
Abstract
Resveratrol and aspirin are known to exert potential chemopreventive effects through modulation of numerous targets. Considering that the CYP450 system is responsible for the activation of environmental procarcinogens, the aim of this study was to design a new class of hybrid resveratrol–aspirin derivatives possessing the stilbene and the salicylate scaffolds. Using HepG2 cells, we evaluated (a) the inhibition of TCDD-mediated induction of CYP1A1 exerted by resveratrol–aspirin derivatives using the EROD assay, and (b) CYP1A1 mRNA in vitro. We observed significant inhibition (84%) of CYP1A1 activity and a substantial decrease in CYP1A1 mRNA with compound 3, compared to control. Resveratrol did not exert inhibition under the same experimental conditions. This inhibitory profile was supported by docking studies using the crystal structure of human CYP1A1. The potential effect exerted by compound 3 (the most active), provide preliminary evidence supporting the design of hybrid molecules combining the chemical features of resveratrol and aspirin.
Introduction
Cytochrome P450 (CYP) is a large family of constitutive and inducible enzymes associated with the metabolic transformation of endogenous molecules, as well as external xenobiotic substratesCitation1. CYP450 plays a significant role in carrying out phase I metabolism reactions, which include (but are not limited to) oxidative dehalogenation, oxidative cleavage, hydroxylation, epoxidation, dealkylation and alcohol/aldehyde oxidationCitation2. However, aside from the physiological detoxification mechanism assisted by CYP enzymes, CYPs are also involved in the activation of procarcinogenic compounds into carcinogenic agentsCitation1. Moreover, the overexpression of different CYP enzymes in cancer tissues has been found to be a crucial step for cancer progressionCitation3.
The CYP1A1 enzyme is a member of the CYP1 family, which is mainly expressed in extra hepatic tissues such as lung, skeletal muscle and the thyroid glandCitation4. CYP1A1 is one of the main CYP enzymes involved in the metabolic activation of several environmental contaminants, such as polycyclic aromatic hydrocarbons (PAH)Citation5. CYP1A1 is primarily regulated by the aryl hydrocarbon receptor (AhR) pathway, and its carcinogenic potential involving the bio-activation of several PAH has been well documentedCitation6. Furthermore, CYP1A1 expression (transcriptional level) is significantly higher in cancer than normal tissuesCitation3, which makes the CYP1A1 isozyme a potential drug target. In this regard, numerous efforts have been made to develop novel CYP1A1 inhibitorsCitation7.
There is a significant body of evidence supporting the potential chemopreventive properties of natural polyphenols and NSAIDs, both in vitro and in vivo. This favorable chemopreventive profile is attributed to the ability of these two classes of compounds to modulate, simultaneously, several cell targets linked to cancer initiation and cancer progression, such as cyclooxygenase (COX)-2, NF-κB, iNOS, 5-LOX, VEGF and CYP450 enzymes, among many othersCitation8. Some of these proteins are targeted by both NSAIDs (particularly aspirin) and polyphenols (specifically resveratrol), and some proteins are targeted by only one type of drug. Consequently, the idea of forming a hybrid molecule combining the pharmacological profile of aspirin and resveratrol is a promising approach. Nevertheless, as far as the inhibitory profile of CYP1A1 by these two molecules is concerned, the background information points to a potential issue. On one hand, it is clear that resveratrol (3,4′,5-trihydroxystilbene, ) is a naturally occurring polyphenol capable of exerting significant in vitro inhibition of the CYP1A1 enzymeCitation9,Citation10, but the data on aspirin (and salicylates in general) is still controversial. For example, salicylic acid only shows CYP1A1 inhibition at relatively high concentrations (≥2.5 mM) when tested in MCF-7 cellsCitation11, while other reports have shown that neither aspirin nor salicylic acid significantly inhibited CYP1A1 activityCitation12. Consequently, the addition of a salicylate moiety to the chemical structure of resveratrol would suggest a significant decrease in this polyphenol’s ability to inhibit the enzymatic activity of CYP1A1. Nevertheless, preliminary results generated in our group with a molecular modeling (docking) study using the recently reported crystal structure of human CYP1A1 (described in the experimental section), seemed to suggest that it might be possible for the new hybrid molecules () to inhibit this enzyme. Encouraged by these in silico (preliminary) results, we decided to synthesize a series of new hybrid resveratrol–aspirin derivatives and test their ability to inhibit CYP1A1 catalytic activity.
Figure 1. General design of resveratrol–salicylate hybrid compounds.
As part of an interdisciplinary research work aimed at developing new anticancer/chemopreventive agents, we present two sequential papers describing the design, synthesis and biological evaluation of a series of new hybrid resveratrol–aspirin agents. In this article, we report the effects of adding a salicylate (or an acetylsalicylate) moiety on resveratrol’s ability to modulate the activity and expression of CYP1A1. In this regard, we synthesized 10 resveratrol derivatives possessing a salicylate-like scaffold via the Wittig reaction, and then we used the 7-ethoxyresorufin-O-de-ethylation (EROD) assay to measure the catalytic activity of CYP1A1 in the presence of each test drug. The results obtained in this experiment showed different degrees of in vitro modulation of enzyme activity, where some compounds inhibited CYP1A1, while others increased it. Among the group of inhibitors, we identified compound 3 as the most promising resveratrol analog, which showed a suitable inhibitory profile by reducing 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-mediated induction of CYP1A1 catalytic activity in HepG2 cells. We also observed that this effect was associated with a reduction of TCDD-mediated induction of CYP1A1 mRNA levels. We submit that compound 3 is a novel hybrid resveratrol–salicylate derivative with promising modulatory effects on CYP1A1, which could protect against CYP1A1 activation of environmental carcinogens.
Methods
Chemistry
General
Melting points were determined with an Electrothermal Mel-Temp® melting point apparatus (Dubuque, IA), and are uncorrected.1H-NMR and 13C-NMR spectra were recorded on either Bruker AVANCE 600 or Bruker AM-300 NMR spectrophotometer; coupling constants (J) are reported in Hertz (Hz), and the corresponding chemical shifts are represented as δ units (ppm), using TMS as internal standard. ESI-MS spectra were recorded using Water’s micromass ZQ-4000 single quadruple mass spectrometer. Elemental analysis (C, H) of compound 3 was within ±0.4 of theoretical values for elements listed (Microanalytical Service Laboratory, Department of Chemistry, University of Alberta, Alberta, Canada). Compounds 3–12 showed a single spot on RediSep® silica gel glass plates (UV254, 0.2 mm) using high, medium and low polarity solvent mixture and no residue remained after combustion, indicating a purity higher than 95%. Column chromatography was performed on a CombiFlash Retrieve, or CombiFlash Rf system using RediSep Rf silica gel® (40–60 µm) cartridges, or pre-packed RediSep Gold® columns. 4-Methoxybenzyltriphenylphosphonium bromide (1a)Citation13, 3,5-dimethoxybenzyltriphenylphosphonium bromide (1b)Citation13 and methyl 5-formyl-2-methoxybenzoate (2a)Citation14, were synthesized according to literature procedures. All other reagents were purchased from either Sigma Aldrich (Milwaukee, IN) or TCI America (Portland, OR) and were used without further purification.
Methyl 2-acetoxy-5-formylbenzoate (2b)
To a solution of methyl 5-formyl-2-hydroxybenzoate (0.75 g, 4.1 mmol) in dry THF (25 ml), triethylamine (0.50 g, 5 mmol) was added and the mixture was stirred for about 2 min before adding acetylchloride (0.39 g, 5.0 mmol) dropwise at 25 °C. The reaction progress was followed by TLC, and after about 2 h, the triethylammonium chloride (white solid) produced as secondary product was filtered off and the solvent was evaporated under vacuum. The product (0.9 g, 97% yield, yellow crystals, m.p. = 34–36 °C) was used in the next step without further purification (only one spot on TLC). 1H-NMR (300 MHz, CDCl3) δ 10.03 (s, 1H, CHO), 8.53 (d, J = 1.83 Hz, 1H, phenyl H-2), 8.09 (dd, J = 1.83, 8.52 Hz, 1H, phenyl H-6), 7.29 (d, J = 8.55 Hz, 1H, H-5), 3.92 (s, 3H, CH3OCO), 2.38 (s, 3H, CH3CO). 13C-NMR (600 MHz, CDCl3) δ 190.09, 169.02, 163.80, 155.09, 134.01, 133.94, 133.85, 124.99, 124.05, 52.49, 20.89. ESI-MS m/z; 245.0 [M + Na]+.
