
Abstract
Two viral proteases of severe acute respiratory syndrome coronavirus (SARS-CoV), a chymotrypsin-like protease (3CLpro) and a papain-like protease (PLpro) are attractive targets for the development of anti-SARS drugs. In this study, nine alkylated chalcones (1–9) and four coumarins (10–13) were isolated from Angelica keiskei, and the inhibitory activities of these constituents against SARS-CoV proteases (3CLpro and PLpro) were determined (cell-free/based). Of the isolated alkylated chalcones, chalcone 6, containing the perhydroxyl group, exhibited the most potent 3CLpro and PLpro inhibitory activity with IC50 values of 11.4 and 1.2 µM. Our detailed protein-inhibitor mechanistic analysis of these species indicated that the chalcones exhibited competitive inhibition characteristics to the SARS-CoV 3CLpro, whereas noncompetitive inhibition was observed with the SARS-CoV PLpro.
Introduction
Severe acute respiratory syndrome coronavirus (SARS-CoV) is a zoonotic single-stranded positive-strand RNA virusCitation1. SARS, a contagious and often fatal respiratory illness, was first reported in the Guangdong Province of China in November 2002. The virus’s rapid and unexpected spread to other Asian countries, North America and Europe alarmed both the public and the World Health Organization (WHO)Citation2. The SARS genome contains 14 open reading frames (ORFs). ORF1a and ORF1b encode the replicase proteins and are processed primarily by the virally encoded chymotrypsin-like protease (3CLpro) and papain-like protease (PLpro)Citation3–5. These two proteases catalyze their own release and liberate other nonstructural proteins (nsps) from the polyproteinCitation4–6. Therefore, the enzymatic activities of both PLpro and 3CLpro are essential for the viral life cycle, and both enzymes are attractive targets for the development of antiviral drugs directed against SARS-CoV and other coronavirus infectionsCitation7. Despite numerous biochemical, structural and inhibitor (peptidomimetics and substrate analogs)-development studies directed at 3CLpro and PLproCitation8, a potent antiviral drug that directly targets the proteases has yet to be developed. To solve these problems, many researchers are interested in developing anti-SARS nutritional foods, food medicines and even new drugs developed from plants, which contain a rich source of bioactive components with fewer side effects. Until now, SARS-CoV PLpro inhibitors have been reported from many synthetic peptidyl compound libraries, but little natural compounds have been shown to exhibit PLpro inhibition. The natural-derived inhibitors include diarylheptanoidsCitation9, tanshinonesCitation10, flavonoidsCitation11 and cinnamic amidesCitation12.
During our search for positional anti-SARS-CoV agents from medicinal plants and foodstuffs, we found that the EtOAc-soluble fraction of an ethanol extract of Angelica keiskei showed significant inhibition against 3CLpro (75% inhibition at 30 µg/ml) and PLpro (88% inhibition at 30 µg/ml). A. keiskei is a large perennial plant belonging to the Umbelliferae family, and its leaves are used as a folk medicine and in health-promoting foods. A. keiskei contains bioactive chalcones, flavanones and coumarinsCitation13–16 that exhibit antioxidant, antidiabeticCitation17, antihypertensiveCitation18 and cancer chemopreventive effectsCitation15. Additionally, our group has succeeded in isolating and characterizing influenza virus neuraminidase inhibitors from this plantCitation19.
In this study, we isolated 13 constituents from the A. keiskei that target the SARS-CoV proteases. We have expressed and purified in Escherichia coli (E. coli) the full-length 3CLpro and PLpro, as well as the truncated forms containing only the catalytic domains. The isolated compounds were evaluated separately for their inhibitory activities against the viral proteases. The compounds’ inhibition mechanisms for the targeted proteases were ascertained by kinetic plots and molecular docking studies. To the best of our knowledge, this report is the first examining the inhibition of the SARS-CoV cysteine proteases by compounds isolated from A. keiskei.
Materials and methods
General apparatus and chemicals
All reagent-grade chemicals were purchased from Sigma Chemical Co. (St. Louis, MO). The chromatographic separations were carried out by thin-layer chromatography using commercially available glass plates pre-coated with silica gel (E. Merck Co., Darmstadt, Germany) and visualized under UV light at 254 and 360 nm or stained with 10% H2SO4. Column chromatography was performed on 230–400 mesh silica gel (Kieselgel 60, Merck, Darmstadt, Germany). RP-18 (ODS-A, 12 μm, S-150 Å, YMC) and Sephadex LH-20 (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) columns were used for column chromatography. 1H, 13C NMR and 2D-NMR data were obtained on a JNM-ECA 400 (Jeol, Tokyo, Japan) spectrometer in CDCl3, acetone-d6, DMSO-d6 or methanol-d4 with tetramethylsilane (TMS) as the internal standard. EIMS and HREIMS data were collected on Jeol JMS-700 spectrometer (JEOL, Tokyo, Japan).
