
Abstract
Context: The inhibition of glutathione S-transferase P1-1 (GSTP1-1) is a sound strategy to overcome drug resistance in oncology practice.
Objective: The nitrobenzoxadiazolyl (NBD) S-conjugate of glutathione and the corresponding γ-oxa-glutamyl isostere (compounds 1 and 5, respectively) have been disclosed as GST inhibitors. The rationale of their design is discussed in juxtaposition to non-peptide NBD thioethers.
Materials and methods: Synthesis of derivatives 1 and 5 and in vitro evaluation on human GSTP1-1 and M2-2 are reported.
Results: Conjugates 1 and 5 were found to be low micromolar inhibitors of both isoforms. Furthermore, they display a threefold reduction in selectivity for GSTM2-2 over the P1-1 isozyme in comparison with the potent non-peptide inhibitor nitrobenzoxadiazolyl-thiohexanol (NBDHEX).
Discussion and conclusions: Spectroscopic data are congruent with the formation of a stable sigma-complex between GSH and the inhibitors in the protein active site. Conjugate 5 is suitable for in vivo modulation of GST activity in cancer treatment.
Introduction
The development of resistance to antineoplastic agents remains a primary concern in cancer therapy. Despite this circumstance, the comprehension of the multiple mechanisms that contribute to cell survival following cytotoxic treatment has remarkably evolved. Thus, alongside the view that makes resistance arising from events limiting intracellular drug accumulation and target interaction, such as minor uptake, enhanced extrusion of the compound by energy-dependent transporters and/or induction of chemical-detoxifying mechanisms, recent insights direct on cell insensitivity to factors that act downstream of the initial drug-induced insult through the apoptotic machinery.
As a result, a renewed interest has been focused on the multifunctional family of glutathione S-transferases (GSTs)Citation1–3, since these predominantly cytosolic enzymes were recognized to play crucial roles in this contextCitation4. The most widely investigated function of GSTs is the metabolic conjugation reaction of electrophilic compounds, including carcinogens and anticancer drugs, to reduced glutathione (tripeptide γ-l-glutamyl-l-cysteinyl-glycine, GSH)Citation5. It has been shown that different GST isoenzymes are overexpressed in many cancer cell lines and that cytosolic GSTs as well as GSH biosynthesis are up-regulated by electrophiles. According to this evidence, it is still widely accepted that activation of the cellular GST/GSH system can contribute directly to drug resistance in some tumor cell types via its detoxifying activityCitation6. More recently, however, a fundamental non-catalytic, ligand binding activity has been proposed for GST isoforms alpha, mu and pi which, to a different extent, are able to associate with the c-Jun N-terminal kinase (JNK) complexCitation7. In addition, tumor necrosis factor receptor-associated factor 2 (TRAF2), an upstream activator of JNK, can be sequestered by the pi class isoenzyme of GST, glutathione S-transferase P1-1 (GSTP1-1)Citation8, thus preventing the MAPK/JNK signaling cascade which is part of the apoptotic event. Although GSTP1-1 is overexpressed in a wide range of treated (in addition to untreated) human solid tumors, its relatively weak affinity for the majority of anticancer drugs indicates that the mentioned interfering role in signal transduction is predominant on the catalytic function. Because of their multi-faced involvement in modulation of cell survival and stress responseCitation9, GSTs keep on to be attractive drug targets for both chemists and biologists, and inhibition of these enzymes, particularly the P1-1 isoform, represents one of the most opportune strategy to sensitize tumor tissues to apoptotic and antiproliferative effects of anticancer drugs.
A variety of GST inhibitors have been reported to date, including glutathione S-conjugates (GS-R), GSH analogs and non-peptide compoundsCitation10–12. In our long-term research in this area, we developed the non-glutathione molecule 6-(7-nitrobenzo[c][1,2,5]-oxadiazol-4-yl)thiohexan-1-ol (NBDHEX), which inhibits human GSTs at micromolar or submicromolar concentrationsCitation13.
Mechanistically, NBDHEX behaves like a mechanism-based inhibitor of the transferase; after conjugation with GSH it forms a tetrahedral intermediate (sigma or Meisenheimer complex) which is strongly stabilized by the GST active site. Tested on several human cancer cell lines, NBDHEX was shown to possess a high antiproliferative activity, which depends on its ability to arrest cell cycle and trigger apoptosis through dissociation of the GSTP1-1 from the complexes with JNK and TRAF2Citation14–16. Unfortunately, NBDHEX seems to be less specific for GSTP1-1 (IC50 = 0.80 µM) than for GSTM2-2 (IC50 = 0.01 µM). Furthermore, its pharmacokinetic profile is hampered by a very low water solubility. Very recently we designed and synthesized various NBDHEX analogs, some of which were endowed with greater water solubility than the parent compoundCitation17.
In the search for potent and selective inhibitors of GSTs much attention has been devoted to glutathione S-conjugates, following the observation that GSTs of the alpha, mu and pi classes are particularly sensitive to product inhibitionCitation18,Citation19. It is well noted that GS-R conjugates use both recognition areas for binding to GSTs, i.e. the GSH-binding site (the G-site) and the hydrophobic binding region for the electrophilic substrate (the H-site). Since the H-site is made up of multiple distinct and partially overlapping hydrophobic subsitesCitation20, conjugation with GSH is considered a valid approach to reduce the mobility of hydrophobic R groups within this large cavity. Therefore, the linking of a glutathionyl moiety to the nitrobenzoxadiazole scaffold may improve the inhibitor selectivity toward GSTP1-1 by exploiting the differences in both G- and H-binding sites of distinct GST isoforms.
