
Abstract
In the present article, we describe the inhibitory potency of nine thiosemicarbazide derivatives against bacterial type IIA topoisomerases, their antibacterial profile and molecular modelling evaluation. We found that one of the tested compounds, compound 7, significantly inhibits activity of Staphylococcus aureus DNA gyrase with an IC50 below 15 μM. Besides, this compound displays antibacterial activity on reference Staphylococuss spp. and Enterococcus faecalis strains as well as clinical S. aureus isolates at non-cytotoxic concentrations in mammalian cells with MIC values ranging from 16 to 32 μg/mL thereby indicating, in some cases, equipotent or even more effective action than standard drugs such as vancomycin, ampicillin and nitrofurantoin. The computational studies showed that both molecular geometry and the electron density distribution have a great impact on antibacterial activity of thiosemicarbazide derivatives.
Introduction
Improper use and overuse of antibiotics have led to evolution of multidrug-resistant bacteria, among which the ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter species) are of utmost concernCitation1. These pathogens cause not only the major part of hospital-acquired infections but also constitute a financial burden to healthcare systems all over the worldCitation2–4. The development of novel small-molecule antibacterial drugs is therefore urgently needed. At the dawn of the twentieth century, it was common belief that genomic and associated new technologies would deliver a plethora of novel antibacterial targetsCitation5,Citation6 and thus, new scaffold classes that lack cross-resistance to clinically used agents. Unfortunately, the efforts have not yet proven successful, and a significant research effort is still dedicated to the “old” but clinically validated targetsCitation7 to maintain a pipeline of effective antibioticsCitation8,Citation9. Of all the validated bacterial targets, bacterial type II topoisomerases (DNA gyrase and topoisomerase IV) are clinically proven attractive targets for antibacterial drug discoveryCitation10–13. Each of these enzymes is a tetrameric A2B2 complex (wherein the A subunits contain the DNA cleavage domain while the B subunits contain the ATP binding and hydrolysis domain) characterized by its ability to alter chromosome structure through breaking and rejoining double stranded DNA. In addition, each enzyme is independently essential for bacterial DNA replication. Gyrase is primarily responsible for introducing negative supercoils into conformationally constrained DNA, while topoisomerase IV primarily resolves linked chromosome dimers at the conclusion of DNA replicationCitation14–17. Inhibitors of DNA gyrase typically also inhibit bacterial topoisomerase IV, although the relative importance for expression of antibacterial activity can vary depending on the inhibitor and the bacterial speciesCitation18–20.
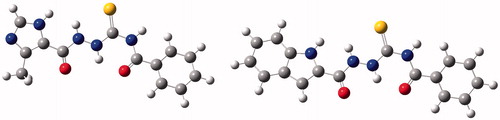
RecentlyCitation21, we showed for the first time inhibitory potency of two 4-benzoylthiosemicarbazides towards bacterial topoisomerase IV (). These compounds have a totally different chemical structure from the known antibiotics, which target bacterial topoisomerase IV such as the quinolones. It even differs structurally from the main types of antibiotics currently used, including β-lactams, glycopeptides, aminoglycosides, lipopeptides, quinolones, etc. Therefore, this class of compounds could offer a new way to combat the problem of infectious diseases induced by drug-resistant bacteria. To further explore the space available at the N1 and N4 positions of thiosemicarbazide core and, thus, to maximize binding to the bacterial topoisomerase target, we undertook additional synthesis in this class of compounds. Specifically, we have designed and prepared series of seven thiosemicarbazide derivatives (compounds 3–9) with geometries very similar to that of initial hits 1 and 2 and, for comparison, two thiosemicarbazide derivatives (compounds 10 and 11) of substantially different geometry. Subsequently, the obtained compounds were screened for inhibition of the topoisomerases DNA gyrase and topoisomerase IV isolated from the Gram-positive bacterium S. aureus and from the Gram-negative bacterium Escherichia coli. We found that one of tested compounds, that is 4-benzoyl-1-(2-methyl-furan-3-ylcarbonyl)thiosemicarbazide 7, significantly inhibits activity of S. aureus DNA gyrase with an IC50 below 15 μM. Moreover, the compound inhibited the growth of clinical isolates of S. aureus at MICs of 16 and 32 µg/mL, thereby indicating, in most cases, equipotent or even more effective action than some standard drugs such as vancomycin, ampicillin and nitrofurantoin. Based on computational studies we propose that both molecular geometry and the electron density distribution have a great impact on antibacterial activity of thiosemicarbazide derivatives.
Figure 1. Structures of previously reportedCitation21 4-benzoylthiosemicarbazides with inhibitory activities against S. aureus topoisomerase IV.
