
Abstract
This work describes the antitrypanocidal activity of two hydroxamic acid derivatives containing o-ethoxy (HAD1) and p-ethoxy (HAD2) as substituent in the aromatic ring linked to the isoxazoline ring. HAD1 and HAD2 induced a significant reduction in the number of intracellular parasites and consequently showed activity on the multiplication of the parasite. Treatment of cardiomyocytes and macrophages with the compounds revealed no significant loss in cell viability. Ultrastructural alterations after treatment of cardiomyocytes or macrophages infected by Trypanosoma cruzi with the IC50 value of HAD1 revealed alterations to amastigotes, showing initial damage seen as swelling of the kinetoplast. This gave a good indication of the ability of the drug to permeate through the host cell membrane as well as its selectivity to the parasite target. Both compounds HAD1 and 2 were able to reduce the cysteine peptidases and decrease the activity of metallopeptidases
Introduction
Chagas disease, a zoonosis caused by the protozoan Trypanosoma cruzi, is currently a global public health challenge. The disease has two distinct clinical phases: acute, mostly asymptomatic with patent parasitemia, and, chronic, which can be asymptomatic (indeterminate stage) or symptomatic (determinate stage), showing severe cardiomyopathy, megacolon, or megaoesophagousCitation1. This disease affects 7–8 million people mostly in Latin AmericanCitation2 and it is currently the leading cause of heart failure in South American countriesCitation3. However, there is a new epidemiological scenario showing a growing number of infected individuals in non-endemic countriesCitation4 that is drawing attention to this silent and neglected disease. There are a large number of immigrants with Chagas disease in the United States and European countriesCitation5,Citation6 and also there is the emerging infection by the congenital and oral transmission in endemic areasCitation7 all of which alert the need to implement strategies to control the transmission and burden of this infectious diseaseCitation8.
The current chemotherapies for clinical practice are benznidazole (BZ) and nifurtimox. The medications are predominantly active during the acute phase of the disease. Resistance to benznidazole (BZ) has been observed with some parasite strainsCitation9. Additionally, adverse side effects of these drugs, such as digestive and dermatological intolerance, have been implicated in the treatment complianceCitation10. Also it is important to highlight that the T. cruzi polyparasitism and co-infection with other pathogens may interfere with the failure of the present therapeutic interventionsCitation11. The failure of promising drugs undergoing clinical trialsCitation12 and also the unsuccessful stem cell therapyCitation13 reinforce the need for new strategies in the treatment and management of Chagas' diseaseCitation14. These strategies should include an analysis of the advantage of combination therapyCitation15 and eventual benefits from vaccine developmentCitation16, as well as the identification of new drug targets and biomarkers to monitor the treatment response and cure prediction. Thus, the lack of effective drugs for the treatment of this worldwide, emerging disease encourages initiatives on the rational drug development of new trypanocidal drugs.
Given the key role of peptidases in biological systems, these enzymes have been considered a potential therapeutic target against infectious agentsCitation17,Citation18. Different classes of peptidases participate in important biological events in the T. cruzi life cycle, including processes of invasion, differentiation, proliferation as well as mechanisms of escape from host immune responsesCitation19. Thus, peptidase inhibitors are a potential to design trypanocidal drugs. For example, K777, a peptidase inhibitor of the major cysteine proteinase of T. cruzi known as cruzain or cruzipain, became a potent candidate to advance into clinical trials in view of its effective activity against different strains of T. cruziCitation20, including its efficiency in protecting against cardiac damage in a canine experimental modelCitation21 and in response to multi-drug therapyCitation22. Aspartic peptidase from T. cruzi has also been proposed as a drug target since treatment of epimastigotes with pepstatin A, an aspartic peptidase inhibitor, inhibited parasite proliferation, induced morphological changes with detachment of flagellum, and increased expression of gp63Citation23. Moreover, matrix metallopeptidase-9 homologues detected in T. cruzi may be a potential therapeutic target, since they could be able to induce a reduction in the expression of matrix metallopeptidase-9 (MMP-9) during interaction with host cellsCitation24,Citation25.
A series of novel 4,5-dihydroisoxazoles incorporating hydroxamate moieties were synthesized and evaluated against Chagas disease. These hydroxamic acid derivatives (HAD) proved to be very effective against T. cruzi. The assays carried out revealed molecules with low cytotoxicity and efficient action against T. cruzi carbonic anhydrase (TcCA) and metallopeptidaseCitation26. Carbonic anhydrases (CA, EC 4.2.1.1) are metalloenzymes which catalyze the conversion of CO2 to the bicarbonate ion and protons. CAs are involved in numerous physiological and pathological processes including microbial diseases caused by fungi and bacteriaCitation27. The identification of a gene encoding α-carbonic anhydrase in T. cruzi (TcCa) raised interest in the application of this enzyme as a potent therapeutic target in Chagas diseaseCitation26,Citation28.
Thus, this study aimed to investigate the detailed trypanocidal action of the two derivatives of this new series, since they showed promising results against TcCACitation26, and their trypanocidal activity has not been investigated until now. Our results showed that these hydroxamic acid derivatives (HAD1 and 2) demonstrated a good activity against intracellular amastigotes, the clinical form found in the chronic phase. An inhibitory effect against parasite peptidases was also observed. Altogether, our data suggest that the hydroxamates scaffold can be considered as interesting candidates to move forward towards the development of more effective, selective and efficient Chagas disease drugs.