As a matter of fact, in cells glutathione S-conjugates are exposed to degradation by γ-glutamyl transpeptidase (γ-GT), which selectively cleaves the isopeptidic γ-Glu-Cys bond of GSH and its S-linked derivatives, and to recognition by efflux pumps belonging to the ATP-binding cassette (ABC) superfamily, such as the multidrug resistance-associated protein I (MRP1/ABCC1)Citation21,Citation22. MRP1 plays an important role in extruding cytotoxic drugs from the cell and its overexpression has long been considered a cause of failure of anticancer chemotherapyCitation23,Citation24. Despite this, it is worthy to note that one of the most selective GSTP1-1 inhibitor to date is TER 117, the S-benzyl conjugate of the GSH mimic containing the lipophilic C-terminal phenylglycyl residue in place of glycine. This peptide competitively inhibits the human transferases with an approximately one to two orders of magnitude increase in the affinity for GST P1-1 compared with other GST isoformsCitation10,Citation25. Furthermore, its diethyl ester derivative TER199, which is proceeding in phase II clinical trials as an antimyelodysplastic agent, was shown to act as an effective inhibitor of the MRPI transporter. The anticancer potential is therefore augmented as a result of the combined inhibition of GSTP1-1 activity and MRP1 transportCitation26.
For the purpose to obtain further insights on this topic, we were then interested in investigating 7-nitrobenzoxadiazol-4-yl (NBD) S-conjugates of the GS-R type. Of necessity we considered some structural modifications of the GSH peptide moiety to achieve stability toward γ-GT and possibly ameliorate bioavailability. Over the years we designed a variety of GSH mimics and derivatives characterized by amino acid substitution and/or bioisosteric replacements of the γ-glutamyl linkage, addressed to enhance both metabolic resistance and affinity to GST binding sites, through the attribution of specific chemical and conformational properties to the modified GSH backboneCitation27–34. Among these, the γ-oxa-glutamyl (Glo) analog of GSH, H-Glo[Cys-Gly-OH]-OH, deserves particular attention for more than one reason: (1) as expected, the replacement of the scissile γ-glutamyl-cysteinyl peptide bond with the OCONH unit assures resistance to γ-GT-mediated hydrolysisCitation29,Citation35; (2) the substitution does not sensibly alter the physico-chemical properties of the tripeptide; (3) groups crucial for binding to the G-site resemble those of the natural ligand, with the H-bond donor/acceptor potential of the GSH backbone fairly maintained and (4) more importantly, S-conjugates of this urethane mimic of glutathione have been shown to inhibit MRP1Citation36.
Thus we report here a straight preparation of the glutathione conjugate H-Glu[Cys(NBD)-Gly-OH]-OH (GS-NBD, 1) and the synthesis of its new analog H-Glo[Cys(NBD)-Gly-OH]-OH or ψ(OCONH) GS-NBD (5), to be comparatively evaluated as inhibitors of human GSTP1-1 and M2-2.
The structural relationship between compounds 1, 5 and NBDHEX is evident (): this latter may be considered as a molecular simplification, lacking the polar and H-bond donor/acceptor groups, of the Cys(NBD)-Gly moiety obtained from removal of the (pseudo)glutamyl recognition determinant that characterizes the full peptide conjugates.
Figure 1. Backbone-side chain structural analogies connecting NBDHEX and the dipeptide unit in conjugates 1 (X = CH2) and 5 (X = O).
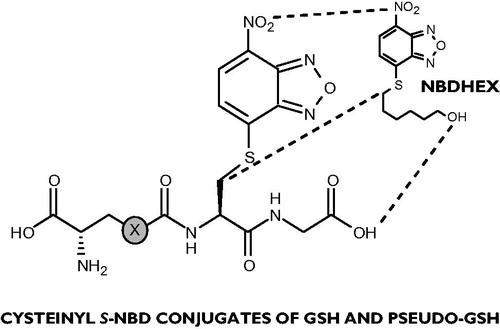
A further interesting feature of these ligands lies in their fluorescent properties that enable indicative spectral characterization and monitoring in biological media.
Methods
Chemistry
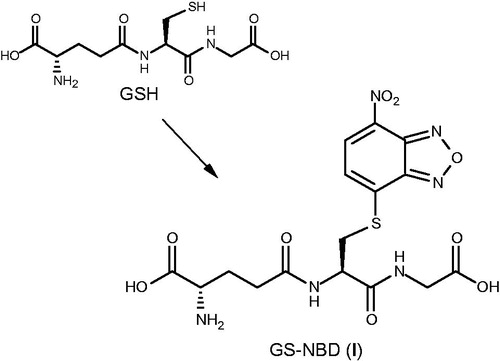
S-conjugates 1 and 5 were synthesized in good overall yields employing solution phase procedures, as outlined in Schemes 1 and 2. A minor problem associated with the preparation of the target peptides was the chemical (and photochemical) instability of 4-chloro-7-nitrobenzo[c][1,2,5]-oxadiazole (NBD-Cl), therefore the arylation reactions were preferably conducted with the exclusion of light. Compound 1 was easily prepared by treating an hydroalcoholic solution of glutathione with NBD-Cl in the presence of pyridine (2.5 equiv.) to keep the pH value at 5.5 (Scheme 1). In these conditions, reaction was complete in 1 h and resulted in a pure and abundant crop of the expected S-heteroaryl-conjugate 1 as a yellow precipitate. When sodium phosphate buffer (pH 7.0) was used as an alternative medium, the formation of 1 proceeded with lengthy times.