Methods
Inhibition of bacterial type IIA topoisomerases
Supercoiling assays: the assays were performed using S. aureus, and E. coli Gyrase Supercoiling Assay Kits (Inspiralis, Norwich, UK), respectively. Briefly, supercoiled pBR322 plasmid DNA (0.5 µg) was incubated with 1 unit of gyrase, in the dedicated supercoiling assay buffer supplied by the manufacturer, in the presence of varying concentrations of the compounds tested. Reactions were carried out at 37 °C for 1 h and then terminated by the addition of equal volume of 2 × STOP Buffer (40% sucrose, 100 mM Tris–Cl pH 7.5, 1 mM EDTA and 0.5 mg/mL bromophenol blue) and chloroform/isoamyl alcohol. Samples were vortexed, centrifuged and run through a 15 cm 1% agarose gel in TAE buffer (40 mM Trisacetate, 2 mM EDTA) for 3 h at 50 V. Gels were stained with ethidium bromide, visualized and photographed under UV light.
Decatenation assays: the assays were performed using S. aureus, and E. coli topoisomerase IV decatenation kits (Inspiralis), respectively. Interlinked kDNA substrate (0.5 µg) was incubated with 1 unit of topoisomerase IV (Inspiralis), in the dedicated decatenation assay buffer supplied by the manufacturer, in the presence of varying concentrations of the compounds tested. Reactions were carried out at 37 °C for 1 h and then terminated by the addition of equal volume of 2 × STOP Buffer (40% sucrose, 100 mM Tris–Cl pH 7.5, 1 mM EDTA, 0.5 mg/mL bromophenol blue) and chloroform/isoamyl alcohol. Samples were vortexed, centrifuged and run through a 15 cm 1% agarose gel in TAE buffer for 1.5 h at 80 V. Gels were stained with ethidium bromide, visualized and photographed under UV light. In order to determine concentrations of the compound that inhibited activity of the gyrase and topoisomerase IV by 50% (IC50 values), the intensity of the bands corresponding, respectively, to supercoiled DNA and decatenated minicircles was analyzed densitometrically using Quantity One software (BioRad, Hercules, CA). Briefly, bands of interest were detected manually and the width of analyzed area was adjusted to the band size. The background was then subtracted for each line using rolling disc method and the density for each band was reported as a peak density function. GraphPad Prism software (GraphPad Prism Software, Inc, La Jolla, CA) was used for plotting the results and calculation of IC50 values.
Antibacterial assay
The following reference microorganisms were used in this study: S. aureus ATCC 6538, S. aureus ATCC 29213, S. epidermidis ATCC 12228, E. faecalis ATCC 29212, E. coli NCTC 8192, P. vulgaris ATCC 49990, P. mirabilis ATCC 29906, P. aeruginosa NCTC 6249. Additionally, the analyzed compounds were tested against 12 clinical isolates of S. aureus from the collection belonging to the Chair of Immunology and Infectious Biology, University of Łódź, two multidrug-resistant MRSA (MDR-MRSA) pathogens isolated from hospitalized patients and, for comparison reasons, one reference MRSA ATCC 43300 strain from the collection belonging to the Department of Pharmaceutical Microbiology, Medical University of Lublin. Initially, antibacterial activity of thiosemicarbazide derivatives was screened on the basis of growth inhibition zone (giz) utilizing the disc diffusion method, according to the Clinical and Laboratory Standards Institute guidelinesCitation22. For compounds showing the inhibitory effect on the growth of tested bacteria, monitored as an appearance of giz, the minimal inhibitory concentrations (MICs) were determined using agar dilution method, according to the Clinical and Laboratory Standards Institute guidelinesCitation23. The MICs were defined as the lowest concentration of the compound preventing growth of the tested microorganism. In both methods, recommended Mueller-Hinton II agar medium (Becton Dickinson, Franklin Lakes, NJ) was used. Solutions containing the tested agents were prepared in methanol or DMSO. Ciprofloxacin, vancomycin, nitrofurantoin, ampicillin and streptomycin were used as control antimicrobial agents. Characteristics and handling of clinical S. aureus strains were described elsewhereCitation24.
Cytotoxic assay
Cytotoxic effect of tested compounds on host cells was detected by determining cellular viability using MTT reduction assay. Murine fibroblast cells L929 (ATTC® catalog no. CCL-1™, recommended by the International Standard ISO 10993:2009 for evaluation of cytotoxic activities) or human HeLa cells (ATTC® catalog no. CCL-2™) were plated in 96-well microplates at a density of 1 × 104 cells/mL (100 μL per well) and cultivated in Iscove’s modified Dulbecco’s medium (IMDM), supplemented with 100.0 U/mL penicillin and streptomycin, 5 × 10−5 M 2-mercaptoethanol and enriched with 10% fetal bovine serum (FBS). After overnight incubation at 37 °C, the growth medium was removed and 100 μL of medium supplemented with different concentrations of compounds in the range of 5–300 µg/mL were added. Cells were further incubated for 24 h with tested agents. At the end of the incubation time, the medium was removed and MTT was added to each well at a final concentration of 0.5 mg/mL and plates were incubated for the next 2 h at 37 °C. Then, formazan crystals were solubilized in 150 μL DMSO. The optical density was measured at 550 nm. The results of the experiments were shown as mean arithmetic values of eight repeats (two experiments) and CC30 values were determined.