3-Methoxy-4-acetoxybenzaldehyde (2c)
This compound was synthesized from vanillin (0.5 g, 3.3 mmol) using the same experimental procedure described for intermediate 2b. The product (0.59 g, 92% yield) was obtained as pale yellow crystals, m.p. = 77–79 °C (reported 77–78 °C)Citation15. 1H-NMR (300 MHz, CDCl3) δ 9.95 (s, 1H, CHO), 7.50 (d, J = 1.83 Hz, 1H, phenyl H-2), 7.48 (dd, J = 1.83, 7.32 Hz, 1H, phenyl H-6), 7.22 (d, J = 7.32 Hz, 1H, phenyl H-5), 3.91 (s, 3H, CH3O), 2.35 (s, 3H, CH3CO).
General procedure for the synthesis of stilbenes 3–7 (Scheme 1)
In a heat-dried three neck round bottom flask, the corresponding phosphonium bromide (1a or 1b, 1.1 Eq) was added, and the flask was immediately flushed with a stream of dry nitrogen to remove oxygen and moisture. Using a bath of dry ice and acetone, the system was cooled down to about −60 °C, time at which 50 ml of freshly distilled THF was added; after stirring for a few minutes, n-butyllithium (1.1 Eq) was added. The resulting red suspension was stirred for 30 min before adding a solution of the corresponding aldehyde (2a–2c, 1.0 Eq) in (3–6 ml) of dry THF, drop-wise, over 20–30 min; the dry ice bath was removed to allow the reaction mixture to reach room temperature, and then stirred for about 12–18 h (until complete consumption of the aldehyde as monitored by TLC). After this, the reaction was quenched by adding water (90 ml); followed by extraction with ethyl acetate (30 ml) three times. The combined organic layers were dried using sodium sulfate, and the solvent was evaporated under vacuum to obtain the crude stilbene, which was then purified by silica gel column chromatography, using a solvent mixture of hexanes/ethyl acetate (6:4) to afford the E/Z mixture of stilbenes as an oily liquid.
Scheme 1. Chemical synthesis of resveratrol–salicylate derivatives 3–12. Reagents and conditions: (i) n-butyllithium, dry THF, −78 °C for 2 h then room temperature for 12–18 h; (ii) Ph2S2, dry THF, reflux 4 h; (iii) BBr3, dry CH2Cl2, −60 °C then room temperature for 2 h; (iv) acetic anhydride, pyridine room temperature for 4 h.
To obtain the pure E isomer from the pre-purified mixture obtained above, the stilbene (1 Eq) was dissolved in dry THF (70 ml), and diphenyl disulfide (0.5 Eq) was added as described previouslyCitation16. This reaction mixture was stirred under nitrogen atmosphere until all the Z stilbene re-isomerized to the E isomer (on average, Z isomer Rf value was 0.64, while that of the E isomer was 0.51). Finally, the solvent was evaporated under vacuum and the product was purified by column chromatography using a mixture of petroleum ether/ethyl acetate (7:3) to afford the pure E isomer.
(E)-3,4′,5-Trimethoxystilbene (TMS)
In a 250 ml three neck round bottom flask, the phosphonium bromide 1b (1.8 g, 3.6 mmol) and 4-methoxybenzaldehyde (0.5 g, 3.6 mmol) were mixed in dry THF according to the general procedure described above. The title compound was obtained (0.65 g, 64% yield), as a colorless needles, m.p. = 55–56 °C (reported 55–57 °C)Citation17. 1H-NMR (600 MHz, CDCl3) δ 7.44 (d, J = 8.4 Hz, 2H, H-2′ & H-6′), 7.04 (d, J = 16.2 Hz, 1H, alkene), 6.90 (d, J = 16.8 Hz, 1H, alkene), 6.89 (d, J = 8.4 Hz, 2H, H-3′ & H-5′), 6.65 (d, J = 2.4 Hz, 2H, H-2 & H-6), 6.37 (d, J = 2.4 Hz, 1H, H-4), 3.83 (s, 9H, CH3O). 13C-NMR (300 MHz, CDCl3) δ 160.98, 159.42, 139.72, 129.97, 128.76, 127.77, 126.62, 114.16, 104.39, 99.68, 55.36.
(E)-4,4′-Dimethoxy-3-(methoxycarbonyl)stilbene (3)
In a 250 ml three neck round bottom flask, the phosphonium bromide 1a (3.0 g, 6.5 mmol) and the aldehyde 2a (1.15 g, 5.9 mmol) were mixed in dry THF according to the general procedure described above. The title compound was obtained (1.25 g, 71% yield), as a white solid, m.p. = 133–134 °C. 1H-NMR (600 MHz, CDCl3) δ 7.93 (d, J = 2.4 Hz, 1H, H-2′), 7.58 (dd, J = 8.4, 2.4 Hz, 1H, H-6′), 7.43 (d, J = 9 Hz, 2H, H-2 and H-6), 6.98 (d, J = 16.2 Hz, 1H, alkene), 6.97 (d, J = 8.4 Hz, 1H, H-5′), 6.91 (d, J = 18.3 Hz, 1H, alkene), 6.89 (d, J = 9 Hz, 2H, H-3 and H-5), 3.92 (s, 3H, CH3O), 3.92 (s, 3H, CH3OCO), 3.83 (s, 3H, CH3O). 13C-NMR (300 MHz, CDCl3) δ 166.61, 159.20, 158.25, 131.03, 130.07, 130.04, 129.36, 129.20, 127.52, 127.38, 125.01, 120.16, 114.13, 112.30, 56.16, 55.32, 52.09. ESI-MS m/z; 321.0 [M + Na]+. Anal. calcd for C18H18O4: C 72.47, H 6.08, found: C 72.49, H 6.19.
(E)-4-Acetoxy-4′-methoxy-3-(methoxycarbonyl)stilbene (4)
In a 250 ml three neck round bottom flask, the phosphonium bromide 1a (1.25 g, 2.7 mmol) and the aldehyde 2b (0.42 g, 1.9 mmol) were mixed in dry THF according to the general procedure described above. The title compound was obtained (0.43 g, 70% yield), as a white solid, mp 123–125 °C. 1H-NMR (300 MHz, CDCl3) δ 8.11 (d, J = 2.43 Hz, 1H, H-2′), 7.62 (dd, J = 8.55, 2.43 Hz, 1H, H-6′), 7.44 (d, J = 9.15 Hz, 2H, H-2 and H-6), 7.07 (d, J = 7.95 Hz, 1H, H-5′), 7.06 (d, J = 15.87 Hz, 1H, alkene), 6.93 (d, J = 16.47 Hz, 1H, alkene), 6.90 (d, J = 9.15 Hz, 2H, H-3 and H-5), 3.90 (s, 3H, CH3OCO), 3.81 (s, 3H, CH3O), 2.36 (s, 3H, CH3CO). 13C-NMR (300 MHz, CDCl3): δ 169.55, 164.72, 159.43, 149.24, 135.71, 130.87, 129.47, 129.39, 129.14, 127.74, 124.29, 123.88, 123.05, 114.03, 55.13, 52.06, 20.83). ESI-MS m/z; 348.9 [M + Na]+.
(E)-4′-Acetoxy-3,3′,5-trimethoxystilbene (5)
In a 250 ml three neck round bottom flask, the phosphonium bromide 1b (1.2 g, 2.4 mmol) and vanillin acetate 2c (0.45 g, 2.3 mmol) were mixed in dry THF according to the general procedure described above. The title compound was obtained (0.5 g, 65% yield) as white crystals, m.p. = 118–120 °C (reported 120–122 °C)Citation18. 1H-NMR (600 MHz, CDCl3) δ 7.09 (dd, J = 5.4 & 1.8 Hz, 1H, H-6′), 7.07 (d, J = 1.8 Hz, 1H, H-2′), 7.04 (d, J = 16.2 Hz, 1H, alkene), 7.02 (d, J = 8.4 Hz, 1H, H-5′), 6.97 (d, J = 16.8 Hz, 1H, alkene), 6.66 (d, J = 1.8 Hz, 2H, H-2 & H-6), 6.40 (t, J = 2.4 Hz, 1H, H-4), 3.89 (s, 6H, CH3O), 3.83 (s, 3H, CH3O), 2.32 (s, 3H, CH3CO). 13C-NMR (300 MHz, CDCl3) δ 169.00, 160.96, 151.17, 139.36, 139.07, 136.20, 128.95, 128.53, 122.91, 119.30, 110.11, 104.58, 100.11, 55.89, 55.38, 20.70). ESI-MS m/z; 351.0 [M + Na]+.