Materials and methods
Chemicals
Medium constituents were obtained from Oxoid Ltd. (Hampshire, England). Reagents used in electrophoresis were purchased from Amersham Life Science (Little Chalfont, England). Gelatin and casein were purchased from Merck (Darmstadt, Germany). The metallopeptidase (1,10-PHE – 1,10-phenanthroline), the cysteine peptidase (trans-epoxysuccinyl-l-leucylamido-(4-guanidino)butane [E-64]) inhibitors and all reagents used in the synthesis of the 4,5-dihydroisoxazole derivatives were obtained from Sigma-Aldrich (St. Louis, MO). All other reagents were of analytical grade.
Synthesis of 3-(2-ethoxyphenyl)-N-hydroxy-4,5-dihydroisoxazole-5-carboxamide (HAD1) and 3-(4-ethoxyphenyl)-N-hydroxy-4,5-dihydroisoxazole-5-carboxamide (HAD2)
The hydroxamic acid derivatives were synthesized as described by Rodrigues et al.Citation26 Briefly, the 2-ethoxy and 4-ethoxy-phenyl-aldoxime were synthesized from 20 mmol of the respective aldehydes and 60 mmol of NH2OH.HCl contained in 20 mL of solvent. The reaction medium was maintained for 2 h at a temperature of 27 °C under constant agitation. After which, the respective aldoximes were reacted with trichloroisocyanuric acid (10 mmol) and triethylamine (20 mmol) for 30 min under stirring at room temperature to produce the corresponding hydroximoyl chlorides. Then, dipolarophile (ethyl acrylate) was added to the solutions and, after 24 h, the 4,5-dihydroisoxazol derivatives were generated by 1,3-cycloaddiction reactions. Finally, a solution containing NH2OH (20 mmol) was added to the 4,5-dihydroisoxazol derivatives formed and kept at reflux for 24 h. The respective hydroxamic acid derivatives generated were recrystallized from hot water and filtered. The yields were 40% and 50%, respectively.
Parasites
Trypomastigote forms of T. cruzi, Y strain, were isolated from the blood of infected mice at the peak of parasitemia (7 d after infection). To increase the yield of T. cruzi trypomastigotes per mouse and reduce the number of animals used in the procedure, mice were immunosuppressed with intraperitoneal (ip) administration of cyclophosphamide 500 mg/kg on the 4th day after infection. To isolate the parasites, blood was collected by cardiac puncture with 3.8% sodium citrate as an anticoagulant and centrifuged at 1200 rpm for 26 min to separate them from the blood cells. The supernatant containing the parasites was then harvested and centrifuged at 4500 rpm for 14 min to pellet the trypomastigotes. The parasites were resuspended in culture medium DMEM-F12 and the concentration of parasites was assessed using a hemocytometer. The amastigote forms were obtained and analyzed through infection of mammalian cultures with trypomastigotes.
Primary culture
Murine peritoneal macrophages were obtained from Swiss Webster mice (18–20 g). Cold Dulbecco’s modified Eagle medium (DMEM; Sigma, St. Louis, MO) was injected into the peritoneal cavity and then removed by fluid aspiration with a syringe. Peritoneal exudate cells were centrifuged at 400 × g for 10 min and the pellet resuspended in culture medium supplemented with 10% fetal bovine serum (FBS) followed by determination of cell number/mL using a Neubauer chamber. The cells were plated at a density of 1 × 105 and 4 × 105 cells per well in black, clear bottom 96-well (Greiner Bio-One, #655090, Frickenhausen, Germany) and 24-well tissue culture plates, respectively, and maintained at 37 °C with 5% CO2 atmosphere.
Primary culture of cardiac muscle cells was obtained as previously describedCitation29. Briefly, the heart was removed from 18-d old mouse embryos and fragmented ventricles were dissociated with trypsin and collagenase. Isolated cells were resuspended in DMEM-F12 (Sigma, St. Louis, MO) supplemented with 7% FBS, 2% chicken embryo extract, 2.5 mM of CaCl2, 1 mM l-glutamine and antibiotics. The cells were plated at a density of 5 × 104 and 1.2 × 105 cells per well in 96-well and 24-well tissue culture plates, respectively, and maintained at 37 °C at 5% CO2 atmosphere. All procedures with animals were performed according to the Oswaldo Cruz Foundation committee of ethics for the use of experimental animals (LW-37/13).
Cell cytotoxicity assay
Primary cardiomyocyte and macrophage cultures were used to evaluate the cytotoxic effect of the compounds. After 24 h of culture, the cells were treated with the synthetic derivatives of 4,5-dihydroisoxazoles incorporating hydroxamate moieties (HAD1 and HAD2) synthesized as describedCitation26. A wide range of concentrations, ranging from 0.22 to 500 µg/mL, were tested to evaluate toxicity to mammalian cells. The cell viability analysis (CC50 value = the cytotoxic concentration corresponding to 50% reduction of cell viability) was performed after 3 d of treatment with HAD1 and HAD2 compounds using resazurin 0.005% (4 h at 37 °C) as the metabolic function indicator, followed by fluorescence intensity measurements at 530 nm and 630 nm in a Molecular Devices Flexstation 3 Multi-Mode Microplate Reader (Molecular Devices, Sunnyvale, CA). Negative controls were performed with the vehicle medium with or without DMSO (≤1%) and untreated cell cultures under similar conditions.