Results and discussion
Rationale and chemistry
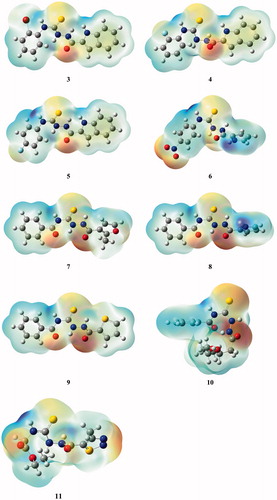
As mentioned in the Introduction, recentlyCitation21, we described initial prototype of a novel class of bacterial topoisomerase IV inhibitors, that is 4-benzoyl-1-(4-methyl-imidazol-5-ylcarbonyl)-thiosemicarbazide 1 () with promising antibacterial activity. This compound showed inhibitory potency against topoisomerase IV from the Gram-positive bacterium S. aureus with an IC50 of 90 μM and had potent antibacterial activity against a panel of Gram-positive organisms, especially against S. epidermidis, M. luteus and B. subtilis, with MIC range of 0.98–7.81 μg/mL. Using a molecular modelling approach, its new derivative with very similar geometry was designed and prepared. The resulting 4-benzoyl-1-(indol-2-ylcarbonyl)thiosemicarbazide, 2, () was found to be much more potent inhibitor of S. aureus topoisomerase IV with an IC50 of 14 μM. Unfortunately, the improvement in enzyme potency did not resulted in an improvement in antibacterial potency; compound 2 exhibited inhibiting effects against the growth of S. epidermidis, M. luteus and B. subtilis at a concentration of 50 μg/mL. Factors that may limit its antibacterial potency could include lack of penetration of the cell wall or membrane, removal of compound by active efflux mechanisms and alteration of the sensitivity of the target enzyme in its “biophase” in the bacteriumCitation25. In order to determine if any further improvements could be obtained in inhibitory potency and, consequently, in antibacterial activity of the class of thiosemicarbazides, a series of seven 4-benzoyl(aryl)thiosemicarbazides, 3–9, with molecular geometries very similar to that of initial hits 1 and 2 and, for comparison, two 4-aryl(alkyl)-thiosemicarbazides, 10 and 11, of substantially different geometry ( and ) were designed and subsequently prepared using an easy one-step synthesisCitation26–28 () with commercially available starting materials. Details of experimental procedures and physicochemical characterization of title compounds are given in Supplementary Material.
Figure 2. Overlay of the structures of hit 2 and representative model compounds 7 and 11. Geometries of the remaining thiosemicarbazides 3–6 and 8–10 are given in .
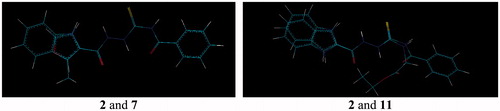
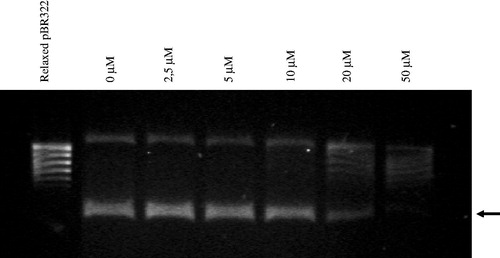
Figure 3. Inhibitory activity of furane derivative 7 against S. aureus gyrase. The arrow shows position of a supercoiled DNA.
Scheme 1. Synthetic route for thiosemicarbazide derivatives 3–11 (for symbols Ri and Rj used to identify studied compounds, see ).
Effects of thiosemicarbazide derivatives 3–11 on the activity of bacterial type IIA topoisomerases
The thiosemicarbazide derivatives 3–11 were screened for inhibition of the DNA gyrase and topoisomerase IV isolated from the Gram-positive bacterium S. aureus and from the Gram-negative bacterium E. coli. Based on the results of enzymatic studies () we have concluded that inhibitory potency of the studied compounds is very enzyme-specific and the following structure–activity trends can be identified:
the best inhibitory activity against DNA gyrase from S. aureus was observed for furane derivative 7 (), with an IC50 of 14.59 µM. The replacement of furane moiety in 7 with two other azole substituents or an alkoxy chain leads to essential changes in bioactivity of 4-benzoylthiosemicarbazides (compounds 7–10) with pyrrole 8 and thiophene 9 derivatives being much worse inhibitors than 7 (IC50 93.30 and 83.63 µM, respectively) and diethoxypropionyl derivative 10 being inactive. The inhibitory activities against S. aureus DNA gyrase were also observed for 4-arylthiosemicarbazides (compounds 3–6) with indole 5 and pyrrole 6 substitution, however, at much higher concentration as compared to 7 (IC50 127.68 and 64.21 µM, respectively).