Plant material and isolation of bioactive components
The plant material for this research was purchased from local market in Jeongup, Korea. The sample used in this study is stored at Korea Research Institute of Bioscience and Biotechnology (KRIBB) as sample No. R-KRIBB-020. The dried A. keiskei leaves (2.4 kg) were pulverized and extracted with 95% ethanol (2 × 10 L) for a week at room temperature. The combined extract was concentrated in vacuo to yield a dark residue (225 g). The crude extract was suspended in water and successively partitioned into organic solvents (hexane and ethyl acetate) based on their polarity differences to afford hexane (20 g), ethyl acetate (19 g) and H2O (150 g) layers. The ethyl acetate fraction (19 g) was chromatographed over a silica gel column (10 × 30 cm, 230–400 mesh; Merk) and eluted with a solvent gradient of n-hexane/ethyl acetate (30:1 to 1:1, v/v) to yield 10 fractions (fr. 1–fr. 10) based on the TLC profile. Fraction 4 (1.0 g) was chromatographed over a Sephadex LH-20 column (2 × 90 cm) using methanol as the eluting solvent to give four fractions (fr. 41–fr. 44); fr. 44 (214 mg) was resubjected to RP-18 (ODS-A, 12 nm, S-150 mM, YMC) chromatography to yield compounds 2 (18 mg) and 4 (12 mg). Fraction 43 (140 mg) was separated through a RP-C18 column and preparative-HPLC to yield compounds 5 (13 mg), 6 (5.2 mg) and 7 (11.2 mg). Fraction 5 (1.9 g) was chromatographed over a Sephadex LH-20 column (2 × 90 cm) using methanol as the eluting solvent to give six fractions (fr. 51–fr. 56); fr. 52 and fr. 53 were separated by subjection them to a RP-C18 chromatography column several times with 50% methanol to give compounds 10 (8 mg) and 12 (5 mg). Fraction 54 was chromatographed on a RP-C18 column using 85% acetonitrile to yield compound 9 (6 mg) and preparative-HPLC to yield compound 8 (16 mg). Fraction. 55 and fraction. 56 were separated on a RP-C18 chromatography column using 50% methanol to give compounds 11 (9 mg) and 13 (12 mg).
Expression and purification of SARS-CoV 3CLpro and PLpro from E. coli
The two genes encoding the 3CL protease (nucleotide residues 9970–10887, GenBank accession no. AY345987) and PL protease (amino acid residues 723–1041, GenBank accession no. NC 828862) of SARS-CoV were synthesized based on the sequence reported by Sun et alCitation20 and Han et alCitation21 (GenScript, Piscataway, NJ). The plasmids pET-23 d(+) containing the 3CLpro or PLpro gene were transformed into E. coli strain BL21(DE3) in CodonPlus-RIL cells (Stratagene, La Jolla, CA) for protein expression. The transformed cells were grown in 50 ml of LB broth medium containing 100 μg/ml ampicillin at 37 °C overnight and then inoculated in 1 L of LB supplemented with antibiotic. The cells were grown to an absorbance of 0.5 at 600 nm and then induced with 0.5 mM isopropyl β-D-thiogalactopyranoside for 12 h at 16 °C. After collection by centrifugation at 6000g at 4 °C for 25 min, the pellet was washed, frozen and suspended in buffer A (20 mM sodium phosphate, 300 mM NaCl, 10 mM imidazole and 0.1% Triton X-100 at pH 7.5). The suspended cells were sonicated and centrifuged at 15 000 g at 4 °C for 30 min. The supernatant was loaded on a 5 ml HisTrap Ni2+ chelating column (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) equilibrated with buffer A, and the recombinant protein was eluted by buffer A with 100–200 mM imidazole. The elution buffer was then changed to 20 mM Tris–HCl buffer (pH 7.5) with 1 mM dithiothreitol, and the purified protein was stored at −80 °C before use in any of the assays. The expression and purification of SARS-CoV PLpro were performed using the same procedure as for SARS-CoV 3CLpro. The purity and molecular weights of SARS-CoV 3CLpro and SARS-CoV PLpro were verified by SDS-PAGE. The purified proteases ran approximately at the calculated size of 33 kDa (3CLpro) and 36.5 kDa (PLpro) on SDS-PAGE.
SARS-CoV 3CLpro cell-free trans-cleavage inhibition assay
The inhibitory effect of isolated compounds on SARS-CoV 3CLpro was measured using a FRET method previously developed and describedCitation10. For the SARS-CoV 3CLpro inhibition assay, the 14-mer fluorogenic peptide Dabcyl-KTSAVLQSGFRKME-Edans (Anygen Co., Republic of Korea) was used as a substrate, and fluorescence was monitored in 96-well plates by a fluorescence plate reader (Flx800, BioTeck Instrument Inc., Winooski, VT) for up to 60 min. The enhanced fluorescence due to protease-catalyzed substrate cleavage was measured at 590/40 nm with excitation at 360 nm. The IC50 values of the isolated compounds were measured in a reaction mixture containing 10 µg/ml of the 3CLpro (final concentration, 2.5 µg), the test compounds (0–200 µM) and 10 µM of the fluorogenic 14-mer peptide substrate in 20 mM Bis–Tris buffer (pH 7.5). The initial velocities of the inhibited reactions were plotted against the concentrations of inhibitor to obtain the IC50 values by properly fitting the data according to the analysis method previously reportedCitation9,Citation10,Citation22.