Scheme 1. Synthesis of GS-NBD (1). Reagents and conditions: NBD-Cl, Py, H2O/EtOH, r.t., 1 h, obscured.
Scheme 2. Synthesis of H-Glo[Cys(NBD)-Gly-OH]-OH (5). Reagents and conditions: (a) DBU, DCM, r.t., 15 min; (b) dioxane, 80 °C, 20 h; (c) I2, MeOH, r.t., 4 h; (d) TFA, r.t., 4 h; (e) P(nBu)3, aq. NH3, n-PrOH/H2O, r.t., 1 h; (f) NBD-Cl, phosphate buffer pH 7.0, r.t., 48 h, obscured.
![Scheme 2. Synthesis of H-Glo[Cys(NBD)-Gly-OH]-OH (5). Reagents and conditions: (a) DBU, DCM, r.t., 15 min; (b) dioxane, 80 °C, 20 h; (c) I2, MeOH, r.t., 4 h; (d) TFA, r.t., 4 h; (e) P(nBu)3, aq. NH3, n-PrOH/H2O, r.t., 1 h; (f) NBD-Cl, phosphate buffer pH 7.0, r.t., 48 h, obscured.](/cms/asset/ab633bd2-5c41-4fc8-bf01-cfb526160deb/ienz_a_1070845_sch0002_c.jpg)
The strategy to the γ-oxa-glutamyl analog 5 required of necessity to assemble the whole tripeptide first, performing the S-arylation in the final step. The pseudopeptide was prepared essentially after our reported protocol, with the urethanic junction made up by convenient functionalization of a protected serine residue (Scheme 2)Citation29. By following the aforementioned conditions for heteroaryl substitution, the S-conjugate was collected in only moderate yields, together with significative amounts of non-reacted starting thiol. Attempts to force the SNAr toward completion in phosphate buffer were successful by using a stoichiometric excess of pseudoglutathione and longer reaction times.
Final compounds were purified to apparent homogeneity by gel-permeation chromatography and fully characterized by 1H and 13C NMR spectroscopy. Spectral data for both compounds 1 and 5 are congruent with the expected structures of NBD conjugates at the sulfur atom and exclude the formation of isomeric N-derivatives due to the presence of a second nucleophilic group in the reactant. This result is in accord with previous note reporting that, differently from cysteine S-NBD derivative, which can easily undergo S → N aryl transfer, glutathione forms only the S-conjugateCitation37.
Biological assays
Peptide conjugates 1 and 5 were assayed in vitro for their ability to inhibit the GSH conjugation reaction with 1-chloro-2,4-dinitro-benzene (CDNB), mediated by human GST P1-1 and M2-2. Inhibition experiments were performed at 25 °C in 0.1 M potassium phosphate buffer pH 6.5 containing constant (1 mM) GSH and acceptor substrate concentrations and variable amounts of the inhibitors.
Experimental
Peptide synthesis
GSH and amino acid derivatives were purchased from Sigma-Aldrich (St. Louis, MO) and Bachem (Bubendorf, Switzerland). All other chemicals and solvents were of analytical grade and were supplied from Sigma-Aldrich and VWR (Radnor, PA). All the reactions were monitored by analytical TLC on Merck (Kenilworth, NJ) 60 F254 plates developed with the following solvents: (a) n-BuOH/AcOH/H2O (2:1:1); (b) CHCl3/MeOH (99:1); (c) CHCl3/MeOH (98:2). Column chromatography was carried out in absorption using Merck 60 silica gel (230–400 mesh). Melting points were determined on a Büchi B-450 apparatus (Uster, Switzerland) and are uncorrected. Elemental analyses (C, H, N and S) were performed on a Carlo Erba 1106 Analyzer (Milano, Italy) and were within ±0.4% of the theoretical values. IR spectra were recorded employing a Perkin-Elmer FTIR 1600 spectrophotometer (Waltham, MA). 1H (300 MHz) and 13C (75.43 MHz) NMR spectra were acquired on a Varian VXR-300 instrument (Palo Alto, CA). Chemical shifts are reported in ppm, referenced to residual solvent peaks and multiplicities are indicated as s (singlet), d (doublet), t (triplet), m (multiplet) and br (broad). Peak assignments were confirmed by 2D 1H–13C hetero-correlated experiments.