(E)-4′-Acetoxy-3,5-dimethoxy-3′-(methoxycarbonyl)stilbene (6)
In a 250 ml three neck round bottom flask, the phosphonium bromide 1b (0.9 g, 1.82 mmol) and the aldehyde 2b (0.4 g, 1.82 mmol) were mixed in dry THF according to the general procedure described above. The title compound was obtained (0.45 g, 70% yield) as colorless needles, mp 97–99 °C. 1H-NMR (300 MHz, CDCl3) δ 8.12 (d, J = 1.83 Hz, 1H, H-2′), 7.80 (dd, J = 8.55, 1.83 Hz, 1H, H-6′), 7.19 (d, J = 16.47 Hz, 1H, alkene), 7.13 (d, J = 8.55 Hz, 1H, H-5′), 7.12 (d, J = 16.47 Hz, 1H, alkene), 6.73 (d, J = 2.46 Hz, 2H, H-2 and H-6), 6.41 (t, J = 2.40 Hz, 1H, H-4), 3.87 (s, 3H, CH3OCO), 3.80 (s, 6H, CH3O), 2.30 (s, 3H, CH3COO). 13C-NMR (300 MHz, CDCl3) δ 169.67, 164.75, 160.96, 149.77, 138.70, 135.30, 131.32, 130.10, 129.66, 127.06, 124.09, 123.26, 104.65, 100.36, 55.36, 52.25, 20.96. ESI-MS m/z; 378.9 [M + Na]+.
(E)-3,4′,5-Trimethoxy-3′-(methoxycarbonyl)stilbene (7)
In a 250 ml three neck round bottom flask, the phosphonium bromide 1b (3.3 g, 6.7 mmol) and the aldehyde 2a (1.17 g, 6.03 mmol) were mixed in dry THF as described in the general procedure described above. The title compound was obtained (1.38 g, 70% yield) as a white solid, m.p. = 102–103 °C. 1H-NMR (300 MHz, CD3OD) δ 7.92 (d, J = 1.83 Hz, 1H, H-2′), 7.72 (dd, J = 8.55, 2.46 Hz, 1H, H-6′), 7.12 (d, J = 9.15 Hz, 1H, H-5′), 7.11 (d, J = 14.01 Hz, 1H, alkene), 7.00 (d, J = 16.47 Hz, 1H, alkene), 6.70 (d, J = 2.43 Hz, 2H, H-2 and H-6), 6.38 (t, J = 2.46 Hz, 1H, H-4), 3.89 (s, 3H, CH3OCO), 3.87 (s, 3H, CH3O), 3.80 (s, 6H, CH3O). 13C-NMR (300 MHz, CDCl3) δ 166.32, 160.80, 158.46, 139.08, 131.21, 129.58, 129.31, 127.58, 127.41, 120.03, 112.15, 104.26, 99.76, 99.65, 55.96, 55.16, 51.91, 29.57. ESI-MS m/z; 351.0 [M + Na]+.
General procedure for synthesis of compounds 8–10
The corresponding E stilbene (3 or 7) was dissolved in dry dichloromethane using a heat-dried three neck round bottom flask equipped with a magnetic stirrer. This solution was cooled down using dry ice and acetone, to about −60 °C; then a solution of boron tribromide (1 M in CH2Cl2) was added dropwise. After adding the deprotecting agent, the ice bath was withdrawn to allow the reaction mixture to warm up to room temperature and it was stirred for 1–4 h. The reaction was quenched by adding water (60 ml; very slowly), and the product was extracted with dichloromethane (20 ml). The combined organic phases were dried using sodium sulfate, and the solvent was evaporated under vacuum. Finally, the product was purified by silica gel column chromatography using a mixture of ethyl acetate/methanol (8:2) to afford the corresponding hydroxylated stilbenes.
(E)-4,4′-Dihydroxy-3′-(hydroxycarbonyl)stilbene (8)
In a 50 ml three neck round bottom flask, compound 3 (0.26 g, 0.87 mmol) and a solution of boron tribromide (5.2 ml; 6 Eq) were mixed in dry dichloromethane according to the general procedure described above. Compound 8 was obtained (0.135 g, 60% yield) as an off-white solid, m.p. = 237–240 °C (reported 241–243 °C)Citation19. 1H-NMR (300 MHz, CD3OD) δ 7.94 (d, J = 2.46 Hz, 1H, H-2′), 7.66 (dd, J = 8.55, 2.43 Hz, 1H, H-6′), 7.35 (d, J = 8.55 Hz, 2H, H-2 and H-6), 6.96 (d, J = 16.4 Hz, 1H, alkene), 6.90 (d, J = 8.52 Hz, 1H, H-5′), 6.89 (d, J = 15.87 Hz, 1H, alkene), 6.75 (d, J = 8.55 Hz, 2H, H-3 & H-5). ESI-MS m/z; 256.8 [M + H]+.
(E)-3,4′,5-Trihydroxy-3′-(hydroxycarbonyl)stilbene (9)
In a 50 ml three neck round bottom flask, compound 7 (0.58 g, 1.7 mmol) and a solution of boron tribromide (13.6 ml; 8 Eq) were mixed in dichloromethane according to the general procedure reported above. The product 9 was obtained (0.2 g, 41% yield), as a yellow solid, m.p. = 213–215 °C. 1H-NMR (300 MHz, CD3OD) δ 7.95 (d, J = 2.46 Hz, 1H, H-2′), 7.68 (dd, J = 8.55, 2.43 Hz, 1H, H-6′), 6.98 (d, J = 16.47 Hz, 1H, alkene), 6.91 (d, J = 8.52 Hz, 1H, H-5′), 6.86 (d, J = 16.47 Hz, 1H, alkene), 6.46 (d, J = 1.83 Hz, 2H, H-2 and H-6), 6.16 (t, J = 2.46 Hz, 1H, H-4). 13C-NMR (300 MHz, CD3OD) δ 173.34, 162.65, 159.69, 140.78, 134.07, 130.24, 129.60, 128.62, 128.17, 118.62, 113.96, 105.93, 103.02. ESI-MS m/z; 271.2 [M − H]−.
(E)-3,4′,5-Trihydroxy-3′-methoxycarbonylstilbene (10)
In a 50 ml three neck round bottom flask, compound 7 (0.28 g, 0.85 mmol) and a solution of boron tribromide (3.8 ml; 4.5 Eq) were mixed in dichloromethane according to the general procedure reported above. Product 10 was obtained (0.17 g, 69% yield) as a pale yellow solid, m.p. = 80–84 °C. 1H-NMR (300 MHz, CD3OD) δ 7.93 (d, J = 2.43 Hz, 1H, H-2′), 7.70 (dd, J = 8.55, 2.43 Hz, 1H, H-6′), 6.97 (d, J = 16.47 Hz, 1H, alkene), 6.94 (d, J = 8.55 Hz, 1H, H-5′), 6.86 (d, J = 15.84 Hz, 1H, alkene), 6.46 (d, J = 1.83 Hz, 2H, H-2 and H-6), 6.17 (t, J = 1.83 Hz, 1H, H-4), 3.97 (s, 3H, CH3OCO). 13C-NMR (300 MHz, CD3OD) δ 171.60, 162.08, 159.69, 140.69, 134.29, 130.50, 129.05, 128.89, 127.98, 118.83, 113.63, 105.96, 103.09, 52.93. ESI-MS m/z; 285.3 [M − H]−.
(E)-4,4′-Diacetoxy-3′-(hydroxycarbonyl)stilbene (11)
In a round-bottom flask, compound 8 (0.061 g, 0.24 mmol) in pyridine (0.5 ml) was dissolved at room temperature, and acetic anhydride (0.3 ml; 13 Eq) was then added drop-wise. This reaction mixture was stirred until the stilbene starting material was not observed (about 3 h); the reaction was quenched by adding water (20 ml), and then the mixture was acidified to pH 3 using 1 N hydrochloric acid. The product was extracted by adding ethyl acetate (8 ml) two times; then the combined organic layers was dried with sodium sulfate, and evaporation of the solvent under vacuum afforded the crude product, which was purified by crystallization from a mixture of ethyl acetate/petroleum ether. Compound 11 (0.062 g, 76%yield) was obtained as a white solid, m.p. = 193–196 °C. 1H NMR (600 MHz, CD3OD) δ 8.16 (d, J = 2.4 Hz, 1H, H-2′), 7.79 (dd, J = 8.4, 2.4 Hz, 1H, H-6′), 7.61 (d, J = 8.4 Hz, 2H, H-2 and H-6), 7.22 (d, J = 16.2 Hz, 1H, alkene), 7.19 (d, J = 16.2 Hz, 1H, alkene), 7.12 (d, J = 8.4 Hz, 1H, H-5′), 7.10 (d, J = 8.4 Hz, 2H, H-3 and H-5), 2.28 (s, 3H, CH3CO), 2.27 (s, 3H, CH3CO).13C-NMR (600 MHz, DMSO-d6) δ 169.72, 169.66, 166.06, 150.56, 149.77, 135.52, 134.95, 131.44, 129.73, 129.10, 128.10, 128.03, 127.28, 124.69, 122.62, 21.36, 21.34. ESI-MS m/z; 363.1 [M + Na]+.