SARS-CoV 3CLpro cell-based cis-cleavage inhibition assay
To determine the cell-based cis-cleavage assay, the in-frame gene of 3CLpro, the substrate (SAVLQSGFRK) and luciferase were synthesized as the plasmid pcDNA3.1-3CLpro-S-Luc, and the plasmid DNA was transfected into Vero cellsCitation23,Citation24. The transfected cells were incubated in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% bovine serum (FBS) for 5 h at 37 °C under a 5% CO2 atmosphere. Then, the media was removed and replaced by DMEM with tested compounds at different concentration. After incubation, the cells were harvested with 200 µl passive lysis buffer (Promega Co., Madison, WI). After clarification (10 000 g, 5 min), the luciferase activity was measured from 50 µl of the supernatant using the dual luciferase reporter assay system (Promega Co., Madison, WI) and a Centro XS3 LB 960 luminometer (Berthold Technology, Inc., Oak Ridge, TN)Citation24.
SARS-CoV PLpro inhibition assay
SARS-CoV PLpro activity was measured by the method previously described by our groupCitation9,Citation10,Citation22. The inhibition assay was optimized in a 96-well plate to establish suitable assay conditions and incubation times. The fluorogenic peptide, Arg–Leu–Arg–Gly–Gly–AMC (ENZO Life Sciences, Farmingdale, NY) and 208 nM purified PLpro in 20 mM Tris–HCl buffer (pH 6.8) were used as the substrate and the enzyme, respectively. For the inhibition studies, 54 nM PLpro and 0–200 µM of the individual compounds were mixed with the substrate (50 µM) at 37 °C, and the fluorescence intensity was monitored at excitation and emission wavelengths of 360 and 460 nm on a SpectraMax M2e Multimode Reader (Molecular Devices Co., Sunnyvale, CA).
Deubiquitination activity assay
For the deubiquitination assay, the purified PLpro (208 nM) was mixed with various concentrations of the compound (0–200 µM) in 20 mM Tris–HCl buffer (pH 6.8) before the substrate (400 nM), Ubiquitin-AMC (Enzo Life Sciences Inc., Farmingdale, NY), was added. All assays were performed at 37 °C using the 96-well plate format. The enzyme activities were determined by monitoring the enhanced fluorescence emission upon substrate cleavage at excitation and emission wavelengths of 360 and 460 nm, respectively, on a SpectraMax M2e Multimode Reader (Molecular Devices Co., Sunnyvale, CA). Release of AMC was measured in the same manner as that described above for the IC50 measurementsCitation25,Citation26.
DeISGylation activity assay
PLpro inhibition assay with ISG15-AMC (Boston Biochem Inc., Cambridge, MA) was performed in a 96-well plate at 37 °C in buffer containing 20 mM Tris–HCl (pH 6.8), 54 nM PLpro and various concentrations of compound ranging from 1 to 200 µM. The ISG15-AMC substrate concentration was 100 nM, and the release of AMC was monitored at excitation and emission wavelengths of 360 and 460 nm, respectivelyCitation25.
Other proteases assays
Chymotrypsin activity was evaluated according to a method described by Francisco et alCitation27, using BTEE as the substrate, where 10 µl of enzyme solution (4.5 U/ml) was mixed with 570 µl of 1 mM BTEE (N-benzoyl-L-tyrosine ethyl ester) in 10 mM Tris–HCl buffer at pH 7.8 with 10 mM CaCl2 at 25 °C. For the inhibition studies, chymotrypsin and 0–200 µM of the individual compounds were mixed with BTEE at 25 °C. The production of benzoyl-tyrosine was measured by monitoring the absorbance at 260 nm every 10 s for 6 min. Trypsin activity was measured by the modified method of Blanco and GuisanCitation28 using BAEE as substrate. In spectrophotometric experiments, trypsin activity was monitored by increasing absorbance at 260 nm every 10 s for 6 min. Papain in a solution of 50 mM sodium phosphate (pH 6.2) was incubated with various concentrations of compounds for 5 min at 25 °C. Then, 20 mM BAPNA (N-α-benzoyl-DL-arginine-p-nitroanilide) was added, and the increase in absorbance at 410 nm with time was monitored.
In silico molecular simulation study
Docking experiments were conducted using Autodock 3.0.5 software with a Lamarckian genetic algorithm (LGA)Citation29. A three-dimensional coordinate in the X-ray crystal structures of SARS-CoV 3CLpro (PDB accession code 2ZU5)Citation30 and SARS-CoV PLpro (PDB accession code 3MJ5)Citation31 was obtained from the Protein Data Bank (PDB; http://www.pdb.org) and was used for the docking studies. After removing the water molecules, the polar hydrogen (H) atoms were added to the macromolecule, and the Kollman charges were assigned to all of the atomsCitation32. The three-dimensional atomic coordinates of the compounds were generated by the Corina program (Molecular Networks GmbH, Erlangen, Germany). AutoDock version 3.0.5 was used for the computational molecular docking simulation of the flexible small molecules to the rigid proteinsCitation29. The docking results were ranked according to their docking energy scores. The Chimera 1.4.1 software program (University of California, San Francisco, CA) was used to identify the potential H-bonds between residues in the proteins’ active site pockets.