Trypanocidal assay
The effect of the HAD1 and HAD2 hydroxamic derivatives on T. cruzi was evaluated using bloodstream trypomastigotes of the Y strain. Parasites were isolated from infected Swiss Webster mice at the peak of parasitemia (7 d post-infection). Trypomastigotes (3 × 106 parasites/well) were incubated for 24 h at 37 °C in the presence or absence of serial dilutions of HAD1 or HAD2 compounds (0.04–90 µM). After treatment, the resazurin 0.005% was added to the parasites for 4 h at 37 °C and the fluorescence emission measurements were registered at 530 nm and 630 nm in a Molecular Devices Flexstation 3 Multi-Mode Microplate Reader (Molecular Devices, Sunnyvale, CA). The inhibitory concentration (IC50), the drug concentration that reduces 50% of trypomastigotes viability, was determined by fitting the dose–response curve to inhibitory activity compared with the untreated control. A similar curve for treatment with benznidazole (BZ) was also carried out in a parallel assay. Untreated trypomastigotes and cell-free media were used as negative controls.
The efficacy of the compounds was also evaluated, in terms of eliminating the intracellular parasites, by incubating 24 h-infected mammalian cells primary cultures (macrophages and cardiomyocytes) for 72 h at 37 °C with a serial dilution (3.3–90 µM) of HAD1 and HAD2 compounds, followed by fixation with Bouin solution fixative. The percentage of infection was determined by quantification of a minimum of 200 cells in randomly selected microscopic fields using a Zeiss Axioplan microscope (Zeiss, Rochester, NY). The experimental assays were performed at least three times in duplicate.
Transmission electron microcopy (TEM)
Primary cultures of cardiomyocyte and macrophage were infected with Y strain of bloodstream trypomastigotes in a 10:1 host cell–parasite ratio. After 24 h of infection, free trypomastigotes were removed from the culture supernatant and, after washing with Ringer’s solution (154 mM NaCl, 56 mM KCl, 17 mM Na2HPO4, pH 7.0), the cells were treated for 24 h or 48 h with the IC50 concentration (30 µM) of the HAD1 compound. Subsequently, the cultures were fixed for 1 h at 4 °C with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.2, and followed by post-fixation for 1 h at 4 °C with 1% OsO4 in the same buffer. The samples were dehydrated in serial-graded acetone batches impregnated and embedded in Epon 812. Ultra-thin sections, contrasted with uranyl acetate and lead citrate, were observed in a JEOL transmission electron microscope (JEM/1011).
Analysis of peptidase activity
The inhibitory effect on the cellular proteolytic activity was evaluated on 10% SDS–PAGE with 0.1% gelatin incorporated as substrateCitation30. Trypanosoma cruzi epimastigote cultures were harvested by centrifugation at 1500 × g for 15 min at 4 °C, and 1 × 107 cells were then lysed at 0 °C by the addition of 100 µL of sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) sample buffer. The gels were loaded with 25 µL of parasite extract per slot (equivalent to 1 × 107 cells) and after electrophoresis at a constant voltage of 170 V for 2 h at 4 °C; then they were soaked for 1 h in 2.5% Triton X-100 under constant agitation. The gel was incubated for 48 h at 37 °C in 50 mM sodium phosphate buffer, pH 5.5, with 100 µM of the HAD1 and HAD2. In order to determine the class of peptidases, 10 µM E-64 (a cysteine-peptidase inhibitor), 10 mM 1,10-phenanthroline (a non-specific metallopeptidase inhibitor) and 2 mM DTT (a cysteine-peptidase activator) were also testedCitation26.
Statistical analysis
Student’s t-test was used to determine the significance of differences between the means in the amastigote proliferation assays. A p value ≤ 0.05 was considered significant.
Result and discussion
Chemistry
Previous studies have demonstrated promising antitrypanocidal activities of hydroxamic acid derivatives – HADCitation26, acting as effective T. cruzi carbonic anhydrase (TcCA) inhibitors and ineffective as human CA inhibitors. Also, a lead compound containing a p-benzyloxy as a substituent on the aromatic ring (HAD-ph, ) besides its inhibitory activity against TcCA also showed promising features as an antitrypanosomal agentCitation26. This led us to analyze the efficacy of the two analogs containing only o-ethoxy (HAD1) and p-ethoxy (HAD2) moieties as substituents in the aromatic ring () against trypomastigotes and amastigotes.
Figure 1. Hydroxamic acid derivatives (HAD) with potential action against Chagas disease. Carbonic anhydrases human isoforms hCA I and II and T. cruzi enzyme TcCACitation26.