(E)-3,4′,5-Triacetoxy-3′-(hydroxycarbonyl)stilbene (12)
This compound was obtained from compound 9 (0.166 g, 0.61 mmol) using the same procedure described for the synthesis of compound 11. The title compound (0.075 g, 31% yield) was obtained as a pale yellow solid, m.p. = 160–165 °C. 1H-NMR (600 MHz, CD3OD) δ 8.16 (d, J = 1.8 Hz, 1H, H-2′), 7.80 (dd, J = 8.4, 2.4 Hz, 1H, H-6′), 7.25 (d, J = 17.4 Hz, 1H, alkene), 7.24 (d, J = 1.8 Hz, 2H, H-2 and H-6), 7.19 (d, J = 16.8 Hz, 1H, alkene), 7.13 (d, J = 8.4 Hz, 1H, H-5′), 6.84 (t, J = 1.8 Hz, 1H, H-4), 2.29 (s, 3H, CH3CO), 2.28 (s, 6H, CH3CO). 13C-NMR (600 MHz, DMSO-d6) δ 169.71, 169.48, 165.98, 151.65, 150.08, 139.64, 135.08, 131.73, 130.02, 129.18, 128.27, 124.93, 124.79, 117.83, 115.70, 21.33, 21.29. ESI-MS m/z; 421.1 [M + Na]+.
Cell culture
Human hepatoma HepG2 cells (ATCC HB-8065, Manassas, VA) were maintained in Dulbecco's modified Eagle's medium, supplemented with 10% heat-inactivated fetal bovine serum, 2 mM l-glutamine, 100 IU/ml penicillin and 100 μg/ml streptomycin. Cells were grown in 75-cm2 tissue culture flasks at 37 °C in a 5% CO2 humidified incubator.
Cell viability
These experiments were carried out by adding DMSO solutions of the corresponding drug to cells in the appropriate media; the final concentration of DMSO/well was 0.5%, which is lower than that reported (1%) to be safe for use in HepG2 cellsCitation20. Cell viability assay was determined using the MTT colorimetric assay. Briefly, HepG2 cells were seeded in 96-well plates at a density of 3 × 104 per well (200 µL), and were allowed to attach overnight. Then, resveratrol or its derivatives, dissolved in DMSO at concentrations 0.04, 1.0 or 25 mM were added (1 µL) to the corresponding 96-well plate containing serum free media (200 µL), and then were incubated for 24 h at 37 °C. After that time, the media was removed (aspirated out) and the wells were washed one time with PBS before the addition of the MTT solution in PBS (0.5 mg/ml). The plate was then incubated for 2 h at 37 °C. Finally, the insoluble formazan crystals were dissolved in a solution of 0.01 M HCl in isopropanol (100 µL/well), and the absorbance of each well was read at 570 nm. The percentage of cell viability was correlated to that of DMSO-treated wells, which were set to a value of 100 % viability. Each compound was assayed at three concentrations in triplicate and the experiment was performed three times, and the results are expressed as mean ± SD.
Determination of CYP1A1 enzymatic activity (24 h incubation)
The CYP1A1-dependent 7-ethoxyresorufin O-deethylase (EROD) activity was performed on intact living cells, using 7-ethoxyresorufin (7ER) as a substrate, following a previously reported procedureCitation21. Briefly, cells (104 cells per well) were seeded onto 96-well microtiter cell culture plates until they reached 70–80% confluency. Resveratrol or its analogs dissolved in DMSO, was added to the cells (5 µM final concentration) before TCDD addition (1 nM final concentration) and the cells were incubated for 24 h. Thereafter, cells were washed with PBS, and then 200 μl of 7ER solution (2 μM) was added to each well. The amount of resorufin formed in each well at each time-point (every 5 min) was detected by fluorescence spectroscopy for each treatment per minute by comparison with a standard curve of known concentrations (excitation, 545 nm; emission, 575 nm; Baxter 96-well fluorometer)Citation22. The CYP1A1 enzymatic activity was normalized to cellular protein content using a modified fluorescence methodCitation23. Results are presented as the mean ± SEM, and statistical differences between treatment groups were determined using one way ANOVA followed by Student–Newman–Keuls post hoc test, using SigmaStat 3.5 program for Windows, Systat Software Inc. (San Jose, CA).
Determination of CYP1A1 enzymatic activity (1 h incubation)
To test the direct inhibitory effect of resveratrol and its analogs on CYP1A1 enzyme, a method similar to EROD assay was performed, with slight modifications as described previouslyCitation24,Citation25. Briefly, HepG2 cells were incubated with TCDD (1 nM) for 24 h. Thereafter, media was removed, and the cells were washed three times with PBS; and 1 mM of resveratrol or its analogs in assay buffer [Tris (0.05 M), NaCl (0.1 M), pH 7.8] were added to the cells for 60 min prior to the addition of 7ER (2 μM final concentration) as a substrate for the EROD measurement. The CYP1A1 enzymatic activity was normalized to cellular protein content using a modified fluorescence methodCitation23. Results are presented as mean ± SEM, and statistical differences between treatment groups were determined using one way ANOVA, followed by Student–Newman–Keuls post hoc test using SigmaStat 3.5 program for Windows, Systat Software Inc. (San Jose, CA).
CYP1A1 mRNA expressions
RNA extraction and cDNA synthesis
Six hours after incubation with the test compounds, cells were collected and total RNA was isolated using TRIzol reagent (Ambion/RNA, Carlsbad, CA) according to the manufacturer's instructions and quantified by measuring the absorbance at 260 nm. Thereafter, first-strand cDNA synthesis was performed using the High-Capacity cDNA reverse transcription kit (Applied Biosystems, Warrington, United Kingdom) according to the manufacturer's instructions. Briefly, 1.5 μg of total RNA from each sample was added to a mix of 2.0 μl of 10 × reverse transcription (RT) buffer, 0.8 μl of 25 × dNTP mix (100 mM), 2.0 μl of 10 × RT random primers, 1.0 μl of MultiScribe reverse transcriptase and 4.2 μl of nuclease-free water. The final reaction mix was kept at 25 °C for 10 min, heated to 37 °C for 120 min, heated to 85 °C for 5 min and finally cooled to 4 °C.
Quantification by real-time PCR
Quantitative analysis of CYP1A1 mRNA expression was performed by real time PCR by subjecting the resulting cDNA to PCR amplification using 96-well optical reaction plates in the ABI Prism 7500 system (Applied Biosystems). Twenty-five-microliter reactions contained 0.1 μl of 10 μM forward primer and 0.1 μl of 10 μM reverse primer (40 nM final concentration of each primer),12.5 μl of SYBR Green Universal Master mix, 11.05 μl of nuclease-free water and 1.25 μl of cDNA sample (equivalent to 0.20 ng/ml). The primers used in the current study were chosen from previous studyCitation26; human CYP1A1: forward primer 5′-CTATCTGGGCTGTGGGCAA-3′, reverse primer 5′-CTGGCTCAAGCACAACTTGG-3′ and human β-actin: forward primer 5′-CTGGCACCCAGCACAATG-3′, reverse primer 5′-GCCGATCCACACGGAGTACT-3′. Assay controls were incorporated onto the same plate, namely, no-template controls to test for the contamination of any assay reagents. After the plate was sealed with an optical adhesive cover, the thermocycling conditions were initiated at 95 °C for 10 min, followed by 40 PCR cycles of denaturation at 95 °C for 15 s and annealing/extension at 60 °C for 1 min. A melting curve (dissociation stage) was performed by the end of each cycle to ascertain the specificity of the primers and the purity of the final PCR product.