Statistical analysis
All measurements were performed in triplicate. The results were subjected to variance analysis using Sigma plot. The differences were considered significant at p < 0.05.
Results and discussion
During the first step of this study, we investigated the over-expression of SARS-CoV 3CLpro in E. coli host cells (BL21 (DE3), BL21 (DE3) CodonPlus-RIL, BL21 (DE3) pLysS, BL21 Star (DE3), C41 (DE3), Rosetta (DE3), Tuner (DE3) and Origami (DE3)). As a result, the gene encoding the SARS-CoV 3CLpro controlled by the T7 promoter expressed large quantities of recombinant His-tagged SARS-CoV 3CLpro in various E. coli host cells except BL21 (DE3) and Rosetta (DE3). For this study, the SARS-CoV 3CLpro was expressed in E. coli BL21 (DE3) CodonPlus-RIL host cells. By optimizing the expression and purification methods of SARS-CoV 3CLpro from E. coli, the homogeneous protein was successfully isolated by Ni-NTA column chromatography. The purified 3CLpro ran approximately to the calculated size of 33 kDa. The peptide cleavage reaction of 3CLpro was easily monitored in real time using a fluorescence plate reader. For the fluorogenic substrate, Dabcyl-KTSAVLQSGFRKME-Edans, the Km value was 32.0 ± 5.2 µM, and the kcat value was 3.4 ± 0.7 min−19.
Subsequently, the recombinant gene-encoding SARS-CoV PLpro expressed in E. coli BL21 (DE3) CodonPlus-RIL cells and the gene controlled by the T7 promoter yielded large quantities of the recombinant His-tagged SARS-CoV PLpro. The purified PLpro ran approximately to the calculated size of 36.5 kDa on SDS-PAGE. The peptide cleavage reaction of PLpro was easily monitored in real time using a fluorescence plate reader. For the fluorogenic substrate, RLRGG-AMC, the Km value was 38.3 ± 1.8 µM, and the kcat value was 18.5 ± 2.3 min−1.
The dried chip of A. keiskei (2.4 kg, whole plant) was extracted with EtOH and yielded 225 g of crude extract. The EtOH extract was then dissolved in water and subjected to liquid–liquid partitioning successively with n-hexane and EtOAc, resulting in 20 g and 19 g or residue, respectively, and 150 g of crude residue was isolated from the aqueous layer. The EtOAc-soluble fraction remarkably inhibited the SARS-CoV 3CLpro activity (75% inhibition at 30 µg/ml). The EtOAc-soluble fractions isolated from the A. keiskei EtOH extract were subjected to ordinary- and reversed-phase silica gel column chromatography and resulted in nine alkylated chalcones (1–9) and four coumarins (10–13). The resulting compounds were identified as isobavachalcone (1), 4-hydroxyderricin (2), xanthoangelol (3), xanthoangelol F (4), xanthoangelol D (5), xanthoangelol E (6), xanthoangelol B (7), xanthoangelol G (8), xanthokeistal A (9), psoralen (10), bergapten (11), xanthotoxin (12) and isopimpinellin (13) by analyzing their spectroscopic data and comparing it to previous studies ().
Figure 1. Chemical structures of isolated alkylated chalcones from A. keiskei.
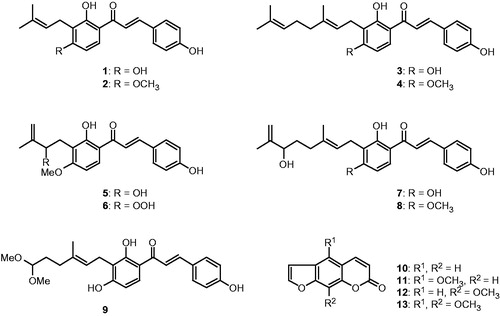
The inhibitory activities against SARS-CoV 3CLpro of the 13 constituents, 1–13, obtained from A. keiskei were tested. We measured the 3CLpro activity in the presence or absence of the test compounds using a fluorescence resonance energy transfer (FRET) method. All of the isolated compounds (1–13) investigated except for the coumarin derivatives (10–13) showed a dose-dependent inhibitory effect towards SARS-CoV 3CLpro activity. As the concentrations of the inhibitors increased, the residual protease activity rapidly diminished. Compounds 10–13, which contain coumarin derivatives, were shown to have IC50 values greater than 200 µM. The inhibitory effects of the isolated alkylated chalcones (1–9) on SARS-CoV 3CLpro activity are summarized in . As shown in , the activity of these compounds ranged from 11.4 to 129.8 µM. We describe below the logic behind the structural identification of compound 6 because it was the most potent 3CLpro inhibitor (IC50 = 11.4 µM) of the 13 constituents and because the location of the perhydroxyl (–OOH) group in this chemotype may play a crucial role in its high activity. Our data demonstrate several interesting facets of the structure–activity relationship. By comparing the 2-hydroxy-3-methyl-3-butenyl (HMB) group of 5 to the 2-perhydroxyl-3-methyl-3-butenyl (PMB) group of 6, the PMB group was more effective than HMB (IC50 = 26.6 µM) because it presumably interacts with the hydrogen bonding site. Similarly, the hydroxyl group (–OH) on the A-ring showed higher activity than when the –OH was capped with a methyl group. However, the length of the terpene (C5–C10 unit) fragment did not affect the IC50 value of the inhibitors. Interestingly, substitution of the A-ring with a HMB group (5) produced 4-fold higher potency than substitution with the dimethylally (DMA) group (2, IC50 = 81.4 µM).