the inhibitory activities of all studied thiosemicarbazide derivatives against DNA gyrase from E. coli were in general lower in comparison to DNA gyrase from S. aureus, with IC50 in the range of 85.46–285.21 µg/mL. As can be seen from the data collected in , of all 4-benzoylthiosemicarbazides, the inhibitory activities was noted for pyrrole derivative 8 (IC50 111.45 µg/mL) and thiophene derivative 9 (IC50 85.46 µg/mL). Within series of 4-arylthiosemicarbazides, compound with indole substitution 3 showed inhibitory action at concentration comparable to 8 (IC50 141.07 µg/mL) while bioactivity of pyrrole derivative 6 was negligible (IC50 285.21 µg/mL).
the best inhibitory activity against topoisomerase IV from S. aureus was observed for 4-benzoylthiosemicarbazide with pyrrole substitution (compound 8, IC50 41.04 µM). Remaining 4-benzoylthiosemicarbazides were inactive. Within series of 4-arylthiosemicarbazides, the inhibitory activity was observed for two indole derivatives with ortho-fluoro 4 (IC50 63.47 µM) and para-fluoro substitution 5 (IC50 267.04 µM).
the inhibitory activity against topoisomerase IV from E. coli was observed only for two compounds with pyrrole substitution, that is, for 6 (IC50 120.48 µg/mL) and for 8 (IC50 114.08 µM).
Table 1. Chemical structures of thiosemicarbazide derivatives 3–11.
Table 2. Inhibitory activity [IC50, µM] against bacterial type IIA topoisomerases for thiosemicarbazide derivatives 3–11.
Correlation of geometric and electronic structure with inhibitory activity against bacterial type IIA topoisomerases
Based on the enzymatic studies we can conclude that of all the thiosemicarbazide derivatives 3–11, only 4-benzoyl-1-(2-methyl-furan-3-ylcarbonyl)thiosemicarbazide 7 with an IC50 below 15 µM against DNA gyrase from S. aureus exhibited satisfactory level of inhibitory potency. In the search for the origin of the inhibitory potency of 7 against S. aureus DNA gyrase, we have studied in details its molecular structure with hope to correlate the structural and/or electronic features with observed bioactivity. Since the Amber force field (as implemented in HyperChem) provides excellent prediction of geometries of the thiosemicarbazide class of compoundsCitation29, it has been used in calculations of conformations of all title thiosemicarbazides. As can be seen from , all thiosemicarbazide derivatives with at least marginal inhibitory potency against bacterial topoisomerase(s) (compounds 3–9) have very similar conformations while geometry of inactive compounds 10 and 11 substantially differed from these of 3–9. However, no direct structure–activity relation for the most potent inhibitor 7 was found when the geometries of active derivatives 3–9 were studied in details. As illustrated in Figure S1, the most stable conformations of 7 and compounds with substantially weaker inhibitory potency against S. aureus DNA gyrase, that is compounds 9 with IC50 at ∼84 μM and 8 with IC50 at ∼93 μM are almost superimposable while 6 (IC50 ∼64 μM) and the weakest S. aureus DNA gyrase inhibitor 5 (IC50 ∼128 μM) show only minor deviations. Thus, it seems that the geometry of molecule is necessary but not sufficient factor for the binding of 7 to S. aureus DNA gyrase and this conclusion is in line with the results of our previous experimentsCitation30. Following these findings, the electrostatic potential maps for title compounds were calculated for the geometries that resulted from RM1 calculations using Gaussian 03 and GaussView 5 at the HF/6-31G level. We have recently showedCitation31 that RM1 parametrization performs very well for compounds with N–N–C(=S)–N core. The generated electrostatic potential maps () reveal that the center for the most negative potential (red region) in all studied thiosemicarbazide derivatives lies in the vicinity of the oxygen and sulphur atoms whereas the center for most positive potential (most blue region) lies near the nitrogen atoms. Again, comparison of the frontier molecular orbitals (Figure S2) leads to the conclusion that there is no relationship between HOMO distributions and inhibitory potency of 7. A similar conclusion can be drawn from analysis of the LUMO distributions.
Figure 4. Comparison of the geometries and electrostatic potential surfaces of thiosemicarbazide derivatives 3–11.
Docking studies
In attempt to identify the binding site of 7 we have performed docking studies using two crystal structures of S. aureus DNA gyrase deposited in the Protein Data Bank, one containing gyrA fragment with the DNA binding pocket (4BUL) and the other containing gyrB fragment with the ATP binding site (3U2K). FlexX and HYDE docking algorithms as implemented in LeadIT platform of BioSolveIT program were used with default parameters. While quantitative conclusions are not possible due to the lack of consistent set of bioactivity indexes of compounds involved in comparison, similarity of scores of 7 and GSK299423, the latter being a potent inhibitor of gyrA siteCitation32, and significant difference from the score of 4-[4-[(3,4-dichloro-5-methyl-1H-pyrrole-2-carbonyl)amino]-1-piperidyl]quinolone-2-carboxylic acid, a potent inhibitor of gyrB siteCitation33, seems to indicate that 7 binds to the DNA binding pocket. This observation is opposite to the conclusion we have arrived at in our previous studiesCitation34 and may indicate that thiosemicarbazide derivatives can inhibit both sites of gyrase. This conclusion is, however, very preliminary, as there is not enough structural and bioactivity data and thus awaits verification in the future.