H-Glu[Cys(NBD)-Gly-OH]-OH (1)
To a stirred solution of GSH (0.31 g, 1.0 mmol) in a (2:1) mixture of H2O/EtOH (6 mL) solid NBD-Cl (0.20 g, 1.0 mmol) and Py (0.2 mL, 2.5 mmol) in EtOH (1 mL) were added at room temperature. The initial solution was kept under vigorous stirring with the exclusion of light for 1 h, during which time pH was maintained to 5.5 by addition of Py. The reaction mixture was diluted with EtOH and filtered in vacuo. The precipitate was recrystallized from acetone/EtOH (2:1) to give 0.33 g of compound 1 as a yellow powder (70% yield). Rf (a) = 0.4; m.p. 199–200 °C (dec.). IR (KBr) νmax: 3350, 3055, 1690, 1645, 1510, 1335 cm−1; 1H NMR ([D6]DMSO): δ 1.7–2.0 (2H, m, Glu β-CH2), 2.2–2.4 (2H, m, Glu γ-CH2), 3.15 (1H, m, Glu α-CH), 3.4 (1H, m, Cys β-CHB), 3.7 (2H, m, Gly CH2), 3.75 (1H, m, Cys β-CHA), 4.85 (1H, m, Cys α-CH), 7.6 (H, d, J = 7.5 Hz, hetArH), 8.6 (1H, d, J = 7.5 Hz, hetArH), 8.8 (1H, d, J = 8.2 Hz, Cys NH), 8.95 (1H, t, J = 5.0 Hz, Gly NH). 13C NMR ([D6]DMSO): δ 27.25 (Glu Cβ), 31.96 (Glu Cγ), 33.68 (Cys Cβ), 41.98 (Gly Cα), 51.75 (Cys Cα), 53.62 (Glu Cα), 123.26, 133.08, 139.79, 143.32, 149.84 (heteroaromatics), 170.42, 171.25, 171.55, 172.73 (CO). Anal. calcd for C16H18N6O9S: C, 40.85; H, 3.86; N, 17.87, S, 6.82. Found: C, 40.68; H, 3.70; N, 18.05, S, 6.98.
Fmoc-Cys(Trt)-Gly-OtBu (2)
Fmoc-Cys(Trt)-OH (1.2 g, 2.1 mmol) was suspended in THF (5 mL) and HOBt (0.28 g, 2.1 mmol) was added under stirring. The solution was cooled to 0 °C and an ice-cold solution containing HCl . H-Gly-OtBu (0.35 g, 2.1 mmol) and NMM (0.21 g, 2.1 mmol) in THF (5 mL) was added, followed by portionwise addition of a solution of DCC (0.43 g, 2.1 mmol) in THF (4 mL). After 6 h at 0 °C and 16 h at 5 °C, the reaction mixture was filtered and the resulting solution evaporated under reduced pressure. The residue was taken up in AcOEt and the organic layer washed with 1 N KHSO4, saturated aqueous NaHCO3 and H2O. The residue obtained after drying and solvent evaporation was chromatographed on silica gel using a CHCl3/MeOH (98:2) mixture as eluent, to give 2 (1.4 g, 94%) as a white foam. Rf (b) = 0.5; IR (neat) νmax: 3410, 1730, 1670 cm−1; 1H NMR (CDCl3): δ 1.4 (9H, s, CH3), 2.55 (1H, m, Cys β-CHB), 2.7 (1H, m, Cys β-CHA), 3.2 (1H, m, Cys α-CH), 3.75 (2H, m, Gly CH2), 4.2 (1H, m, Fmoc CH), 4.35 (2H, m, Fmoc CH2), 6.0 (1H, br d, Cys NH), 7.2–7.4 (15H, m, ArH), 7.5 (1H, br t, Gly NH). Anal. calcd for C43H42N2O5S: C, 73.90; H, 6.06; N, 4.01; S, 4.59. Found: C, 74.12; H, 6.15; N, 3.97; S, 4.46.
H-Cys(Trt)-Gly-OtBu (3)
The protected dipeptide 2 (0.56 g, 0.80 mmol) was dissolved in DCM (3 mL) and DBU (0.12 g, 0.80 mmol) in DCM (1 mL)Citation34 was added in portions at room temperature. After 15 min the solvent was evaporated to dryness and the residue eluted from a silica gel column using a CHCl3/MeOH (97:3) mixture as eluent, to give compound 3 as an oil (0.34 g, 89%). Rf (c) = 0.8; IR (neat) νmax: 3360, 1730, 1650 cm−1; 1H NMR (CDCl3): δ 1.45 (9H, s, CH3), 2.6 (1H, m, Cys β-CHB), 2.8 (1H, m, Cys β-CHA), 3.0 (1H, m, Cys α-CH), 3.8 (2H, m, Gly CH2), 7.2-7.45 (15H, m, ArH), 7.5 (1H, br t, Gly NH). Anal. calcd for C28H32N2O3S: C, 70.56; H, 6.77; N, 5.88, S, 6.73. Found: C, 70.31; H, 6.91; N, 5.65, S, 7.04.