Table 1. Inhibitory effects of isolated alkylated chalcones (1–9) and coumarins (10–13) on cell-free trans-cleavage and cell-based cis-cleavage activities of SARS-CoV 3CLpro.
The cell-based cleavage assay of SARS-CoV 3CLpro for the isolated alkylated chalcones does not require purification of the active 3CLpro. For the cell-based cleavage assay, the in-frame construction of the SARS-CoV 3CLpro, the substrate (S) and the luciferase (Luc) into the designed the plasmid pcDNA3.1-3CLpro-S-Luc was performed, and the plasmid was transfected into Vero cells. The transfected cells were grown, and then the luciferase activity of the cell lysate was measured using the Luciferase Reorter Assay System (Promega). Because a protein greater than 33 kDa was fused at the N-terminus of luciferase causes a dramatic decrease in luciferase activityCitation23,Citation24, the detection of luciferase activity could be used to measure the amount of cis-cleavage by the SARS-CoV 3CLpro. The isolated alkylated chalcones (1–9) inhibited the cis-cleavage activity of the SARS-CoV 3CLpro with IC50 values ranging from 5.8 to 50.8 µM in a dose-dependent manner (). The active chalcones were 2-fold more effective when evaluated with the cis-cleavage assay than when evaluated by the cell-free trans-cleavage assay. However, the isolated active chalcones exhibited 50% cytotoxic concentrations (CC50) lower than 70 µM. Of the compounds examined, compounds 3 and 6 were most efficient in blocking 3CLpro-cleavage activity (3, IC50 = 5.8 µM and 6, IC50 = 7.1 µM). Similar trends as those previously described for the trans-cleavage assay were observed. For example, the compound that contained the perhydroxyl group at PMB (6) exhibited the most potent activity with a selective index (SI = CC50/EC50) value of 9.2.
The numerous functions and requisite roles of SARS-CoV PLpro in viral replication and pathogenesis suggest that PLpro may serve as an attractive target for antiviral drugs. Therefore, to identify potential inhibitors of PLpro, we measured PLpro activity in the absence or presence of the compounds (1–13) isolated from A. keiskei using fluorogenic methods. SARS-CoV PLpro is catalytically more active toward ubiquitin-derived substrates relative to polyprotein-based peptide substratesCitation33. We used a commercially available peptide substrate that representing the 5 C-terminal residues of an ubiquitin (Ub) derivative with a C-terminal 7-amido-4-methylcoumarin (AMC) fluorogenic reporter groupCitation25,Citation34. Additionally, structural and functional studies of SARS-CoV PLpro have revealed that PLpro is homologous to human deubiquitinating enzymes and is capable of cleaving ubiquitin and ubiquitin-like modifiers such as an interferon-induced 15 kDa protein (ISG15)Citation25.
Utilizing the SARS-CoV PLpro inhibition assay, all of the isolated compounds (1–13) except the coumarin derivatives (10–13) showed a dose-dependent inhibitory effect toward PLpro activity. Additionally, increasing the inhibitor concentrations resulted in a decreased slope of the line indicating that these compounds were reversible inhibitors (). The SARS-CoV PLpro inhibitory activity is expressed as IC50 values (). Primary analyses of SAR showed that the –OOH substituted analogue (6) exhibited the most potent inhibitory activity against 3CLpro compared to the other active chalcones; it also showed extremely high inhibition against SARS-CoV PLpro (IC50 = 1.2 µM) with 5- to 40-fold higher activity than the other analogues. These results suggest that the –OOH group on the substituted hemiterpene (PMB) might play a role in enzyme binding and/or contribute to conformational stabilization of the polyhydroxylated chain through intramolecular hydrogen bonding. Additionally, all of the isolated compounds (1–13) were evaluated against the cleavage of ubiquitin (Ub-AMC) and an ubiquitin-like protein (ISG15-AMC). Deubiquitination may have important implications on viral replication and pathogenesis. As shown in , all 13 constituents isolated in this study showed potent activity against both ubiquitin and ubiquitin-like proteins with IC50 values range from 1.1 to 73.3 µM. Therefore, these results suggest the isolated compounds could possibly be developed into antiviral drugs.
