Nabeno M, Akahoshi F, Kishida H, et al. A comparative study of the binding modes of recently launched dipeptidyl peptidase IV inhibitors in the active site. Biochem Biophys Res Commun 2013;434:191–6
- Yoshida T, Akahoshi F, Sakashita H, et al. Fused bicyclic heteroarylpiperazine-substituted L-prolylthiazolidines as highly potent DPP-4 inhibitors lacking the electrophilic nitrile group. Bioorg Med Chem 2012;20:5033–41
- Kato N, Oka M, Murase T, et al. Discovery and pharmacological characterization of N-[2-({2-[(2S)-2-cyanopyrrolidine-1-yl]-2-oxoethyl}amino)-2-methylpropyl]-2-methylpyrazolo[1,5-a]pyrimidine-6-caroxamide hydrochloride (anagliptin hydrochloride salt) as a potent and selective DPP-IV inhibitor. Bioorg Med Chem 2011;19:7221–7
- Kim D, Wang L, Beconi M, et al. (2R)-4-Oxo-4-[3-(trifluoromethyl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl]-1-(2,4,5-trifluorophenyl)butan-2-amine: a potent, orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J Med Chem 2005;48:141–51
- Villhauer EB, Brinkman JA, Naderi GB, et al. 1-[[(3-Hydroxy-1-adamantyl)amino]acetyl]-2-cyano-(S)-pyrrolidine: a potent selective, and orally bioavailable dipeptidyl peptidase IV inhibitor with antihyperglycemic properties. J Med Chem 2003;46:2774–89
- Feng J, Zhang Z, Wallace MB, et al. Discovery of alogliptin: a potent, selective, bioavailable, and efficacious inhibitor of dipeptidyl peptidase IV. J Med Chem 2007;50:2297–300
- Adachi H, Takano K, Hosokawa Y, et al. Laser irradiated growth of protein crystal. Jpn J Appl Phys 2003;42:L798–800
- Lebedev AA, Vagin AA, Murshudov GN. Model preparation in MOLREP and examples of model improvement using X-ray data. Acta Crystallogr 2008;D64:33–9
- Metzler WJ, Yanchunas J, Weigelt C, et al. Involvement of DPP-IV catalytic residues in enzyme–saxagliptin complex formation. Protein Sci 2008;17:240–50
- Winn MD, Murshudov GN, Papiz MZ. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol 2003;374:300–21
- Glide version 6.1. New York (NY): Schrödinger, LLC; 2013
- Flack HD. On enantiomorph-polarity estimation. Acta Crystallogr 1983;A39:876–81
- Tsuzuki S, Honda K, Uchimaru T, et al. Origin of attraction and directionality of the π/π interaction: model chemistry calculations of benzene dimer interaction. J Am Chem Soc 2002;124:104–12
- Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science 2012;336:1030–33
- Lovell SC, Davis IW, Arendall WB III, et al. Structure validation by Cα geometry: φ, ψ and Cβ deviation. Proteins 2003;50:437–50
- Burkey BF, Russell M, Wang K, et al. Vildagliptin displays slow tight-binding to dipeptidyl peptidase (DPP)-4, but not DPP-8 or DPP-9. Presented at 42nd EASD meeting, Copenhagen, Denmark 2006; September 14–17:abstract0788
- Furuta S, Smart C, Hackett A, et al. Pharmacokinetics and metabolism of [14C]anagliptin, a novel dipeptidyl peptidase-4 inhibitor, in humans. Xenobiotica 2013;43:432–42
- Yoshida T, Akahoshi F, Sakashita H, et al. Discovery and preclinical profile of teneligliptin (3-[(2S,4S)-4-[4-(3-methyl-1-phenyl-1H-pyrazol-5-yl)piperazin-1-yl]pyrrolidin-2-ylcarbonyl]thiazolidine): a highly potent, selective, long-lasting and orally active dipeptidyl peptidase IV inhibitor for the treatment 2 diabetes. Bioorg Med Chem 2012;20:5705–19
- Lankas GR, Leiting B, Roy RS, et al. Dipeptidyl peptidase IV inhibition for the treatment of type 2 diabetes. Diabetes 2005;54:2988–94
- Rummey C, Metz G. Homology models of dipeptidyl peptidases 8 and 9 with a focus on loop predictions near the active site. Proteins 2007;66:160–71
- Ninković DB, Andrić JM, Malkov SN, Zarić SD. What are the preferred horizontal displacements of aromatic–aromatic interactions in proteins? Comparison with the calculated benzene–benzene potential energy surface. Phys Chem Chem Phys 2014;16:11173–7
- Ahn JH, Park WS, Jun MA, et al. Synthesis and biological evaluation of homopiperazine derivatives with β-aminoacyl group as dipeptidyl peptidase IV inhibitors. Bioorg Med Chem Lett 2008;18:6525–9