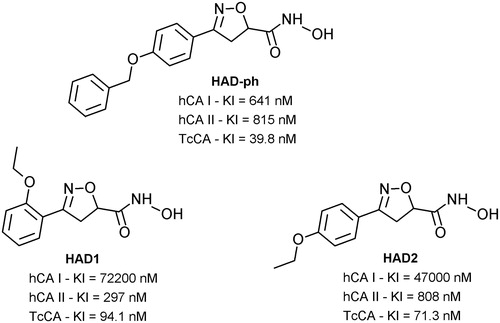
Effect of the hydroxamic acid derivatives (HAD1-2) against T. cruzi
We first analyzed the effect of the synthetic drugs (HAD1 and 2) on the viability of the bloodstream trypomastigotes (Y strain). Our findings demonstrated that even the highest concentration (90 µM) of both compounds (HAD1 and 2) was not effective against the trypomastigote forms of T. cruzi, while the IC50 values for benznidazole (BZ), reference drug, was 12.24 µM ± 3.42 (). In fact, the synthetic hydroxamic derivatives tested here (HAD1 and 2) were not as effective as their analog (HAD-ph) investigated earlier by Rodrigues et al.Citation26 This drug demonstrated a potent activity (greater than BZ) against all evolutive forms of T. cruzi, reaching an IC50 value from 7 to <1 μM for different strains of the parasite. The absence of a third aromatic ring in the HAD1-2 structures decreased their inhibitory activity against TcCA and trypanocidal activity trypomastigote forms. Additionally, it was possible to observe that the ortho or para ethoxy positions of the substituent on the aromatic ring linked to the heterocyclic ring did not influence the trypanocidal activity. This fact highlighted the importance of the p-benzyloxy as the substituent on the aromatic ring linked to the heterocyclic ring (HAD-ph) for trypanocidal activity, either by a steric effect or by increasing the liposolubility of the moleculeCitation26.
Figure 2. Effect of hydroxamic acid derivatives (HAD1-2) on bloodstream trypomastigotes (Y strain) compared with BZ activity. Trypomastigotes were treated at 37 °C for 24 h with different concentrations of the compounds. HAD1 and HAD2 compounds were inactive against the classical invasive form of T. cruzi. In contrast, the reference drug, BZ, showed an IC50 value of 12.2 µM.
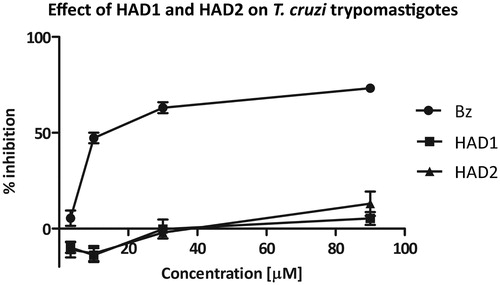
Screening of cytotoxicity in mammalian cells
Prior to analyze the effect of the synthetic compounds on intracellular amastigotes, we established the non-toxicity concentrations of the compounds to mammalian cells. The cellular toxicity assay was estimated on cardiomyocyte and macrophage cultures using resazurin as a viability indicator. Low toxicity was seen for both compounds tested. Treatment of cardiomyocytes with the HAD1 compound revealed no significant loss in viability up to 166 µM, resulting in a CC50 value = 329.8 μM (). Also against macrophages, HAD1 and 2 did not demonstrate any cytotoxicity effect, reaching a CC50 value of 349 µM and 221 µM, respectively (). The lack of toxicity in both cell types was clearly demonstrated by light microscopy, where even the highest concentration of HAD1 (90 µM) did not alter the morphology and viability of the macrophages (data not shown). The absence of p-benzyloxy as the substituent on the aromatic ring decreased the antitrypanosomal activity as well as the cellular cytotoxicity, which could be influenced by lower liposolubility of the HAD1 and 2 molecules than the HAD-ph prototype.
Figure 3. (A) Dose-dependent cardiomyocyte viability curve detected by resazurin in response to HAD1 treatment, resulting in CC50 value = 329.8 μM. (B) HAD1 and 2 cytotoxicity were also assessed in macrophages in the same fashion, resulting in similar viability curves and a CC50 value of 349 µM and 221 µM, respectively.
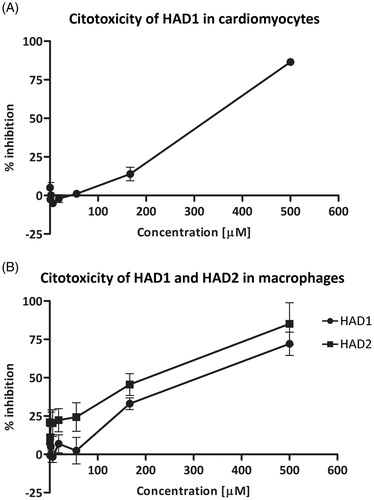
Efficacy of compounds against intracellular amastigotes
Considering the previous observations that TcCA inhibitors affect parasite growthCitation28 and that both compounds were effective against epimastigotesCitation26, we hypothesized that these derivatives could have a potential activity on the multiplication of the parasite. Therefore, we investigated the effectiveness of the drugs in clearing the infection. The activity against intracellular amastigotes was assessed by treating T. cruzi-infected cardiomyocyte cultures with the HAD1 and 2 compounds. Both drugs induced a significant reduction in the number of intracellular parasites in concentrations of 30 μM and 90 μM, but HAD1 was two-fold more effective than HAD2 at 30 μM (). The IC50 values, a concentration that reduces 50% of intracellular parasites, in T. cruzi-infected cardiomyocytes, were 30 ± 10.7 μM and 66 ± 19.3 μM for HAD1 and 2, respectively. This indicated that although these molecules have a lower liposolubility than the lead compound they are able to act against the intracellular forms of the parasite (amastigotes). Furthermore, it was observed that the intracellular activity was influenced by the ethoxy substituent position in the aromatic ring from the two hydroxamic acids, an important observation for future strategy of rational drug design. summarizes the results of the antitrypanocidal effect of hydroxamic acid derivatives
Figure 4. Analyses of the effect of HAD1 and 2 against intracellular amastigotes in cardiomyocyte culture. (A) Treatment with both synthetic compounds, HAD1 and 2, resulted in a dose-dependent inhibition of amastigote proliferation in cardiomyocytes. The lower IC50 value was achieved by HAD1 (30 μM). A significant decrease in the total intracellular amastigotes was observed after HAD1 (≥30 μM) and HAD2 (90 μM) treatment (B). *Statistically significant, p ≤ 0.05.