Real-time PCR data analysis
The real-time PCR data were analyzed using relative gene expression, i.e. the ΔΔCT method, as described in Applied Biosystems User Bulletin No. 2 and explained further by Livak and SchmittgenCitation27. In brief, the primers used in this study were tested to avoid primer dimers, self-priming formation or non-specific amplification. To ensure the quality of the measurements, each plate included, for each gene, a negative control and a positive control. For each sample, a threshold cycle (CT) was calculated based on the time (measured by the number of PCR cycles) at which the reporter fluorescence emission increased beyond a threshold level (based on the background fluorescence of the system). The triplicate measurements for each sample were averaged to give an average CT value for each group, after removing the outliersCitation28. The samples were diluted in such a manner that the CT value was observed between15 and 30 cycles. Results were expressed using the comparative CT method as described in User Bulletin No. 2 (Applied Biosystems). Briefly, the ΔCT values were calculated in every sample for each gene of interest as CT gene of interest − CT reporter gene, with β-actin as the reporter gene. Calculation of relative changes in the expression level of one specific gene (ΔΔCT) was performed by subtraction of the ΔCT of the control (untreated cells) from the ΔCT of the corresponding treatment groups. The values and ranges given in the figure were determined as follows: 2−ΔΔCT with ΔΔCT + SE and ΔΔCT − SE, where SE is the standard error of the mean of the ΔΔCT value (User Bulletin No. 2; Applied Biosystems).
Data are presented as the mean ± SEM. Control and treatment measurements were compared using a one way ANOVA followed by a Student–Newman–Keuls post hoc comparison. A result was considered statistically significant when p < 0.05.
Molecular modeling
Docking study
Protein
The crystal structure of the human CYP1A1 (PDB ID: 4I8V)Citation29 was downloaded from the Protein Data Bank (PDB)Citation30. The chains B and C, crystallographic water molecules, as well as 2-phenyl-4H-benzo[H]chromen-4-one (inhibitor co-crystallized with the protein) were removed manually. Subsequently, all hydrogens and electrostatic charges were added, and protonation sites were corrected. The atomic partial charges of the heme group were computed using the Gasteiger charge methodCitation31. Finally, the structure was submitted to a geometry optimization with 500 steepest descent steps, and 100 conjugate gradient steps, using the AMBER99SB force field implemented in UCSF Chimera 1.9Citation32.
Ligands
The compounds C3–C12, TMS and resveratrol were constructed and submitted to a geometry optimization employing the AMBER99SB force field in UCSF Chimera 1.9.
Docking
Docking calculations were carried out using the AutoDock 4.2 software (La Jolla, CA)Citation33. A grid box of 70 × 70 × 70 pints with a grid spacing of 0.375 Å, and centered at the heme group was used to calculate the atom types needed for the calculation. The Lamarckian genetic algorithm was used as a search method with a total of 30 runs (maximum of 20 000 000 energy evaluations; 27 000 generations; initial populations of 150 conformers). The best binding mode of each molecule was selected based on both the lowest binding free energy and the largest cluster size.
Discussion
Chemistry
We conducted a literature search for synthetic pathways previously used to synthesize similar stilbene derivatives. We found five different strategies by which we could obtain the target compounds 3–12; namely the Heck-couplingCitation19, Wittig reactionCitation34, Horne–Wadsworth–EmmonsCitation35, PerkinCitation36 and the McMurry reactionsCitation37. In this regard, our first attempt to obtain the target molecules 3–12 by the Wittig reaction involved the use of aldehydes possessing a free carboxylic acid group; however, all reactions produced a complex mixture of products with minimal yields of the target molecules. These results are in agreement with previous reports describing such reactions to be “sluggish”Citation34, and affording low yields of the desired stilbene. Therefore, we decided to change our strategy by reacting the corresponding aldehyde precursors as the corresponding methyl esters (2a–2c), with the triphenylphosphonium bromide 1a or 1b, following a classical Wittig reaction (Scheme 1). In all cases, we obtained a mixture of cis and trans isomers, and consequently, to obtain only the desired trans compounds, we reacted the isomeric mixtures with diphenyl disulfide in dry THF according to a reported procedureCitation16, affording the pure trans isomers 3–7 as identified by TLC and subsequent 1H-NMR analysis.
The next synthetic step involved the use of boron tribromide in dichloromethane solution, to obtain the free phenol groups (8–10), and finally, compounds 8 and 9 were acetylated with acetic anhydride in pyridine at room temperature to afford the corresponding aspirin-like resveratrol derivatives (11 and 12). In total, we synthesized 10 different hybrid compounds possessing both the stilbene scaffold present in resveratrol, and a salicylate (or acetylsalicylate) moiety. Derivatives having methoxylated moieties (3–7) are expected to be more lipophilic than those possessing a free phenol group (8–10), whereas those compounds in which the 4′-phenol group is acetylated (11 and 12) are expected to exert aspirin-like properties.
Determination of CYP1A1 enzymatic activity
We measured the induction of the CYP1A1 enzyme by TCDD using HepG2 cells; this model has been extensively utilized to study the effects of resveratrol on CYP1A1 activity and expressionCitation38. In this regard, Beedanagari et al.Citation38 reported that TCDD induced the expression of CYP1A1 mRNA in HepG2 cells, but not CYP1B1. Accordingly, we selected the EROD assay to test the ability of compounds 3–12 to inhibit the catalytic activity of the CYP1A1 enzyme in HepG2 cells, but first, we conducted a concentration-dependent cell viability assay (MTT assay) to determine the maximum concentration at which enzymatic inhibition occurred without causing significant cell death. Based on this assay, we determined that the final concentration to be used for subsequent screening assays with compounds 3–12 was 5 µM, at which we consistently observed cell viabilities higher than 80%.
CYP1A1 catalytic inhibition (24 h incubation)
As expected, we did not observe any CYP1A1 activity in DMSO-treated cells (negative control), whereas cells treated with TCDD (positive control) showed a significantly higher enzyme activity, which was about 700 pmol/min/mg protein, indicating TCDD’s ability to induce the expression and activity of this enzyme in HepG2 cells (). In the same experiment, resveratrol and its methylated derivative, trimethoxystilbene (TMS), showed 15 and 98% inhibition of TCDD-mediated induction of CYP1A1, respectively. This dramatic difference between the hydroxylated and methoxylated stilbenes is likely due to the enhanced lipophilicity and improved cell membrane permeability of TMS. This is in agreement with previous reports which have demonstrated that replacing hydroxyl groups by methoxy groups in resveratrol’s structure, resulted in an increased inhibitory potency on CYP1A1 catalytic activity, as well as on other CYP450 enzymesCitation39,Citation40.
Figure 2. Effect of resveratrol and its analogs on TCDD-mediated induction of CYP1A1 catalytic activity in HepG2 cells. (A) Cells were pre-incubated with 5 μM of resveratrol (Res) or its analogs for 30 min before the addition of TCDD (1 nM) for an additional 24 h. The CYP1A1 activity level was determined using CYP1A1-dependent EROD assay. Values represent mean activity ± SEM (n = 8). (+) p < 0.05 compared with control (C), (*) p < 0.05 compared with TCDD. (B) The direct inhibitory effects of resveratrol and its analogs on CYP1A1 enzyme. HepG2 cells were pre-treated with TCDD (1 nM) for 24 h, thereafter, media were removed, washed thrice with PBS and 5 μM of the tested compounds in assay buffer [Tris (0.05 M), NaCl (0.1 M), pH 7.8] were added for 60 min prior to the addition of 7ER (2 μM final concentration) for the EROD measurement. Results are expressed as percentage of remaining EROD activity (mean ± SEM, n = 8). (*) p < 0.05 compared with control (C). ND: not detected.
![Figure 2. Effect of resveratrol and its analogs on TCDD-mediated induction of CYP1A1 catalytic activity in HepG2 cells. (A) Cells were pre-incubated with 5 μM of resveratrol (Res) or its analogs for 30 min before the addition of TCDD (1 nM) for an additional 24 h. The CYP1A1 activity level was determined using CYP1A1-dependent EROD assay. Values represent mean activity ± SEM (n = 8). (+) p < 0.05 compared with control (C), (*) p < 0.05 compared with TCDD. (B) The direct inhibitory effects of resveratrol and its analogs on CYP1A1 enzyme. HepG2 cells were pre-treated with TCDD (1 nM) for 24 h, thereafter, media were removed, washed thrice with PBS and 5 μM of the tested compounds in assay buffer [Tris (0.05 M), NaCl (0.1 M), pH 7.8] were added for 60 min prior to the addition of 7ER (2 μM final concentration) for the EROD measurement. Results are expressed as percentage of remaining EROD activity (mean ± SEM, n = 8). (*) p < 0.05 compared with control (C). ND: not detected.](/cms/asset/967c847a-18dd-434f-9ade-5ef6d7d1982c/ienz_a_979347_f0002_b.jpg)
also shows that there are four hybrid resveratrol–salicylate compounds that significantly decreased TCDD-induced CYP1A1 activity, namely, compounds 3, 4, 6 and 7 which inhibited CYP1A1 activity by 84, 29, 20 and 43%, respectively (). These derivatives share two common structural features; first, they possess a methyl ester at the carboxyl group in ring A (); second, they have either a methoxy group at position 4, or two methoxy groups at positions 3 and 5 on ring B. These observations are in agreement with previous reports in which it was observed that methoxylation of the stilbene scaffold resulted in an enhanced CYP1A1 inhibitionCitation39,Citation41. Additional structure–activity relationship (SAR) analysis shows that hybrid compounds having an acetoxy group at position 4 of ring A (compounds 4 and 6), are weaker CYP1A1 inhibitors compared to those having methoxy groups (compounds 3 and 7). Apparently, the acetyl group decreases the ability of these stilbenes to inhibit CYP1A1’s catalytic activity.