Correlation of physicochemical parameters with inhibitory activity against bacterial type IIA topoisomerases
Finally, we have calculated several parameters, such as lipophilicity (clogP), hydratation energy (HE), surface area (SA), volume (V), refractivity (Rf), polarizabilitiy (α), dipole moment (μ), the highest occupied molecular orbital energy (EHOMO), the lowest unoccupied molecular orbital energy (ELUMO), the difference between HOMO and LUMO energy levels (HLG) and heat of formation (HF) that are frequently used in SAR studies. An examination of results collected in Table S1 showed that the total dipole moment might be directly related to inhibition of S. aureus DNA gyrase activity, meaning that the most potent inhibitor of this study 7 possess relatively lower value of the dipole moment than remaining compounds. The dipole moment is a global molecular property, which is related to the molecule’s polarity. As known, hydrophobic/hydrophilic properties of molecule are quite important to the diffusion of compound through the biological system. Additionally, the environment of a binding site in enzyme is generally hydrophobicCitation35 and the partition complementarity with the respective ligand is crucial for the biological response. In this regard, compounds devoid of any inhibitory activity, that is, compounds 10 and 11 in our studies, should also present higher value of total dipole moment than compounds with at least marginal inhibitory potency and the data presented in Table S1 confirmed this assumption. The second important result obtained from the physicochemical parameters analysis is that the most potent inhibitor 7 possesses the highest value of energy of hydration that is also deeply related to hydrophobic/hydrophilic balance of the molecule. Of course, our observations on the impact of dipole moment and energy of hydration on inhibitory potency of thiosemicarbazides against DNA gyrase are very preliminary, as there is not enough large set of compounds involved in comparison and thus awaits verification in the future. On the other hand, however, the analysis of physicochemical parameters allowed to eliminate those that apparently do not influence with bioactivity of the studied compounds.
Antibacterial activity of thiosemicarbazide derivatives 3–11 against a panel of reference strains and clinical isolates of S. aureus
In order to correlate the observed enzyme potency with the cellular activity, the enzymatic studies were completed by the evaluation of antibacterial activity of compounds 7, 9 and 10 against a panel of Gram-positive and Gram-negative reference bacterial strains. The antimicrobial results for the MIC, defined as the lowest concentration of the compound preventing the growth of the tested microorganism, are reported in . The results of antibacterial screening for remaining compounds 3–6, 8 and 11 were presented in our previous contributionsCitation30,Citation34,Citation36 and, for comparison reasons, are also included in . As the graph indicates, with the exception of compounds 8 with MIC values ranging from 25 to 50 μg/mL against Gram-positive bacteria and 5 with MICs at 25 and 50 μg/mL against M. luteus, all tested thiosemicarbazide derivatives had very limited antibacterial activity (MIC ≥ 100 μg/mL) or even did not show it at all.
Table 3. In vitro antibacterial activity [MIC, µg/mL] of thiosemicarbazides 3–11 against a panel of reference strains.
Screening of the antibacterial activity of 7, 9 and 10 against reference strains revealed that compound 7 was effective against S. aureus, S. epidermidis and E. faecalis with MICs at 25 µg/mL while compounds 9 and 10 inhibited growth of only S. aureus, however, at high concentrations (MICs at 200 µg/mL). Thus, we can conclude that of all studied thiosemicarbazide derivatives 3–11, compound 7 with the most potent inhibitory activity against S. aureus DNA gyrase and compound 8 sharing dual inhibition of DNA gyrase and topoisomerase IV isolated from S. aureus were recognized in our antimicrobial studies as the most potent antibacterial agents.
Subsequently, compounds 3–5 and 7–10, all of which showed at least marginal antibacterial response against reference strains, were also examined for their inhibitory activity against 12 S. aureus clinical isolates. The vancomycin, ampicillin, nitrofurantoin and streptomycin were included as a control antibiotics.
As is widely known, the misuse and overuse of antibiotics promoted the rise of multi-antibiotic resistant bacteriaCitation37. These include methicillin-resistant S. aureus (MRSA) that, according to European Centre for Disease Prevention and Control, are the most frequently identified antibiotic resistant pathogens in hospitals in Europe, the Americas, North Africa and the Middle and Far East, largely responsible for both minor and serious to even life threatening infectionsCitation38. At present, the most potent antibiotic that is frequently used to treat invasive MRSA infections is vancomycin. However, the escalation of vancomycin MICs for MRSA, poor tissues penetrationCitation39 and loss of accessory gene-regulator function in MRSACitation40 are major reasons for clinical failure of vancomycin in treating deep-seated infections. Consequently, there is an urgent need to discover new antibacterial classes with activity against drug-resistant pathogens that do not rapidly succumb to resistance.