Table 1. Antitrypanocidal effect of hydroxamic acid derivatives (HAD).
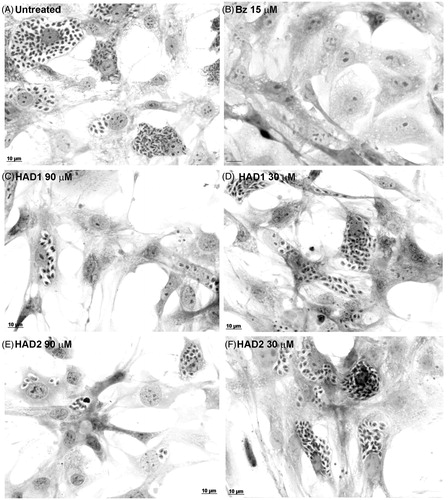
Furthermore, the treatment of T. cruzi-infected cardiomyocyte cultures with 90 μM of HAD1 reduced the number of intracellular amastigotes 7-fold when compared with untreated cultures (). This phenomenon was clearly observed by light microscopy (), which demonstrates a low level of infection in the highest concentration (90 μM) of the HAD1 and 2 compounds compared with control. It is important to highlight that treatment of the T. cruzi-infected cardiomyocyte cultures with BZ at 15 μM eliminated all the intracellular parasites.
Figure 5. Light microscopy of cardiomyocytes treated with HAD1 and 2 compounds. (A) Untreated cells displaying high number of intracellular amastigotes; (B) benznidazole treatment at 15 µM eliminated the parasites. HAD1 treatment resulted in an expressive reduction of amastigote proliferation (C, 90 µM; D, 30 µM). Addition of HAD2 in T. cruzi-infected cardiomyocyte cultures inhibited amastigote proliferation at high concentrations (90 µM, E), but lower concentrations did not produce the same effect (30 µM, F). Bar = 10 µM.
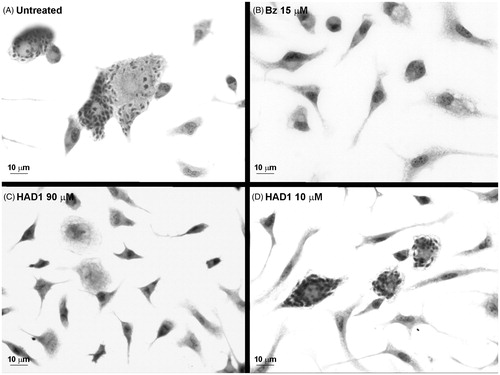
The fact that HAD1 displayed the highest selectivity index (SI ≥ 11) and the lowest IC50 prompted us to analyze its activity against T. cruzi-infected macrophages, also an important cell involved in the infection by this parasite. Although the IC50 value was higher in macrophages (75 µM), the treatment with 90 µM of HAD1 also induced a 5.5-fold reduction of intracellular parasites ( and ). At the highest concentration (90 µM), the macrophages demonstrated normal morphology and spreading, showing the low toxicity of the drug (). Although the drugs have shown activity against T. cruzi, they were not as efficient in the elimination of parasites as benznidazole, which revealed a reduction of 99% of intracellular amastigotes with 15 μM.
Figure 6. HAD1 compound effect on T. cruzi infection in macrophages. (A) Treatment of infected macrophages with HAD1 resulted in a dose-dependent inhibition of amastigote proliferation, achieving an IC50 value of 75 µM. (B) A significant decrease in the total number of intracellular amastigotes was detected with 90 μM of HAD1. *Statistically significant, p ≤ 0.05.
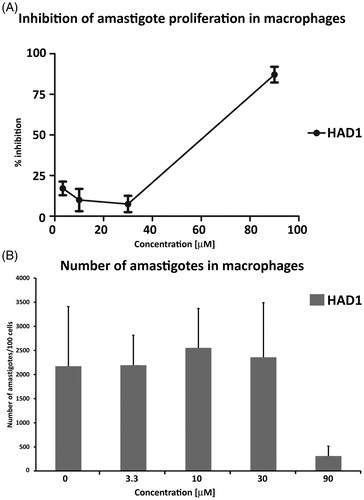
Figure 7. Light microscopy images of T. cruzi-infected macrophage cultures treated with HAD1. The cultures were treated 24 h after the establishment of T. cruzi infection. (A) General aspect of macrophages cultures after 72 h of infection by T. cruzi. (B) Clearance of intracellular parasites after 72 h treatment with 15 µM of benznidazole. Note the effectiveness of HAD1 compound against amastigotes after 72 h of treatment with 90 µM (C) and 10 µM (D).