Additional data about the effect of acetylation on the inhibitory activity exerted by stilbenes is provided by the results obtained with derivative 5, which is structurally similar to 3,3′,4,5′-tetramethoxystilbene, a synthetic analog of piceatannol (CYP1A1 IC50 = 750 nMCitation42). Compound 5 showed about 60% inhibition of CYP1A1 (), and the structural difference between this molecule and 3,3′,4,5′-tetramethoxystilbene (not tested), is the presence of an acetoxy group at position 4 instead of a methoxy group. This supports the observation that acetoxy groups decrease the CYP1A1 inhibitory activity exerted by methoxystilbenes.
It is noteworthy to mention that compounds having a free carboxylic acid (8, 9, 11 and 12) did not inhibit CYP1A1 activity at all. These compounds possess either a salicylic acid or an acetylsalicylic acid (aspirin) moiety. In fact, we observed that compounds 8 and 9 produced a slight increase in CYP1A1 activity as measured by the EROD assay. These findings seem to be in agreement with previous reports showing that both aspirin and salicylic acid failed to decrease EROD levels even when tested at concentrations as high as 1 mM in HepG2 cellsCitation43. Consequently, we observed that stilbenes acted as modulators of CYP1A1 activity; in other words, depending on the chemical structure of the stilbene, some of them inhibited CYP1A1 activity while others increased it. From a therapeutic point of view, an increased CYP1A1 activity in cells with significantly increased metabolic rates (such as cancer cells), might be regarded as an unwanted side effect, because an increased CYP1A1 activity would potentially increase the rate and the extent at which some pro-carcinogens can be converted to carcinogens.
When we analyzed the structure–activity relationship for derivatives 3, 4, 6 and 7, we observed that compounds having a single methoxy group at position 4 are better CYP1A1 inhibitors than those having a 3,5-dimethoxy moiety on ring B. This observation is not in agreement with a previous report published by Mikstacka et al.Citation44 in which this research group reported the synthesis and biological evaluation of a series of methylthiostilbenes as CYP1A1, CYP1A2 and CYP1B1 inhibitors. In this regard, authors reported that a 3,5-disubstituted compounds were more potent than those having only a 4-methoxy groupCitation44. Nevertheless, we realize that this observation requires further validation by testing a more expanded library of compounds using the same assay conditions.
CYP1A1 catalytic inhibition after (1 h incubation)
The main mechanism of TCDD-mediated carcinogenicity involves the activation of the AhR and the subsequent induction of CYP1A1 through transcriptional and translational mechanismsCitation45. Resveratrol is a well-known AhR antagonistCitation46, which has the ability to inhibit CYP1A1 activity by direct inhibition of CYP1A1 activityCitation7. Therefore, to investigate whether our hybrid resveratrol–salicylate analogs exhibit “direct” CYP1A1 inhibition, we tested the inhibitory effects of the most active compounds (3 and 5) by incubating HepG2 cells in the presence of these compounds for 1 h. Briefly, first we treated cells with TCDD for 24 h to induce the expression of the CYP1A1 enzyme (as described above). Then, after changing the medium, we incubated the cells with the corresponding test compound for 1 h (prior to the addition of the substrate for CYP1A1); finally, we measured the catalytic activity of CYP1A1 as described in the “Methods” section.
Our results demonstrate that, unlike the 24 h incubation experiment described previously, resveratrol did not significantly inhibit CYP1A1 activity. Similarly, compound 5 had no direct inhibitory effect on the CYP1A1 enzyme (), even though this compound exerted about 60% enzyme inhibition when tested on a 24-h incubation period (). On the other hand, the new compound 3 showed a moderate inhibitory effect on CYP1A1 enzyme activity (about 38% inhibition), which was very similar to that obtained with TMS (42%, ). Furthermore, we also observed that compound 3 was a more potent CYP1A1 inhibitor than the parent compound resveratrol, which suggests that the hybrid resveratrol–salicylate derivative 3 has the ability to significantly inhibit the carcinogen-activating enzyme CYP1A1 at both 24 h and 1 h incubation.
The significance of the 1 h incubation, compared to the 24 h incubation experiment, is the fact that the assay carried out by incubating cells for 1 h represents the effects of the drug on CYP1A1 at the post-translational level (direct inhibition), rather than (or in addition to) any potential effects at the transcriptional level (modulation of CYP1A1 mRNA expression).
Effects on the expression of CYP1A1 mRNA
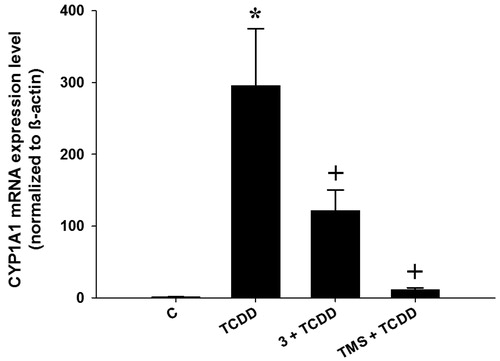
To investigate if the decrease in enzyme activity exerted by the test compounds was due to a classical inhibition of the CYP1A1 active site, or due to a decrease in CYP1A1 transcription, we also measured CYP1A1 mRNA levels in HepG2 cells in the presence of the test compounds (5 µM), for 6 h as described previouslyCitation24. We selected compound 3 (the most promising hybrid stilbene–salicylate analog), and we compared it with TMS under the same experimental conditions. The results of this experiment are shown in . DMSO-treated cells (control) showed negligible induction of CYP1A1 mRNA, whereas TCDD-treated cells exhibited a significant increase in CYP1A1 mRNA levels (about 295-fold increase compared to control cells). Cells co-treated with both TCDD and TMS showed a dramatic decrease of CYP1A1 mRNA expression compared to those treated with TCDD alone, which is in agreement with the results observed in both (1 and 24 h incubation) EROD assays. Similarly, cells co-incubated with TCDD and compound 3 also showed a significant decrease in the expression of CYP1A1 mRNA levels, although this effect was not as strong as that obtained with TMS. This observation suggests that compound 3 exerts its modulatory effect on CYP1A1 by both, inhibiting CYP1A1’s catalytic activity and by decreasing its mRNA levels.
Figure 3. Effects of stilbenes on CYP1A1 mRNA level. HepG2 cells were co-treated with 1 nM TCDD plus compound 3 (3) or 3,4′,5-trimethoxystilbene (TMS) at 5 µM for 6 h. First-strand cDNA was synthesized from total RNA (1.5 μg) extracted from HepG2 cells. cDNA fragments were amplified and quantitated using the ABI 7500 real-time PCR system as described in the experimental section. Duplicate reactions were performed for each experiment, and the values presented as the mean ± SE (n = 6). (*) p < 0.05 compared to control (DMSO; C). (+) p < 0.05 compared to TCDD alone.
Molecular modeling
To evaluate the ability of the test compounds to interact with the catalytic site of human CYP1A1, we carried out a molecular modeling (docking) experiment, in which we assessed the ability of all compounds to exert binding interactions with key amino acid residues in the CYP1A1 active site. It is noteworthy to mention that similar studies have been reported previously, but these had used the CYP1A2 template (PDB code: 2HI4) instead, and from these predictions, authors assumed potential binding interactions between experimental drugs, and the CYP1A1 active site (homology models)Citation7. Nevertheless, considering that the crystal structure of human CYP1A1 was recently reported (PDB code: 4I8VCitation29), we decided to use this template in our docking study. To the best of our knowledge, our work is the first one correlating in vitro CYP1A1 activity data with docking studies using the crystal structure of human CYP1A1.
shows the binding mode for the reference compound TMS within the active site of human CYP1A1. This molecule exerts a π–π interaction with Phe224, which has been reported by other groups in previous in silico studies with resveratrol analogs. shows the binding modes calculated for all the test compounds (3–12); in this regard, it is interesting to note that compounds 3, 4, 5 and 6 exert a similar binding profile () to that observed for the reference stilbene TMS, which may account for their in vitro inhibitory activity. On the other hand, compound 7 showed a slight shift within the active site of CYP1A1 that allows its carbonyl group to interact with the iron atom present in the heme group, and at the same time, to produce a π–π interaction with both Phe224 and Phe258.