Boc-Glo[Cys(Trt)-Gly-OtBu]-OtBu (4)
To a stirred solution of dipeptide ester 3 (3.3 g, 7.0 mmol) in dioxane (10 mL) Boc-Glo(ONp)-OtBuCitation29 (3.0 g, 7.0 mmol) in dioxane (5 mL) was added. The mixture was warmed to 80 °C and left to stir for 20 h. The solution was evaporated under vacuum to give an oily residue which was taken up in CHCl3. The organic layer was washed with 0.5 N HCl, saturated aqueous Na2CO3 and H2O, dried and filtered. The filtrate was evaporated under reduced pressure, and the residue chromatographed on a silica gel column using a CHCl3/MeOH (98:2) mixture as eluent, to give the protected tripeptide 4 as a white foam (3.8 g, 70%). Rf (c) = 0.5; IR (KBr) νmax: 3400 br, 3100, 1735, 1705, 1660, 1540 cm−1; 1H NMR (CDCl3): δ 1.4 (27H, s, CH3), 2.65 (1H, m, Cys β-CHB), 2.85 (1H, m, Cys β-CHA), 3.05 (1H, m, Gly CHB) 3.2 (1H, m, Gly CHA), 4.1 (2H, m, Cys and Glo α-CH), 4.3 (2H, m, Glo β-CH2), 5.65 (1H, br t, Glo NH), 6.6 (1H, br d, Cys NH), 7.2–7.35 (15H, m, ArH), 7.5 (1H, br t, Gly NH). Anal. calcd for C41H53N3O9S: C, 64.46; H, 6.99; N, 5.50, S, 4.20. Found: C, 64.24; H, 7.05; N, 5.72, S, 4.31.
H-Glo[Cys(NBD)-Gly-OH]-OH (5)
Compounds {Boc-Glo[Cys-Gly-OtBu]-OtBu}2, {H-Glo[Cys-Gly-OH]-OH}2 and H-Glo[Cys-Gly-OH]-OH were prepared by following the already published protocolCitation29, and characterized by NMR spectroscopy.
H-Glo[Cys-Gly-OH]-OH (0.42 g, 1.3 mmol) was dissolved in 0.1 M sodium phosphate buffer (pH 7.0) (4 mL) under stirring at room temperature before the addition of solid NBD-Cl (0.13 g, 0.65 mmol). After 48 h with the exclusion of light, during which time the reaction course was followed by TLC, the resulting suspension was evaporated under reduced pressure and the crude material taken up in H2O. Recrystallization from H2O/MeOH afforded conjugate 5 as a powdery yellow solid (0.37 g, 60%). Rf (a) = 0.45; IR (KBr) νmax: 3320 br, 3050, 1690–1615, 1510, 1335 cm−1; 1H NMR (D2O): δ 2.8 (1H, m, Cys β-CHB), 3.1 (1H, m, Cys β-CHA), 3.6 (1H, m, Gly CH2), 3.8 (2H, m, Glo and Cys α-CH), 4.2 (2H, m, Glo β-CH2), 7.4 (1H, d, J = 7.0 Hz, hetArH), 8.4 (1H, d, J = 7.0 Hz, hetArH). 13C NMR (D2O): δ 33.84 (Cys Cβ), 41.59 (Gly Cα), 51.43 (Cys Cα), 54.02 (Glo Cα), 67.92 (Glo Cβ), 123.59, 133.03, 139.53, 143.37, 149.91 (heteroaromatics), 156.84, 174.86, 176.74 (CO). Anal. calcd for C15H16N6O10S: C, 38.14; H, 3.41; N, 17.79, S, 6.79. Found: C, 38.36; H, 3.48; N, 17.63, S, 6.80.
GST inhibition
Human GSTM2-2 and GSTP1-1 were expressed in Escherichia coli and purified as previously describedCitation38. The GST activity was measured in 0.1 M potassium phosphate buffer (pH 6.5) containing 1 mM GSH, 1 mM CDNB as co-substrate and 0.1 M EDTACitation39. Inhibition experiments were performed by adding variable amounts of either GS-NBD 1 or its analog 5, ranging from 0.1 to 50 µM, to the assay mixture. IC50 is defined as the inhibitor concentration which fulfils 50% of catalytic activity inhibition. IC50 values were obtained by the best fit of the experimental data to a hyperbolic binding equation. The selectivity index (SI) toward GSTP1-1 is calculated from the IC50 GSTP1-1/IC50 GSTM2-2 ratio, therefore a decrease of SI compared with that of NBDHEX (SI = 80) indicates an increase of compound selectivity toward GSTP1-1. Activity was recorded at 340 nm, where the NBD conjugate absorbs (ɛ = 9.6 mM−1 cm−1); the UV–vis spectrum of GS-NBD (1) (50 µM), in 0.1 M potassium phosphate buffer pH 7.0, was recorded, before and after the addition of either 1 mM GSH or stoichiometric amounts of GSTP1-1 and 1 mM GSH, using a Kontron double-beam Uvikon 940 spectrophotometer (Redwood City, CA) thermostatically operating at 25 °C.
Results
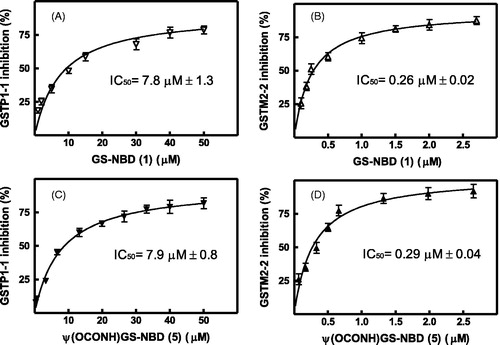
Conjugate 1 inhibits GSTP1-1 and GSTM2-2 with IC50 values of 7.8 and 0.26 µM, respectively (, panels A and B). The inhibitory profile of pseudoglutathione derivative 5 on the target enzymes is very similar, with IC50 values of 7.9 and 0.3 µM for the GSTP1-1 and the GSTM2-2 isoforms (panels C and D, respectively, in ).