Curiously, HAD1 and 2 were less effective than the HAD-ph from the series of hydroxamic acid derivatives investigated here. The substitution of the p-benzyloxy (HAD-ph) with o-ethoxy and p-ethoxy in HAD1 and HAD2 reduced the efficacy of these compounds to eliminate the intracellular amastigotes in mammalian cells by three-fold and seven-fold, respectively. The strategy of rational design of drugs has been encouraged in the search for new active drugs for the treatment of neglected diseasesCitation31. Lead optimization of compounds involving structure–activity relationships (SAR) may improve compound potency and physicochemical properties. However, the synthesis of more active compounds is a challenge and slight structural changes can reduce the compound activity. Therefore, our findings can guide the design of new molecules since the presence of p-benzyloxy instead of ethoxy impacts the activity against T. cruzi.
Ultrastructural analysis of the effect of HAD1 inhibitor against intracellular parasites
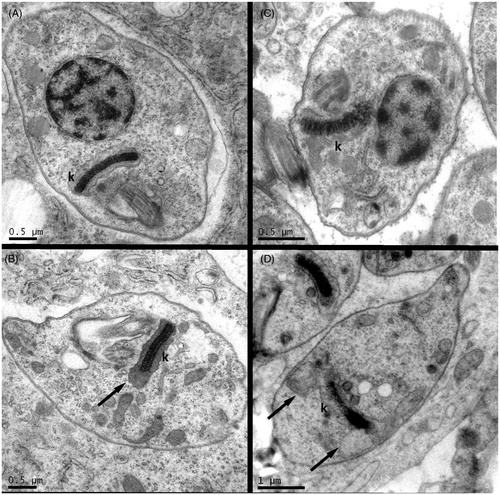
To analyze possible ultrastructural alterations induced by the synthetic compounds, transmission electron microscopy of T. cruzi-infected cardiomyocytes and macrophages after HAD1 treatment was performed (). Treatment of cardiomyocytes or macrophages infected by T. cruzi with the IC50 value of HAD1 revealed initial damage in the amastigotes, visualized by swelling of the kinetoplast. One important fact is that both cells, macrophages and cardiomyocytes, did not undergo structural change with this treatment, demonstrating no cytotoxic effect of the compounds for mammalian cells. Thus, our ultrastructural analysis associated with the cytotoxicity findings provided a good indication of the ability of the drug to permeate through the host cell membrane and its selectivity to the parasite target. The ultrastructural analysis of the effect of nonazoleCYP51 inhibitors against intracellular amastigotes showed blebbling of cellular and nuclear membranes and autophagic vesicles, in parallel with the accumulation of intermediate metabolites from the ergosterol biosynthesis pathwayCitation32. Most morphological studies of drug treatment emphasize the direct effect of compounds on the isolated parasites, mainly T. cruzi epimastigotes. Ultrastructural changes were also evidenced in T. cruzi treated with serine and cysteine protease inhibitorsCitation33,Citation34. A Kunitz-type serine protease inhibitor from Caribbean Sea anemone Stichodactyla helianthus induced the formation of concentric membrane structures and endoplasmic reticulum surrounding vesicles in the cytoplasm of epimastigotes, suggesting the autophagic cell death as its mechanism of actionCitation33. Cysteine protease inhibitors (morpholinecarbonyl–phenylalanine–homophenylalanine–vinyl sulphone phenyl and morpholineureaphenylalanine–homophenylalanine–fluoromethyl ketone) induced severe alterations in the Golgi complex with chagasin accumulation in enlarged cisternae, suggesting a disturbance in the transport within the ER–Golgi systemCitation34. Multiple intracellular compartment damage has been reported with other active drugs against T. cruziCitation35–37. Mitochondrial swelling and the presence of concentric structures were frequently noticed in parasites treated with lysophospholipid analoguesCitation38 and naphthoquinone derivativesCitation39. Remarkable changes were also induced in the kinetoplast–mitochondrial system, microtubule, and reservosomes organization after treatment of T. cruzi with DNA-intercalating drugs and sterol biosynthesis inhibitors, leading to kinetoplast collapse and cell deathCitation36,Citation37,Citation40.
Figure 8. Transmission electron microscopy of mammalian cells infected by T. cruzi and treated with HAD1 compound. Untreated controls showing normal general amastigote structure and regular kinetoplasts (k) from cardiomyocyte (A) and macrophage (B) infected cultures. Forty-eight hours treatment of HAD1 at IC50 concentration (30 µM) induced swelling of kinetoplasts (black arrows) both in cardiomyocyte (B) and in macrophage (D)-infected cultures.
Effect of HAD1 and 2 on the peptidase activity of T. cruzi
Cell-associated peptidases from T. cruzi displayed two distinct proteolytic classes: cysteine-(60 kDa) and metallopeptidase (70 and 80 kDa) activities inhibited, respectively, by E-64 and 1,10-phenanthroline, as shown in . Both compounds HAD1 and 2 were able to reduce the cysteine peptidases and decrease the activity of metallopeptidases. Cruzain/cruzipain is a cysteine peptidase required for the survival of T. cruzi. This enzyme, present in all life stages of the trypanosome, has been the focus of several studies and patents and is implicated in parasite cellular entry and digestion of immunoglobulin, immune system evasion, and other cellular mechanismsCitation19,Citation41.
Figure 9. Zymography of T. cruzi cellular peptidases after treatment with the HAD1 and HAD2 inhibitors. CTL, control – without treatment; E-64, cysteine peptidase inhibitor; DTT, cysteine peptidase activator; 1,10-PHE, metallopeptidase inhibitor.