Figure 2. (A) Effects of compounds 1–9 on the activity of SARS-CoV PLpro for proteolysis of substrate. (B) Relationship of the hydrolytic activity of SARS-CoV PLpro as function of enzyme concentrations at different concentration of compound 1.
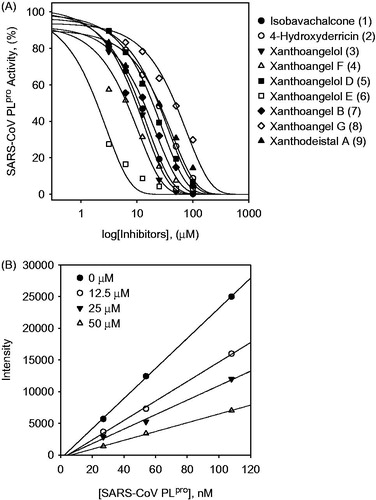
Table 2. Inhibitory effects of isolated alkylated chalcones (1–9) and coumarins (10–13) on SARS-CoV PLpro.
The inhibition mechanisms of the isolated alkylated chalcones (1–9) were studied. This study began by analyzing the mode of inhibition by globally fitting the data to all of the possible kinetic models using Lineweaver–Burk and Dixon plots. As shown in , the kinetic plots show that compound 6 has competitive inhibition profiles because the Lineweaver–Burk plot resulted in a family of straight lines with the same y-axis intercept, Vmax (37.3 ± 4.6 intensity min−1). All of the isolated active chalcones manifested the same inhibition mode of action. The predominant inhibition mode of the naturally occurring viral enzyme inhibitors was noncompetitive. Interestingly, the inhibition of SARS-CoV 3CLpro by the alkylated chalcones isolated from A. keiskei was rarely competitive. The inhibition constant (Ki) of the alkylated chalcones is displayed in and is determined from the common x-axis intercept of the lines on the corresponding Dixon plot (). However, the chalcones (1–9) that inhibit SARS-CoV 3CLpro displayed a different inhibition profile for PLpro ( for compound 6 representatively).
Figure 3. Graphical determination of the type of inhibition for compound 6. (A) Lineweaver–Burk plot for inhibition of compound 6 on SARS-CoV 3CLpro. (B) Dixon plot for inhibition of compound 6 on SARS-CoV 3CLpro. (C) Lineweaver–Burk plot for inhibition of compound 6 on SARS-CoV PLpro. (D) Dixon plot for inhibition of compound 6 on SARS-CoV PLpro.
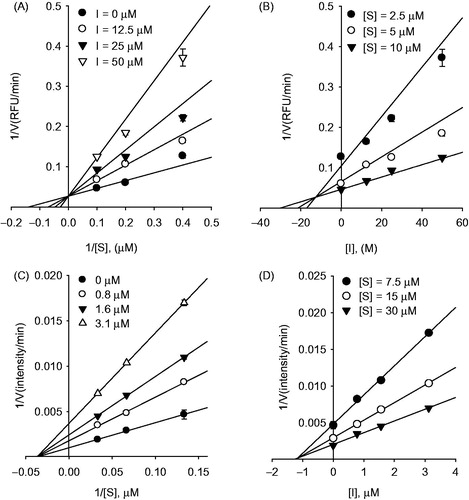
To further elucidate the interaction of SARS-CoV cysteine proteases with the most potent inhibitor, 6, isolated from A. keiskei, a docking simulation was performed in silico. The three-dimensional structure of SARS-CoV 3CLpro complexed to a substrate-analogue inhibitor (coded 2ZU5) obtained from the Protein Data Bank (PDB; http://www.rcsb.org/pdb/) was used for the modeling analysis. The computer docking analysis revealed that most chalcones (1–9) fit nicely into the substrate-binding pocket of 3CLpro (see Supporting Information S7). The overall structure obtained by docking compound 6 to 3CLpro is shown in , where the SARS-CoV 3CLpro is drawn as a ribbon, and the compound is represented as a ball-and-stick model. As shown in , compound 6 formed hydrogen bonds (H-bond) with SARS-CoV 3CLpro. The oxygen atom of the carbonyl group of compound 6 forms hydrogen bond with the nitrogen atom of His163 (2.57 Å); the hydroxyl group of C′2 formed H-bonds with Ser144 (3.48 and 3.59 Å). Interestingly, the perhydroxyl group (PMB) of the substituted compound 6 formed strong H-bonds with Cys145 (3.45 and 2.72 Å) as one of the key motifs in this inhibitor.
Figure 4. In silico molecular docking analysis of compound 6 binding to SARS-CoV 3CLpro (A) and PLpro (B).