Figure 2. Effects of NBD S-conjugates 1 and 5 on the catalytic activity of GSTP1-1 and GSTM2-2. The GST activity was reported as percent of GST activity inhibition. The solid line is the best fit of the experimental data to a hyperbolic binding equation which fulfils the IC50 values of 7.8 ± 1.3 and 0.26 ± 0.02 µM for the reaction catalyzed by GSTP1-1 (panel A) and GSTM2-2 (panel B), respectively, in the presence of GS-NBD 1 as inhibitor. When the enzymatic activity was measured in the presence of NBD conjugate 5, the IC50 values found were 7.9 ± 0.8 and 0.29 ± 0.04 µM for GSTP1-1 (panel C) and GSTM2-2 (panel D), respectively. Data represent means ± SD of three independent experiments.
collects data for GST inhibition by compounds 1 and 5 together with representative conjugates reported so far. Inhibitors are listed in descending order of activity toward GSTP1-1, starting from the most potent (and selective) prototypical inhibitor TER 117.
Table 1. Inhibition of human GSTP1-1 and GSTM2-2 isoforms by compounds bearing a peptide, non-peptide or pseudopeptide skeleton.
The analysis of data allows the following considerations: apart from TER 117, the compound in the series that shows the strongest selectivity for the target enzyme with respect to the M2-2 isoform is GS(Hex). However, the latter is a less efficient GSTP1-1 inhibitor compared to a range of compounds, including NBDHEX and the present conjugates 1 and 5. Despite inhibitors 1 and 5 show a tenfold decrease in potency with respect to NBDHEX against the target isoform GSTP1-1, yet the affinity of both peptides for the GSTM2-2 isozyme is even lower; thus the loss in inhibitory activity toward GSTP1-1 is counterbalanced by an almost threefold gain in selectivity toward the same isoform, as evidenced by their SI index (as defined above) (values of 30 and 27 for compounds 1 and 5, respectively, compared with the NBDHEX value of 80). Interestingly, activity data obtained for the two inhibitors are superimposable. The significance of these results is discussed in the next session.
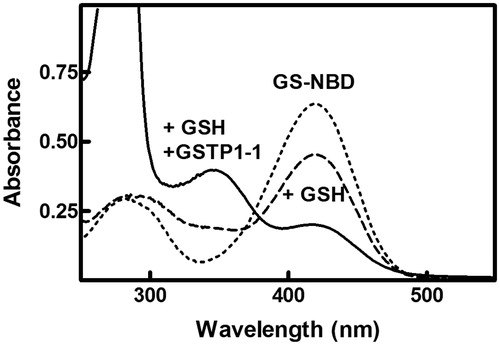
In analogy with the mechanistic behavior of many nitro-substituted aromatic compoundsCitation41, the inhibition mechanism for both NBD derivatives 1 and 5 implies that they bind to the GST active site and form a sigma-complex intermediate with the resident GSH. In fact, the UV–vis spectrum of GS-NBD is minimally affected by the presence of 1 mM GSH (, dashed line). However, the spectral perturbation is significantly higher when GS-NBD is incubated with both GSH and GSTP1-1 (, solid line). In this case, the band of GS-NBD, centered at 419 nm, almost disappears and a new absorption band appears at about 348 nm.
Figure 3. Spectrophotometric analysis of the reaction of GS-NBD (1) with GSTP1-1. The UV–vis spectrum of GS-NBD 1 (50 µM), in 0.1 M potassium-phosphate buffer pH 7.0, was recorded at 25 °C before (dotted line) and after the addition of 1 mM GSH (dashed line) or stoichiometric amounts of GSTP1-1 and 1 mM GSH (solid line).
It is worth mentioning that the two recently reported GSTP1-1 inhibitors Z-Cys(NBD)-OMe and Boc-Cys(NBD)-OMe (), also featuring an albeit minimal peptide nature, share with 1 and 5 the ability to form a sigma-complex inside the active cleft of the transferase; however, to our knowledge, the present NBD S-conjugates 1 and 5 represent the first examples of mechanism-based inhibitors for the human P1-1 isoform characterized by a full (pseudo)glutathione skeleton. Thus, 1 and 5 deserve attention among peptide inhibitors, since their binding mechanism to target enzyme is alternative with regard to TER 117.
Discussion
We have previously reported a series of NBD thioether derivatives as nano/micromolar inhibitors of GSTs, a family of detoxifying enzymes also involved in cancer drug resistance phenomenon. It has been evidenced that the prototype thioether NBDHEX promotes GSTP1-1 dissociation from the GSTP1-1/JNK heterocomplex, resulting in cell death by apoptosisCitation14. A detailed investigation of the interaction between NBDHEX and different human GST isoenzymes revealed that this molecule behaves like a mechanism-based inhibitor: in fact, it is conjugated with GSH in the GST active site leading to a stable sigma-complex that represents the real inhibitor speciesCitation13.