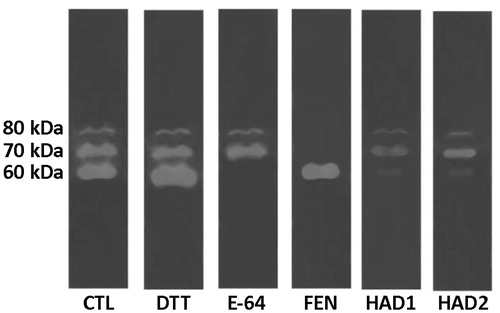
In conclusion, this work demonstrated that hydroxamates are a promising class of compound with anti-T. cruzi activity. Our data showed that hydroxamates HAD1 and 2 were effective against the replicative forms of the parasite, but not against the infective forms. This could be an indication that its mechanism of action involves the inhibition of parasite peptidases and carbonic anhydrase, essential enzymes for the parasite life cycle. HAD1 and 2 partially inhibited the cysteine and metallopeptidases of T. cruzi under the conditions evaluated. Furthermore, previous studies have already demonstrated the selectivity of these compounds against carbonic anhydrase of Trypanosoma cruzi (TcAC). In our assays, we observed that the position of the ethoxy substituent on the aromatic ring linked to the heterocyclic one strongly influenced the biological activity. HAD1, which has an ortho-ethoxy, was 2-fold more effective than HAD2, which has a para-ethoxy moiety, against the intracellular amastigotes. The absence of p-benzyloxy moiety, present in the lead molecule HAD-ph, decreased the lipophilicity of these compounds, but the HAD1 and 2 were still able to be effective against the replicative forms of the parasite. This fact demonstrates that this moiety is important for trypanocidal activity, but not essential. Moreover, new structural changes in the scaffold seem to be important for investigating the trypanocidal activity and the discovery of a more potent agent for the treatment of Chagas disease.
Conclusions
This work demonstrated a promising class of hydroxamates with anti-T. cruzi activity. The hydroxamates were effective against replicative forms of the parasite, but not against infective, trypomastigote forms. Ultrastructural analysis of amastigotes, the replicative, forms showed swelling of the kinetoplast after treatment with HAD1. Both compounds reduce the cysteine and metallopeptidase activities. No cytotoxic effect of the compounds for mammalian cells was observed. HAD1 and HAD2 could be an important scaffold to future rational drug design for anti-chagasic chemotherapy.
Acknowledgements
The authors thank Liliane Mesquita, Alanderson Nogueiraand Maria do Socorro Silva for their technical support in the cardiomyocyte and macrophage cultures.
Declaration of interest
This work was supported by grants from Fundação Oswaldo Cruz (FIOCRUZ), Conselho Nacional de Desenvolvimento Científico e Tecnológico (MCTI-CNPq), Programa Estratégico de Apoio à Pesquisa em Saúde (PAPES) VI), Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Coordenação de Aperfeiçoamento Pessoal de Nível Superior (CAPES) and Instituto Militar de Engenharia (IME),
Related Research Data
References
- Rassi A, Marcondes RJ. American trypanosomiasis (Chagas disease). Infect Dis Clin North Am 2012;26:275–91
- WHO. Chagas disease-factsheet. Wkly Epidemiol Rec 2012;87:519–22
- Bocchi EA. Heart failure in South America. Curr Cardiol Rev 2013;9:147–56
- Strasen J, Williams T, Ertl G, et al. Epidemiology of chagas disease in Europe: many calculations, little knowledge. Clin Res Cardiol 2014;103:1–10
- Jackson Y, Herrera V, Gascon J. Economic crisis and increased immigrant mobility: new challenges in managing Chagas disease in Europe. Bull World Health Organ 2014;92:771–2
- Carlier Y. Globalization of Chagas disease (American trypanosomiasis): the situation in Europe and Belgium. Bull Mem Acad R Med Belg 2011;166:347–55
- Sánchez LV, Ramírez JD. Congenital and oral transmission of American trypanosomiasis: an overview of physiopathogenic aspects. Parasitology 2012;140:1–13
- Montgomery SP, Starr MC, Cantey PT, et al. Neglected parasitic infections in the United States: Chagas disease. Am J Trop Med Hyg 2014;90:814–18
- Mejía-Jaramillo AM, Fernández G, Montilla M, et al. Trypanosoma cruzi strains resistant to benznidazole occurring in Colombia. Biomedica 2012;32:196–205
- Pinazo MJ, Guerrero L, Posada E, et al. Benznidazole-related adverse drug reactions and their relationship with serum drug concentrations in patients with chronic Chagas disease. Antimicrob Agents Chemother 2012;57:390–5
- Perez CJ, Lymbery AJ, Thompson RCA. Chagas disease: the challenge of polyparasitism? Trends Parasitol 2014;30:176–82
- Molina I, Gómez i Prat J, Salvador F, et al. Randomized trial of posaconazole and benznidazole for chronic Chagas’ disease. N Engl J Med 2014;370:1899–908
- De Carvalho KAT, Abdelwahid E, Ferreira RJ, et al. Preclinical stem cell therapy in Chagas disease: perspectives for future research. World J Transplant 2013;3:119–26