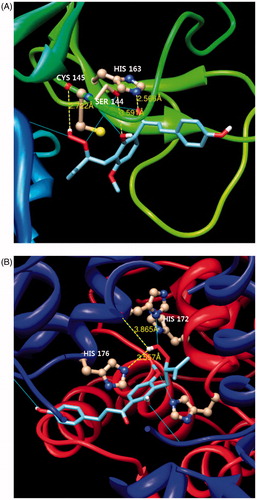
To further elucidate the interaction of SARS-CoV PLpro with the isolated alkylated chalcone 6, which exhibited noncompetitive inhibitory activity, a PLpro complex (pdb coded 3MJ5) was used for modeling analysis. In this study, docking was initially performed using energetically optimized ligands at different potential binding sites throughout the entire protein (blind docking) because this process reduces the simulation time. As shown in , the perhydroxyl group of 6 forms three H-bonds with the nitrogen atom of His176 (3.56 Å), the oxygen atom of His172 (3.87 Å) and the nitrogen atom of His172 (3.40 Å). These docking experiments support the results of the enzymatic assay, and reveal the important inhibitory action of the alkylated chalcones on the activity of the SARS-CoV cysteine proteases.
Conclusion
Currently, research and development of new drugs from natural resources in a systemic and strategic manner has become the global trend. Despite the huge threat SARS continues to pose to global health, no natural products have been shown to affect the two proteolytic enzymes, PLpro and 3CLpro, that are essential for SARS-CoV replication. Our intensive search for effective anti-SARS drugs was performed by screening natural product, and we found that an ethanol extract of A. keiskei inhibited the SARS-CoV cysteine proteases. Angelica keiskei led to the identification of numerous chalcones. Chalcones are ubiquitous substances found in a diversity of plants. Chalcones are precursors to other natural products, such as flavonoids, which have a wide range of biological activities. Additionally, the alkylated chalcones are important derivatives of chalcones, which contain prenyl-(C5 unit terpene) type side chain with different length, including DMA, geranyl or farnesyl chains; these compounds have shown interesting biological activities. In this study, alkylated chalcones 1–9 and coumarins 10–13 were isolated from A. keiskei, and the inhibitory activities of these constituents against both cysteine proteases was examined. To the best of our knowledge, this report is the first to describe the inhibitory effects of alkylated chalcones isolated from A. keiskei against the SARS-CoV proteases. Of the 13 isolated compounds 1–13, the alkylated chalcone substituted with a perhydroxyl group, 6, showed the most potent 3CLpro and PLpro inhibitory activities with IC50 values of 11.4 and 1.2 µM, respectively. Also, compound 6 exhibited more potent inhibition for cysteine protease such as papain than for other serine proteases such as trypsin and chymotrypsin (). The results suggested that compound 6 has specific inhibitory activity against cysteine protease specifically. In particular, the most active chalcone 6 demonstrated inhibitory activity using the cell-based 3CLpro cis-cleavage assay. In addition, our detailed protein-inhibitor mechanistic analysis of these species has unveiled that the chalcones exhibited competitive inhibition of the SARS-CoV 3CLpro, whereas noncompetitive inhibition was observed with SARS-CoV PLpro. Moreover, we proceeded to investigate the interaction of this potent compound with the protein by molecular modeling, which provided important information regarding the principal interactions that cause the activity of this chemotype. Given their inhibitory effects in the cell-free/based assay, we believe that our data may be encouraging for the development of therapeutic reagents for SARS-CoV. Additionally, the results of these studies will be used to aid additional natural product and food chemistry efforts toward the development of protease inhibitors, which is an ongoing concern in our laboratories.
Table 3. Inhibitory effects of isolated alkylated chalcones (1–9) and coumarins (10–13) on other proteases.
Supplementary material available online
Supplementary information
IENZ_1003215_SupplemetaryFile.pdf
Download PDF (2.2 MB)Declaration of interest
This research was supported by KRIBB Research Initiative Program and Fishery Commercialization Technology Development Program through KIMST (Korea Institute of Marine Science & Technology promotion) funded by Ministry of Oceans and Fisheries (MOF), Republic of Korea.