In this work we tested the inhibitory properties on GSTP1-1 and GSTM2-2 of NBD thioethers 1 and 5, in which the aliphatic chain of 6-mercapto-hexanol has been replaced by the peptidyl moiety of GSH and its γ-oxa-analog, respectively. Compounds 1 and 5 are good inactivators of both isoforms, with IC50s in the low micromolar range. As previously observed with NBDHEXCitation13, the GST isoenzyme M2-2 shows much higher sensitivity toward the inhibitory effect of both derivatives compared to the P1-1 isoform. It is interesting to note, however, that, despite inhibitors 1 and 5 inhibit both GSTs to a lesser extent in comparison with NBDHEX, they display a threefold reduction in selectivity for the M2-2 isozyme over GSTP1-1 compared with the aforementioned inhibitor.
Moreover, the present data reveal that replacement of a peptide unit with a urethane bond does not affect at all the inhibition properties of the NBD conjugate; in fact, identical IC50 values are found for GS-NBD 1 and thioether analog 5.
The inhibition mechanism of conjugates 1 and 5 suggests that the main binding determinant for the interaction with the GST active site is the benzoxadiazole ring of NBD, while the peptidyl portion of the inhibitor may be easily displaced from the G-site by free GSH. This is followed by the deprotonation/activation of GSH, enabling the nucleophilic attack of GSH on the C4 of NBD to form a sigma-complex. This hypothesis is confirmed by the UV–vis spectra of GS-NBD, showing that the sigma complex intermediate is mainly formed into the GST active site. This evidence, obtained for the first time with NBDHEXCitation13, has been recently confirmed with all compounds bearing substituted linear or branched alkyl chains at the C4-sulfur atomCitation17.
Moreover, the evidence that the NBD- derivative 5 may fully replace GS-NBD 1, may have important effects in cytotoxicity experiments performed with tumour cell lines, increasing the intracellular stability of this powerful GST inhibitor.
Conclusion
The present study reports synthesis and biological evaluation of glutathione and pseudoglutathione nitrobenzoxadiazole S-conjugates as peptide inhibitors of human P1-1 and M2-2 glutathione transferases.
Activity data show that the tested compounds are good inactivators of both isoforms, with IC50s in the micromolar range. Furthermore, the selectivity profile of compounds 1 and 5, albeit invariantly shifted in favor of the mu isoform, indicates a significant decrease in affinity for this isozyme in comparison with the prototype inhibitor NBDHEX.
The backbone-modified analog 5, which represents the γ-GT resistant mimic of 1, combines resistance to γ-GT-mediated hydrolysis with the potential to behave as a dual targeting agent for GSTP1-1 and MRP1 export pump. Therefore, analog 5 may be suitable for the treatment of drug resistant tumors characterized by an elevated expression of both GSTP1-1 and MRP1.
Declaration of interest
The authors report no declaration of interest. This study was supported by grants from Ministry of Education, University, and Research of Italy (University “Gabriele d’Annunzio”, ex 60% 2014, Luisi G.) and from Associazione Italiana per la Ricerca sul Cancro (AIRC), project IG-10598 (Caccuri A.M.)
References
- Wu B, Dong D. Human cytosolic glutathione transferases: structure, function, and drug discovery. Trends Pharmacol Sci 2012;33:656–68
- Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol 2005;45:51–88
- Salinas AE, Wong MG. Glutathione S-transferases – a review. Curr Med Chem 1999;6:279–309
- Tew KD, Townsend DM. Glutathione-S-transferases as determinants of cell survival and death. Antioxid Redox Signal 2012;17:1728–37
- Armstrong RN. Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem Res Toxicol 1997;10:2–18
- Tew KD. Glutathione-associated enzymes in anticancer drug resistance. Cancer Res 1994;54:4313–20
- Adler V, Yin Z, Fuchs SY, et al. Regulation of JNK signaling by GSTp. EMBO J 1999;18:1321–34
- Wu Y, Fan Y, Xue B, et al. Human glutathione S-transferase P1-1 interacts with TRAF2 and regulates TRAF2-ASK1 signals. Oncogene 2006;25:5787–800
- Singh S. Cytoprotective and regulatory functions of glutathione S-transferases in cancer cell proliferation and cell death. Cancer Chemother Pharmacol 2015;75:1–15
- Lyttle MH, Hocker MD, Hui HC, et al. Isozyme-specific glutathione-S-transferase inhibitors: design and synthesis. J Med Chem 1994;37:189–94
- Klotz P, Slaoui-Hasnaoui A, Banères JL, et al. Synthesis and glutathione S-transferase structure–affinity relationships of nonpeptide and peptidase-stable glutathione analogues. J Med Chem 1998;41:2278–88
- Musdal Y, Hegazy UM, Aksoy Y, Mannervik B. FDA-approved drugs and other compounds tested as inhibitors of human glutathione transferase P1-1. Chem Biol Interact 2013;205:53–62