Fortunately, the results of antibacterial assay for compounds 3–5 and 7–10 against S. aureus clinical isolates are really promising. As seen from data collected in , compound 7 inhibited the growth of clinical isolates in populations originating from the nasopharynx, ulcers/furuncles and bones at MICs of 16 and 32 µg/mL, thereby indicating, with five exceptions, equipotent or even more effective action than those standard drugs vancomycin, ampicillin and nitrofurantoin. Also two other compounds, that is 8 and 9, with MICs at 32 and 64 µg/mL proved, in the great majority of cases, to be as effective or even more than vancomycin and ampicillin. Moreover, compound 8 with MICs at 32 µg/mL displayed activity equipotent to control antibiotic nitrofurantoin against two ulcers/furuncles, one nasopharynx and one bone isolates. Although bioactivity of remaining compounds 3–5 and 10 with MICs at 128 and 256 µg/mL was generally negligible, the compounds in most cases were still just as active or even more so than vancomycin and ampicillin.
Table 4. In vitro antibacterial activity [MIC, µg/mL] of thiosemicarbazides 3–5 and 7–10 against S. aureus clinical isolates.
Finally, the inhibitory effect of compounds 3–5 and 7–10 on two multidrug-resistant MRSA (MDR-MRSA) pathogens isolated from hospitalized patients and, for comparison reasons, one reference MRSA strain was tested. Again, 4-benzoyl-1-(2-methyl-furan-3-ylcarbonyl)thiosemicarbazide 7 with MICs at 16 µg/mL had the most effective antibacterial action. Two other benzoylthiosemicarbazide derivatives 8 and 9 showed inhibitory action at a somewhat higher concentrations (MICs at 16 and 32 µg/mL) while bioactivity of remaining compounds was negligible; MICs at 128 or even at 256 µg/mL.
Based on detailed analysis of molecular structure of studied compounds it can be concluded that there is a connection between antimicrobial activity and chemical properties of the studied compounds. Firstly, the structure of the most active thiosemicarbazides (7–9) differs from the two derivatives 10 and 11 that have an extra methyl group that causes these compounds to be more globular. Compounds 3–6, on the other hand, are missing second carbonyl group . As illustrated in this results in a different electron density distribution. Its analysis for the studied compounds seems to indicate that two negative charges of carbonyl oxygen atoms located on the same side of the molecule are required for the bioactivity.
Cytotoxicity of thiosemicarbazide derivatives 3–5 and 7–10 against L929 and HeLa cells
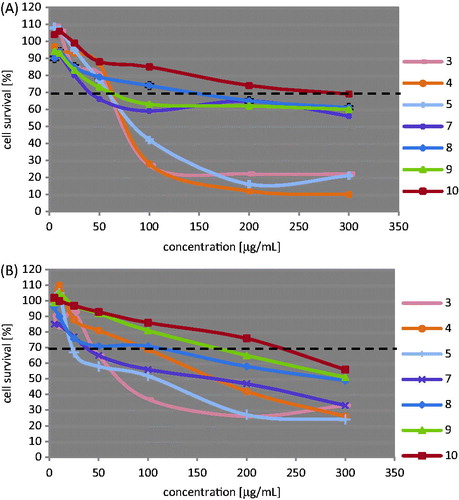
Compounds 3–5 and 7–10, all of which showed at least marginal antibacterial response, were also examined for their initial in vitro toxicity effects against mammalian L929 and HeLa cells. Cytotoxicity, expressed as CC30, was defined as the highest dilution of test samples that causes 30% or greater destruction of cells.
As known, successful antibacterial compounds must have physicochemical properties suitable for achieving relatively high doses with acceptable safety margins, otherwise the activity might just be due to general toxicity, which disqualifies them as a drug or lead molecule candidate. From a comparison of the results of toxicity collected in and antibacterial activity tests it can be seen that two the most promising antibacterial agents found in our antimicrobial studies, that is compounds 7 (CC30 42 μg/mL against L929 and 37 μg/mL against HeLa cells) and 8 (CC30 145 and 109 μg/mL, respectively) display antibacterial activity at non-cytotoxic concentrations for mammalian cells. The second important result taken from the cytotoxic studies is that the remaining compounds with the CC30 values in the range of 59–273 μg/mL for L929, and 23–234 μg/mL for HeLa cells were even less toxic than lead 7 under the same conditions. Unfortunately, in contrast to 7–9, they showed only marginal level of antibacterial activity against all tested microorganisms.
Figure 5. Concentration-dependent cytotoxic activity of thiosemicarbazide derivatives 3–5 and 7–10 against L929 (A) and HeLa (B) cell lines.