Figure 4. Binding mode for 2,4′,5-trimethylstilbene (TMS) in the active site of CYP1A1. 3D figures were generated using PyMOL Molecular Graphics System (DeLano Scientific LLC, Palo Alto, CA, 2007); 2D figures were generated according to a procedure described in the literatureCitation47.
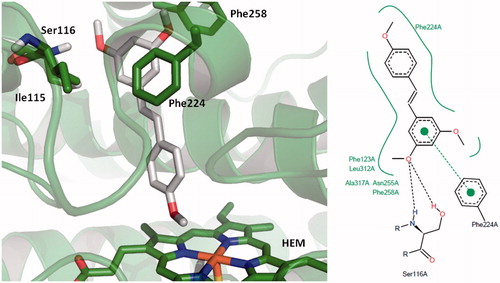
Figure 5. Comparison of the binding mode calculated for trimethylstilbene (TMS; shown in grey), with that observed for compounds (A) 3 (cyan), 4 (purple), 5 (orange), 6 (yellow); (B) 7; (C) 8 (purple), 9 (orange); (D) 10 (purple), 11 (blue) and 12 (yellow) in the active site of human CYP1A1. 3D figures were generated using PyMOL Molecular Graphics System (DeLano Scientific LLC, Palo Alto, CA, 2007).
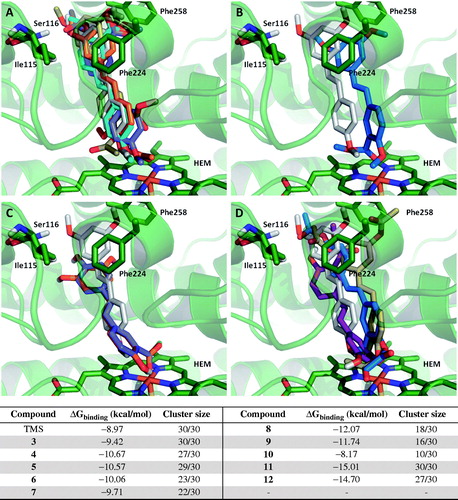
It is important to note that compounds 3–7 showed low free energy values and at the same time, showed high cluster size values, suggesting a high probability of exerting binding interactions with the CYP1A1 active site, having the actual conformation calculated and reported for these drugs. However, compounds 8 and 9 showed a significant binding interaction between their corresponding carboxylate groups and the iron atom present in the heme group of human CYP1A1, which results in loss of the binding interaction with Phe224 ().
Figure 6. Two-dimensional representation of the binding interactions observed between the active site of CYP1A1 and compounds (A) 3; (B) 7, (C) 8 and (D) 12. 2D figures were generated according to a procedure described in the literatureCitation47.
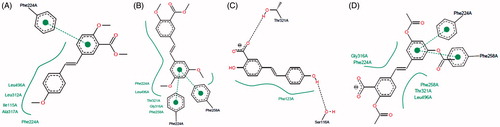
However, despite the low free binding energy values calculated for compounds 8 and 9, their low cluster size values suggests that these two hybrid salicylate–resveratrol derivatives would likely adopt a slightly different conformation than that determined by our molecular modeling protocol; in other words, there is a significant probability for these compounds to bind the CYP1A1 active site with a different conformation than that predicted theoretically. Compound 10 showed the highest free binding energy and the lowest cluster size among the tested compounds, and the predicted binding mode for this particular molecule does not show the π–π interaction with Phe224 observed for some of its analogs. This would explain why this compound was practically inactive in vitro. Finally, compounds 11 and 12 showed the lowest free binding energies and the highest cluster size values, which would account for their high in vitro inhibitory profile. However, the observed interaction of the carboxylic acid group present in these molecules, with the iron atom in the heme group of CYP1A1, could be related to the observed increased in enzymatic activity (activation), rather than enzyme inhibition. This observation could also be related to the increased enzymatic activity of CYP1A1 in the presence of compounds 8 and 9 ().
Finally, it is noteworthy to point out that the vast majority of previous literature reports have only described the inhibitory profile of trans-stilbenes on CYP1A1, and not much attention has been put in the corresponding cis-isomers. Consequently, one of the future directions in this regard could involve the biological evaluation of hybrid resveratrol–salicylate derivatives possessing the cis configuration, especially because cis-isomers have been shown to exert significant anticancer activityCitation48, even though this therapeutic profile has not been correlated with in vitro CYP1A1 inhibition studies. In this regard, we have identified (by molecular modeling), at least one hybrid derivative that could exert potent CYP1A1 inhibition (results not shown), and constitutes the topic of one of our current research projects.
Conclusion
The addition of a carboxylate group to ring A of resveratrol yielded a new series of aspirin–resveratrol analogs, which modulate the in vitro activity of the carcinogen-activating CYP1A1 enzyme. Compounds possessing a methyl esters moiety showed a moderate inhibitory profile of CYP1A1 activity, compared to the parent resveratrol; however, compounds having a free carboxylic acid group (including those possessing an acetylsalicylic acid group) increased the catalytic activity of the enzyme. The potential chemopreventive effect exerted by compound 3 (the most active compound) obtained in this series, is supported by the observation that it also exerts a decrease in the expression of CYP1A1 mRNA in HepG2 cells at concentrations as low as 5 µM. The implications of these results are significant, considering the potential synergistic profile of the new hybrid resveratrol–salicylate analog(s) on many other targets relevant to chemoprevention (some of them are studied and discussed in the proceeding paper). Nevertheless, our study also showed that differences in the chemical structure of different stilbene derivatives may potentially switch CYP1A1 inhibition, to CYP1A1 induction, which may not be useful in a long-term chemopreventive setting, but it might find potential applications in short-term states where CYP1A1 activity is required but compromised.
Importantly, the results obtained in this study provide preliminary evidence supporting the design of hybrid molecules combining the chemical features of two well-known chemopreventive agents, namely resveratrol and aspirin (salicylates). According to our findings, the previously reported unfavorable effects of a salicylate moiety did not void the ability of some hybrid resveratrol analogs to inhibit the catalytic activity of CYP1A1, or its effects on CYP1A1 mRNA in vitro. Finally, compound 3 represents a new potential hybrid chemopreventive agent which combines the chemical structures of two well-known chemopreventive compounds.
Notice of Correction:
The version of this article published online ahead of print on 19 November 2014 contained an error in Figure 1. Figure 1, compound 5, R5 should be “CH3” not “OCH3”. The error has been corrected for this version.
Declaration of interest
The authors report no conflict of interest. The authors alone are responsible for the content and writing of this article. The authors gratefully acknowledge the financial support provided by the Saudi Cultural Bureau in Canada, for a graduate scholarship to F.S.A.; O.H.E. is the recipient of Alberta Cancer Foundation Graduate Studentship Award and Alberta Innovates Technology Futures Scholarship; M.A.M. E. is the recipient of Alberta Innovates-Health Solutions post-graduate fellowship and Canadian Institute of Health Research post-PhD fellowship. C.A.V-M is supported in part by AI-HS; S.B. is supported by AI-HS, CBCF and Department of Pediatrics/Stollery Children’s Hospital Hair Massacure Donation Fund; and Finally, R.A.O. is supported by the National Council of Science and Technology (CONACYT), Mexico.