References
- Peiris JS, Guan Y, Yuen KY. Severe acute respiratory syndrome. Nat Med 2004;10:S88–97
- Stadler K, Masignani V, Eickmann M, et al. SARS – beginning to understand a new virus. Nat Rev Microbiol 2003;1:209–18
- Chou K-C, Wei D-Q, Zhong W-Z. Binding mechanism of coronavirus main proteinase with ligands and its implication to drug design against SARS. Biochem Biophys Res Commun 2003;308:148–51
- Thiel V, Ivanov KA, Putics A, et al. Mechanisms and enzymes involved in SARS coronavirus genome expression. J Gen Virol 2003;84:2305–15
- Ghosh AK, Xi K, Johnson ME, et al. Progress in anti-SARS coronavirus chemistry, biology and chemotherapy. Annu Rep Med Chem 2006;41:183–96
- Harcourt BH, Jukneliene D, Kanjanahaluethai A, et al. Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity. J Virol 2004;78:13600–12
- Anand K, Ziebuhr J, Wadhwani P, et al. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 2003;300:1763–7
- Fear G, Komarnytsky S, Raskin I. Protease inhibitors and their peptidomimetic derivatives as potential drugs. Pharmacol Ther 2007;113:354–68
- Park J-Y, Jeong HJ, Kim JH, et al. Diarylheptanoids from Alnus japonica inhibit papain-like protease of severe acute respiratory syndrome coronavirus. Biol Pharm Bull 2012;35:2036–42
- Park J-Y, Kim JH, Kim YM, et al. Tanshinones as selective and slow-binding inhibitors for SARS-CoV cysteine proteases. Bioorg Med Chem 2012;20:5928–35
- Kim DW, Seo KH, Curtis-Long MJ, et al. Phenolic phytochemical displaying SARS-CoV papain-like protease inhibition from the seeds of Psoralea corylifolia. J Enzyme Inhib Med Chem 2014;29:59–63
- Song SH, Kim DW, Curtis-Long MJ, et al. Papain-like protease (PLpro) inhibitory effects of cinnamic amides from Tribulus terrestris fruits. Biol Pharm Bull 2014;37:1021–8
- Baba K, Nakata K, Taniguchi M, et al. Chalcones from Angelica keiskei. Phytochemistry 1990;29:3907–10
- Akihisa T, Tokuda H, Ukiya M, et al. Chalcones, coumarins, and flavanones from the exudate of Angelica keiskei and their chemopreventive effects. Cancer Lett 2003;201:133–7
- Akihisa T, Tokuda H, Hasegawa D, et al. Chalcones and other compounds from the exudates of Angelica keiskei and their cancer chemopreventive effects. J Nat Prod 2006;69:38–42
- Nishimura R, Tabata K, Arakawa M, et al. Isobavachalcone, a chalcone constituent of Angelica keiskei, induces apoptosis in neuroblastoma. Biol Pharm Bull 2007;30:1878–83
- Enoki T, Ohnogi H, Nagamine K, et al. Antidiabetic activities of chalcones isolated from a Japanese herb, Angelica keieskei. J Agric Food Chem 2007;55:6013–17
- Shimizu E, Hayashi A, Takahashi R, et al. Effects of angiotensin I-converting enzyme inhibitor from Ashitaba (Angelica keiskei) on blood pressure of spontaneously hypertensive rats. J Nutr Sci Vitaminol 1999;45:375–83
- Park J-Y, Jeong HJ, Kim YM, et al. Characteristic of alkylated chalcones from Angelica keiskei on influenza virus neuraminidase inhibition. Bioorg Med Chem Lett 2011;21:5602–4
- Sun H, Luo H, Yu C, et al. Molecular cloning, expression, purification, and mass spectrometric characterization of 3C-like protease of SARS coronavirus. Protein Expr Purif 2003;32:302–8
- Han YS, Chang GG, Juo CG, et al. Papain-like protease 2 (PLP2) from severe acute respiratory syndrome coronavirus (SARS-CoV)-expression, purification, characterization, and inhibition. Biochemistry 2005;44:349–59
- Park J-Y, Kim JH, Kwon JM, et al. Dieckol, SARS-CoV 3CLpro inhibitor, isolated from the edible brown algae Ecklonia cava. Bioorg Med Chem 2013;21:3730–7
- Lin CW, Tsai CH, Tsai FJ, et al. Characterization of trans- and cis-cleavage activity of the SARS coronavirus 3CLpro protease: basis for the in vitro screening of anti-SARS drugs. FEBS Lett 2004;574:131–7
- Lin CW, Tsai FJ, Tsai CH, et al. Anti-SARS coronavirus 3C-like protease effects of Isatis indigotica root and plant-derived phenolic compounds. Antiviral Res 2005;68:36–42
- Ratia K, Pegan S, Takayama J, et al. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc Natl Acad Sci USA 2008;105:16119–24
- Chou CY, Chien CH, Han YS, et al. Thiopurine analogues inhibit papain-like protease of severe acute respiratory syndrome coronavirus. Biochem Pharmacol 2008;75:1601–9
- Castillo-Yáñez FJ, Pacheco-Aguilar R, García-Carreño FL, et al. Purification and biochemical characterization of chymotrypsin from the viscera of Monterey sardine (Sardinops sagax caeruleus). Food Chem 2006;99:252–9
- Guisan JM, Blanco RM. Stabilization of trypsin by multiple-point attachment to aldehyde-agarose gels. Ann N Y Acad Sci 1987;501:67–72
- Morris GM, Goodsell DS, Halliday RS, et al. Automated docking using a Lamarckian genetic algorithm and and empirical binding free energy function. Comput Chem 1998;19:1639–62
- Lee CC, Kuo CJ, Ko TP, et al. Structural basis of inhibition specificities of 3C and 3C-like proteases by zinc-coordinating and peptidomimetic compounds. J Biol Chem 2009;284:7646–55
- Ghosh AK, Takayama J, Rao KV, et al. Severe acute respiratory syndrome coronavirus papain-like novel protease inhibitors: design, synthesis, protein-ligand X-ray structure and biological evaluation. J Med Chem 2010;53:4968–79
- Sanner MF, Olson AJ, Spehner JC. Reduced surface: an efficient way to compute molecular surfaces. Biopolymers 1996;38:305–20
- Barretto N, Jukneliene D, Ratia K, et al. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J Virol 2005;79:15189–98
- Lindner HA, Fotouhi-Ardakani N, Lytvyn V, et al. The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J Virol 2005;79:15199–208