Conclusions
In conclusion, we have evaluated a series of thiosemicarbzide derivatives as inhibitors of four bacterial type IIA topoisomerases from S. aureus and E. coli. All these enzymes were inhibited with micromolar efficacies, depending on the substitution pattern at the nitrogen atoms from the thiosemicarbazide group. The presence of furane moiety at N1 position of 4-benzoyltiosemicarbazide led to potent S. aureus DNA gyrase inhibitor with IC50 below 15 μM. In order to correlate the observed enzyme potency with the cellular activity, the enzymatic studies were completed by the evaluation of antibacterial activity of title tiosemicarbazides against a panel of reference strains and clinical isolates of S. aureus and MDR-MRSA. New lead structure 7 with activity equipotent or even more effective than those standard drugs vancomycin, ampicillin and nitrofurantoin was identified that will direct the production of future structural analogues. We consider that at least three important aspects can be derived from these studies. First, although bacterial topoisomerases share several structural features, the inhibitory potency of thiosemicarbazide derivatives seems to be very enzyme-specific. Second, at least two factors, i.e. geometry of molecule and hydrophobic/hydrophilic balance are important molecular properties for developing thiosemicarbazides as potent S. aureus DNA gyrase inhibitors. Third, both molecular geometry and the electron density distribution have a great impact on antibacterial activity of this class of compounds.
Supplementary material available online
Experimental and computational procedures, Figures S1, S2 and Table S1.
Supplemental Material.pdf
Download PDF (598.1 KB)Declaration of interest
The authors declare no conflict of interest.
The project was funded by the National Science Centre, Poland (decision numbers: 2012/05/D/NZ7/02278 and UMO-2013/11/N/NZ7/00765). Part of the experiments was performed using equipment of the Laboratory of Microscopic Imaging and Specialized Biological Techniques, Faculty of Biology and Environmental Protection, University of Łódź.
References
- Rice LB. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J Infect Dis 2008;197:1079–81
- Spellberg B, Blaser M, Guidos RJ, et al. Combating antimicrobial resistance: policy recommendations to save lives. Clin Infect Dis 2011;52:S397–428
- Klevens RM, Edwards JR, Richards Jr CL, et al. Estimating health care-associated infections and deaths in US hospitals, 2002. Public Health Rep 2007;122:160–6
- Eber MR, Laxminarayan R, Perencevich EN, Malani A. Clinical and economic outcomes attributable to health care-associated sepsis and pneumonia. Arch Intern Med 2010;170:347–53
- Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov 2007;6:29–40
- Silver LL. Challenges of antibacterial discovery. Clin Microbiol Rev 2011;24:71–109
- East SP, Silver LL. Multitarget ligands in antibacterial research: progress and opportunities. Exp Opin Drug Discov 2013;8:143–56
- Butler MS, Cooper MA. Antibiotics in the clinical pipeline in 2011. J Antibiot 2011;64:413–25
- Boucher HW, Talbot GH, Benjamin Jr DK, et al. 10 × ’20 Progress-development of new drugs active against gram-negative bacilli: an update from the Infectious Diseases Society of America. Clin Infect Dis 2013;56:1685–94
- Maxwell A. DNA gyrase as a drug target. Trends Microbiol 1997;5:102–9
- Maxwell A, Lawson DM. The ATP binding site of type II topoisomerases as a target for antibacterial drugs. Curr Top Med Chem 2003;3:283–303
- Drlica K, Zhao X. DNA gyrase, topisomerase IV and the 4-quinolones. Microbiol Mol Biol Rev 1997;61:377–92
- Heisig P. Inhibitors of bacterial topoisomerases: mechanisms of action and resistance and clinical aspects. Planta Med 2001;67:3–12
- Ferrero L, Cameron B, Manse B, et al. Cloning and primary structure of Staphylococcus aureus DNA topoisomerase IV: a primary target of fluoroquinolones. Mol Microbiol 1994;13:641–53
- Kato J-I, Nishimura Y, Imamura R, et al. New topoisomerases essential for chromosome segregation in E. coli. Cell 1990;63:393–404
- Deibler RW, Rahmati S, Zechiedrich EL. Topoisomerase IV, alone, unknots DNA in E. coli. Genes Dev 2001;15:748–61
- Zechiedrich EL, Khodrusky AB, Bachellier S, et al. Roles of topoisomerases in maintaining steady-state DNA supercoiling in Escherichia coli. J Biol Chem 2000;275:8103–13
- Fabrega A, Madurga S, Giralt E, Vila J. Mechanism of action of and resistance to quinolones. Microb Biotechnol 2009;2:40–61
- Bolon MK. The newer fluoroquinolones. Med Clin North Am 2011;95:793–817