References
- Guengerich FP, Shimada T. Activation of procarcinogens by human cytochrome P450 enzymes. Mutat Res-Fundam Mol Mech Mutag 1998;400:201–13
- Sakaki T. Practical application of cytochrome P450. Biol Pharm Bull 2012;35:844–9
- Oyama T, Kagawa N, Kunugita N, et al. Expression of cytochrome P450 in tumor tissues and its association with cancer development. Front Biosci 2004;9:1967–76
- Bieche I, Narjoz C, Asselah T, et al. Reverse transcriptase-PCR quantification of mRNA levels from cytochrome (CYP)1, CYP2 and CYP3 families in 22 different human tissues. Pharmacogenet Genomics 2007;17:731–42
- Shimada T, Fujii-Kuriyama Y. Metabolic activation of polycyclic aromatic hydrocarbons to carcinogens by cytochromes P450 1A1 and 1B1. Cancer Sci 2004;95:1–6
- Androutsopoulos VP, Tsatsakis AM, Spandidos DA. Cytochrome P450 CYP1A1: wider roles in cancer progression and prevention. BMC Cancer 2009;9:187
- Nandekar PP, Sangamwar AT. Cytochrome P450 1A1-mediated anticancer drug discovery: in silico findings. Exp Opin Drug Discov 2012;7:771–89
- Shanmugam MK, Kannaiyan R, Sethi G. Targeting cell signaling and apoptotic pathways by dietary agents: role in the prevention and treatment of cancer. Nutr Cancer Int J 2011;63:161–73
- Schwarz D, Roots I. In vitro assessment of inhibition by natural polyphenols of metabolic activation of procarcinogens by human CYP1A1. Biochem Biophys Res Commun 2003;303:902–7
- Chun YJ, Kim MY, Guengerich FP. Resveratrol is a selective human cytochrome P450 1A1 inhibitor. Biochem Biophys Res Commun 1999;262:20–4
- Abbadessa G, Spaccamiglio A, Sartori ML, et al. The aspirin metabolite, salicylate, inhibits 7,12-dimethylbenz[a]anthracene-DNA adduct formation in breast cancer cells. Int J Oncol 2006;28:1131–40
- MacDonald CJ, Cheng RYS, Roberts DD, et al. Modulation of carcinogen metabolism by nitric oxide-aspirin 2 is associated with suppression of DNA damage and DNA adduct formation. J Biol Chem 2009;284:22099–107
- Pettit GR, Melody N, Thornhill A, et al. Antineoplastic agents. 579. Synthesis and cancer cell growth evaluation of E-stilstatin 3: a resveratrol structural modification. J Nat Prod 2009;72:1637–42
- Flaherty DP, Walsh SM, Kiyota T, et al. Polyfluorinated bis-styrylbenzene beta-Amyloid plaque binding ligands. J Med Chem 2007;50:4986–92
- Mao JL, Ran XK, Tian JZ, et al. Design, synthesis and biological evaluation of novel 4-hydroxybenzene acrylic acid derivatives. Bioorg Med Chem Lett 2011;21:1549–53
- Gabriele B, Benabdelkamel H, Plastina P, et al. Trans-resveratrol-d(4), a molecular tracer of the wild-type phytoalexin; Synthesis and spectroscopic properties. Synthesis-Stuttgart 2008;18:2953–6
- Murias M, Handler N, Erker T, et al. Resveratrol analogues as selective cyclooxygenase-2 inhibitors: synthesis and structure-activity relationship. Biorg Med Chem 2004;12:5571–78
- Gill MT, Bajaj R, Chang CJ, et al. 3,3′,5′-Tri-O-methylpiceatannol and 4,3′,5′-tri-O-methylpiceatannol – improvements over piceatannol in bioactivity. J Nat Prod 1987;50:36–40
- Hansen HC, Chiacchia FS, Patel R, et al. Stilbene analogs as inducers of apolipoprotein-I transcription. Eur J Med Chem 2010;45:2018–23
- Sudan S, Rupasinghe HP. Flavonoid-enriched apple fraction AF4 induces cell cycle arrest, DNA topoisomerase II inhibition and apoptosis in human liver cancer HepG2 cells. Nutr Cancer 2014;66:1–10
- El Gendy MAM, El-Kadi AOS. Harman induces CYP1A1 enzyme through an aryl hydrocarbon receptor mechanism. Toxicol Appl Pharmacol 2010;249:55–64
- El Gendy MAM, El-Kadi AOS. Peganum harmala L. differentially modulates cytochrome P450 gene expression in human hepatoma HepG2 cells. Drug Metab Lett 2009;3:212–16
- Lorenzen A, Kennedy SW. A fluorescence-based protein assay for use with a microplate reader. Anal Biochem 1993;214:346–8
- El Gendy MAM, Soshilov AA, Denison MS, El-Kadi AOS. Transcriptional and posttranslational inhibition of dioxin-mediated induction of CYP1A1 by harmine and harmol. Toxicol Lett 2012;208:51–61
- El Gendy MAM, Soshilov AA, Denison MS, El-Kadi AOS. Harmaline and harmalol inhibit the carcinogen-activating enzyme CYP1A1 via transcriptional and posttranslational mechanisms. Food Chem Toxicol 2012;50:353–62
- Anwar-Mohamed A, Elshenawy OH, Soshilov AA, et al. Methylated pentavalent arsenic metabolites are bifunctional inducers, as they induce cytochrome P450 1A1 and NAD(P)H:quinone oxidoreductase through AhR- and Nrf2-dependent mechanisms. Free Radic Biol Med 2014;67:171–187
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 2001;25:402–8
- Aparicio O, Geisberg JV, Sekinger E, et al. Chromatin immunoprecipitation for determining the association of proteins with specific genomic sequences in vivo. Los Angeles: John Willey & Sons; 2005: chapter 21–3
- Walsh AA, Szklarz GD, Scott EE. Human cytochrome P450 1A1 structure and utility in understanding drug and xenobiotic metabolism. J Biol Chem 2013;288:12932–43
- Bernstein FC, Koetzle TF, Williams GJB, et al. The protein data bank: a computer-based archival file for macromolecular structures. J Mol Biol 1977;112:535–42
- Wang JM, Wang W, Kollman PA, Case DA. Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model 2006;25:247–60
- Pettersen EF, Goddard TD, Huang CC, et al. UCSF chimera – a visualization system for exploratory research and analysis. J Comput Chem 2004;25:1605–12
- Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 2009;30:2785–91
- Murphy PJ, Brennan J. The Wittig olefination reaction with carbonyl-compounds other than aldehydes and ketones. Chem Soc Rev 1988;17:1–30
- Gester S, Wuest F, Pawelke B, et al. Synthesis and biodistribution of an 18F-labelled resveratrol derivative for small animal positron emission tomography. Amino Acids 2005;29:415–28
- Solladie G, Pasturel-Jacope Y, Maignan J. A re-investigation of resveratrol synthesis by Perkins reaction. Application to the synthesis of aryl cinnamic acids. Tetrahedron 2003;59:3315–21
- Dhyani MV, Kameswaran M, Korde AG, et al. Stereoselective synthesis of an iodinated resveratrol analog: preliminary bioevaluation studies of the radioiodinated species. Appl Radiat Isot 2011;69:996–1001
- Beedanagari SR, Bebenek I, Bui P, Hankinson O. Resveratrol inhibits dioxin-induced expression of human CYP1A1 and CYP1B1 by inhibiting recruitment of the aryl hydrocarbon receptor complex and RNA polymerase II to the regulatory regions of the corresponding genes. Toxicol Sci 2009;110:61–7
- Mikstacka R, Baer-Dubowska W, Wieczorek M, Sobiak S. Thiomethylstilbenes as inhibitors of CYP1A1, CYP1A2 and CYP1B1 activities. Mol Nutr Food Res 2008;52:S77–83
- Regev-Shoshani G, Shoseyov O, Kerem Z. Influence of lipophilicity on the interactions of hydroxy stilbenes with cytochrome P450 3A4. Biochem Biophys Res Commun 2004;323:668–73
- Mikstacka R, Przybylska D, Rimando AM, Baer-Dubowska W. Inhibition of human recombinant cytochromes P450 CYP1A1 and CYP1B1 by trans-resveratrol methyl ethers. Mol Nutr Food Res 2007;51:517–24
- Kim S, Ko H, Park JE, et al. Design, synthesis, and discovery of novel trans-stilbene analogues as potent and selective human cytochrome P450 1B1 inhibitors. J Med Chem 2001;45:160–4
- MacDonald CJ, Ciolino HP, Yeh GC. The drug salicylamide is an antagonist of the aryl hydrocarbon receptor that inhibits signal transduction induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Cancer Res 2004;64:429–34
- Mikstacka R, Rimando AM, Dutkiewicz Z, et al. Design, synthesis and evaluation of the inhibitory selectivity of novel trans-resveratrol analogues on human recombinant CYP1A1, CYP1A2 and CYP1B1. Biorg Med Chem 2012;20:5117–26
- Shimada T, Inoue K, Suzuki Y, et al. Arylhydrocarbon receptor-dependent induction of liver and lung cytochromes P450 1A1, 1A2, and 1B1 by polycyclic aromatic hydrocarbons and polychlorinated biphenyls in genetically engineered C57BL/6J mice. Carcinogenesis 2002;23:1199–207
- de Medina P, Casper R, Savouret JF, Poirot M. Synthesis and biological properties of new stilbene derivatives of resveratrol as new selective aryl hydrocarbon modulators. J Med Chem 2005;48:287–91
- Stierand K, Rarey M. Drawing the PDB: protein-ligand complexes in two dimensions. ACS Med Chem Lett 2010;1:540–5
- Paul S, Mizuno CS, Lee HJ, et al. In vitro and in vivo studies on stilbene analogs as potential treatment agents for colon cancer. Eur J Med Chem 2010;45:3702–8