- Ricci G, De Maria F, Antonini G, et al. 7-Nitro-2,1,3-benzoxadiazole derivatives, a new class of suicide inhibitors for glutathione S-transferases. Mechanism of action of potential anticancer drugs. J Biol Chem 2005;280:26397–405
- Turella P, Cerella C, Filomeni G, et al. Proapoptotic activity of new glutathione S-transferase inhibitors. Cancer Res 2005;65:3751–61
- De Luca A, Federici L, De Canio M, et al. New insights into the mechanism of JNK1 inhibition by glutathione transferase P1-1. Biochemistry 2012;51:7304–12
- De Luca A, Mei G, Rosato N, et al. The fine-tuning of TRAF2-GSTP1-1 interaction: effect of ligand binding and in situ detection of the complex. Cell Death Dis 2014;5:e1015
- Rotili D, De Luca A, Tarantino D, et al. Synthesis and structure–activity relationship of new cytotoxic agents targeting human glutathione-S-transferases. Eur J Med Chem 2015;89:156–71
- Burg D, Mulder GJ. Glutathione conjugates and their synthetic derivatives as inhibitors of glutathione-dependent enzymes involved in cancer and drug resistance. Drug Metab Rev 2002;34:821–63
- Meyer DJ. Significance of an unusually low Km for glutathione in glutathione transferases of the alpha, mu and pi classes. Xenobiotica 1993;23:823–34
- Oakley AJ, Lo Bello M, Mazzetti AP, et al. The glutathione conjugate of ethacrynic acid can bind to human pi class glutathione transferase P1-1 in two different modes. FEBS Lett 1997;419:32–6
- Borst P, Evers R, Kool M, Wijnholds J. A family of drug transporters: the multidrug resistance-associated proteins. J Natl Cancer Inst 2000;92:1295–302
- Keppler D. Export pumps for glutathione S-conjugates. Free Radic Biol Med 1999;27:985–91
- Bellamy WT, Dalton WS. Multidrug resistance in the laboratory and clinic. Adv Clin Chem 1994;31:1–61
- Mahjoubi F, Golalipour M, Ghavamzadeh A, Alimoghaddam K. Expression of MRP1 gene in acute leukemia. Sao Paulo Med J 2008;126:172–9
- Johansson A-S, Ridderström M, Mannervik B. The human glutathione transferase P1-1 specific inhibitor TER 117 designed for overcoming cytostatic-drug resistance is also a strong inhibitor of glyoxalase I. Mol Pharm 2000;57:619–24
- O'Brien ML, Vulevic B, Freer S, et al. Glutathione peptidomimetic drug modulator of multidrug resistance-associated protein. J Pharmacol Exp Ther 1999;291:1348–55
- Luisi G, Calcagni A, Pinnen F. ψ(SO2NH) transition state isosteres of peptides. Synthesis of the glutathione disulfide analogue (Glu-ψ(SO2NH)-Cys-Gly)2. Tetrahedron Lett 1993;34:2391–2
- Calcagni A, Duprè S, Lucente G, et al. Synthesis and activity of the glutathione analogue γ-(l-γ-azaglutamyl)-l-cisteinyl-glycine. Int J Peptide Protein Res 1995;46:434–9
- Calcagni A, Duprè S, Lucente G, et al. Synthesis and activity of the glutathione analogue γ-(l-γ-oxaglutamyl)-l-cysteinyl-glycine. Arch Pharm Pharm Med Chem 1996;329:498–502
- Cacciatore I, Di Stefano A, Luisi G, et al. Transition state isosteres of the γ-glutamyl peptide bond hydrolysis: synthesis and characterization of the ψ(CH2NH) pseudopeptide analogue of glutathione. J Peptide Sci 2004;10:109–14
- Cacciatore I, Caccuri AM, Di Stefano A, et al. Synthesis and activity of novel glutathione analogues containing an urethane backbone linkage. Il Farmaco 2003;58:787–93
- Cacciatore I, Caccuri AM, Cocco A, et al. Potent isozyme-selective inhibition of human glutathione S-transferase A1-1 by a novel glutathione S-conjugate. Amino Acids 2005;29:255–61
- Paradisi MP, Mollica A, Cacciatore I, et al. Proline-glutamate chimeras in isopeptides. Synthesis and biological evaluation of conformationally restricted glutathione analogues. Bioorg Med Chem 2003;11:1677–83
- Calcagni A, Lucente G, Luisi G, et al. Novel glutathione analogues containing the dithiol and disulfide form of the Cys–Cys dyad. Amino Acids 1999;17:257–65
- Burg D, Filippov DV, Hermanns R, et al. Peptidomimetic glutathione analogues as novel γGT stable GST inhibitors. Bioorg Med Chem 2002;10:195–205
- Burg D, Wielinga P, Zelcer N, et al. Inhibition of the multidrug resistance protein 1 (MRP1) by peptidomimetic glutathione-conjugate analogs. Mol Pharmacol 2002;62:1160–6
- Birkett DJ, Price NC, Radda GK, Salmon AG. The reactivity of SH groups with a fluorogenic reagent. FEBS Lett 1970;6:346–8
- Lo Bello M, Battistoni A, Mazzetti AP, et al. Site-directed mutagenesis of human glutathione transferase P1-1. Spectral, kinetic, and structural properties of Cys-47 and Lys-54 mutants. J Biol Chem 1995;270:1249–53
- Habig WH, Jakoby WB. Assay for differentiation of glutathione S-transferase. Methods Enzymol 1981;77:237–53
- Ploemen JHTM, van Ommen B, van Bladeren PJ. Inhibition of rat and human glutathione S-transferase isoenzymes by ethacrynic acid and its glutathione conjugate. Biochem Pharmacol 1990;40:1631–5
- Baines BS, Allen G, Brocklehurst K. The highly electrophilic character of 4-chloro-7-nitrobenzofurazan and possible consequences for its applications as a protein-labelling reagent. Biochem J 1977;163:189–92