- Neuhauser MM, Weinstein RA, Rydman R, et al. Antibiotic resistance among Gram-negative bacilli in US intensive care units: implications for fluoroquinolone use. JAMA, J Am Med Assoc 2003;289:885–8
- Siwek A, Stączek P, Wujec M, et al. Biological and docking studies of topoisomerase IV inhibition by thiosemicarbazides. J Mol Model 2011;17:2297–303
- Clinical and Laboratory Standards Institute. Performance standards for antimicrobial disc susceptibility tests; approved standard M2-A9. Wayne (PA): Clinical and Laboratory Standards Institute; 2006
- Clinical and Laboratory Standards Institute. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard M7-A7. Wayne (PA): Clinical and Laboratory Standards Institute; 2006
- Pieczonka AM, Strzelczyk A, Sadowska B, et al. 2013. Synthesis and evaluation of antimicrobial activity of 3-oxido-1H-imidazole-4-carbohydrazides hydrazones. Eur J Med Chem 2013;64:389–95
- Zhi Ch, Long Z-y, Manikowski A, et al. Hybrid antibacterials. DNA polymerase–topoisomerase inhibitors. J Med Chem 2006;49:1455–65
- Wujec M, Kędzierska E, Kuśmierz E, et al. Pharmacological and structure–activity relationship evaluation of 4-aryl-1-diphenylacetyl(thio)semicarbazides. Molecules 2014;19:4745–59
- Plech T, Wujec M, Siwek A, et al. Synthesis and antimicrobial activity of thiosemicarbazides, s-triazoles and their Mannich bases bearing 3-chlorophenyl moiety. Eur J Med Chem 2011;46:241–8
- Siwek A, Stefańska J, Dzitko K, Ruszczak A. Antifungal effect of 4-arylthiosemicarbazides against Candida species. Search for molecular basis of antifungal activity of thiosemicarbazide derivatives. J Mol Model 2012;18:4159–70
- Siwek A, Świderek K, Jankowski S. Problems with molecular mechanics implementations on the example of 4-benzoyl-1-(4-methyl-imidazol-5-yl)-carbonylthiosemicarbazide. J Mol Model 2012;18:843–9
- Siwek A, Stączek P, Kosikowska U, et al. Does dehydrocyclization of 4-benzoylthiosemicarbazides in acetic acid lead to s-triazoles or thiadiazoles? Struct Chem 2012;231:1441–8
- Siwek A, Wujec M, Stefańska J, Paneth P. Antimicrobial properties of 4-aryl-3-(2-methyl-furan-3-yl)-Δ2-1,2,4-triazoline-5-thiones. Phosphorus Sulpur 2009;184:3149–59
- Bax BD, Chan PF, Eggleston DS, et al. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 2010;466:935–40
- Eakin AE, Green O, Hales N, et al. Pyrrolamide DNA gyrase inhibitors: fragment-based nuclear magnetic resonance screening to identify antibacterial agents. Antimicrob Agents Chemother 2012;56:1240–6
- Siwek A, Stączek P, Stefańska J. Synthesis and structure–activity relationship studies of 4-arylthiosemicarbazides as topoisomerase IV inhibitors with Gram-positive antibacterial activity. Search for molecular basis of antibacterial activity of thiosemicarbazides. Eur J Med Chem 2011;46:5717–26
- Palace-Berl F, Jorge SD, Pasqualoto KFM, et al. 5-Nitro-2-furfuriliden derivatives as potential anti-trypanosoma cruzi agents: design, synthesis, bioactivity evaluation, cytotoxicity and exploratory data analysis. Bioorg Med Chem 2013;21:5395–406
- Siwek A, Stefańska J, Wawrzycka-Gorczyca I, Wujec M. Synthesis and in vitro antibacterial evaluation of 1-substituted-4-ethoxycarbonylmethylthiosemicarbazides and products of their dehydrocyclization. Heteroatom Chem 2010;21:131–8
- (a) Boucher HW, Talbot GH, Bradley JS, et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis 2009;48:1–12. (b) Spellberg B, Guidos R, Gilbert D, et al. The epidemic of antibiotic-resistant infections: a call to action for the Medical Community from the Infectious Diseases Society of America. Clin Infect Dis 2008;46:155–64
- European Centre for Disease Prevention and Control. Antimicrobial resistance surveillance in Europe 2009. Annual report of the European Antimicrobial Resistance Surveillance Network (EARS-Net). Stockholm: ECDC; 2010
- (a) Albanèse J, Léone M, Bruguerolle B, et al. Cerebrospinal fluid penetration and pharmacokinetics of vancomycin administered by continuous infusion to mechanically ventilated patients in an intensive care unit. Antimicrob Agents Chemother 2000;44:1356–8. (b) Cruciani M, Gatti G, Lazzarini L, et al. Penetration of vancomycin into human lung tissue. J Antimicrob Chemother 1996;38:865–9. (c) Lamer C, de Beco V, Soler P, et al. Analysis of vancomycin entry into pulmonary lining fluid by bronchoalveolar lavage in critically ill patients. Antimicrob Agents Chemother 1993;37:281–6
- Sakoulas G, Moellering Jr RC, Eliopoulos GM. Adaptation of methicillin-resistant Staphylococcus aureus in the face of vancomycin therapy. Clin Infect Dis 2006;42:S40–50