- Bahia MT, Diniz L de F, Mosqueira VCF. Therapeutical approaches under investigation for treatment of Chagas disease. Expert Opin Investig Drugs 2014;23:1225–37
- Cencig S, Coltel N, Truyens C, Carlier Y. Evaluation of benznidazole treatment combined with nifurtimox, posaconazole or AmBisome® in mice infected with Trypanosoma cruzi strains. Int J Antimicrob Agents 2012;40:527–32
- Dumonteil E, Bottazzi ME, Zhan B, et al. Accelerating the development of a therapeutic vaccine for human Chagas disease: rationale and prospects. Expert Rev Vaccines 2012;11:1043–55
- Ndao M, Beaulieu C, Black WC, et al. Reversible cysteine protease inhibitors show promise for a Chagas disease cure. Antimicrob Agents Chemother 2014;58:1167–78
- Branquinha MH, Marinho FA, Sangenito LS, et al. Calpains: potential targets for alternative chemotherapeutic intervention against human pathogenic trypanosomatids. Curr Med Chem 2013;20:3174–85
- Laverrière M, Cazzulo JJ, Alvarez VE. Antagonic activities of Trypanosoma cruzi metacaspases affect the balance between cell proliferation, death and differentiation. Cell Death Differ 2012;19:1358–69
- Cazzulo JJ. Proteinases of Trypanosoma cruzi: patential targets for the chemotherapy of Changas desease. Curr Top Med Chem 2002;2:1261–71
- Barr SC, Warner KL, Kornreic BG, et al. A cysteine protease inhibitor protects dogs from cardiac damage during infection by Trypanosoma cruzi. Antimicrob Agents Chemother 2005;49:5160–2
- McKerrow JH, Doyle PS, Engel JC, et al. Two approaches to discovering and developing new drugs for Chagas disease. Mem Inst Oswaldo Cruz 2009;104:263–9
- Santos LO, Garcia-Gomes AS, Catanho M, et al. Aspartic peptidases of human pathogenic trypanosomatids: perspectives and trends for chemotherapy. Curr Med Chem 2013;20:3116–33
- Nogueira de Melo AC, de Souza EP, Elias CGR, et al. Detection of matrix metallopeptidase-9-like proteins in Trypanosoma cruzi. Exp Parasitol 2010;125:256–63
- Nogueira de Melo AC, Meirelles MNSL, Porrozzi R, et al. Reduced activity of matrix metalloproteinase-9 in Trypanosoma cruzi-infected mouse embryo hepatocyte cell. Hepatol Res 2004;28:49–56
- Rodrigues GC, Feijó DF, Bozza MT, et al. Design, synthesis, and evaluation of hydroxamic acid derivatives as promising agents for the management of Chagas disease. J Med Chem 2014;57:298–308
- McKenna R, Supuran CT. Carbonic anhydrase inhibitors drug design. Subcell Biochem 2014;75:291–323
- Pan P, Vermelho AB, Capaci Rodrigues G, et al. Cloning, characterization, and sulfonamide and thiol inhibition studies of an α-carbonic anhydrase from Trypanosoma cruzi, the causative agent of Chagas disease. J Med Chem 2013;56:1761–71
- Meirelles MNSL, de Araújo-Jorge TC, Miranda CF, et al. Interaction of Trypanosoma cruzi with heart muscle cells: ultrastructural and cytochemical analysis of endocytic vacuole formation and effect upon myogenesis in vitro. Eur J Cell Biol 1986;41:198–206
- Heussen C, Dowdle EB. Electrophoretic analysis of plasminogen activators in polyacrylamide gels containing sodium dodecyl sulfate and copolymerized substrates. Anal Biochem 1980;102:196–202
- Kellici T, Ntountaniotis D, Vrontaki E, et al. Rational drug design paradigms: the Odyssey for designing better drugs. Comb Chem High Throughput Screen 2015;18:238–56
- Doyle PS, Chen C-K, Johnston JB, et al. A nonazole CYP51 inhibitor cures Chagas’ disease in a mouse model of acute infection. Antimicrob Agents Chemother 2010;54:2480–8
- De Almeida Nogueira NP, Morgado-Díaz JA, Menna-Barreto RFS, et al. Effects of a marine serine protease inhibitor on viability and morphology of Trypanosoma cruzi, the agent of Chagas disease. Acta Trop 2013;128:27–35
- Engel JC, Doyle PS, Hsieh I, Mckerrow JH. Cysteine protease inhibitors cure an experimental Trypanosoma cruzi infection. J Exp Med 1998;188:725–34
- Veiga-Santos P, Barrias ES, Santos JFC, et al. Effects of amiodarone and posaconazole on the growth and ultrastructure of Trypanosoma cruzi. Int J Antimicrob Agents 2012;40:61–71
- Kessler RL, Soares MJ, Probst CM, Krieger MA. Trypanosoma cruzi response to sterol biosynthesis inhibitors: morphophysiological alterations leading to cell death. PLoS One 2013;8:e55497
- Soeiro MNC, Werbovetz K, Boykin DW, et al. Novel amidines and analogues as promising agents against intracellular parasites: a systematic review. Parasitology 2013;140:1–23
- Santa-Rita RM, Barbosa HS, de Castro SL. Ultrastructural analysis of edelfosine-treated trypomastigotes and amastigotes of Trypanosoma cruzi. Parasitol Res 2006;100:187–90
- Menna-Barreto RFS, Goncalves RLS, Costa EM, et al. The effects on Trypanosoma cruzi of novel synthetic naphthoquinones are mediated by mitochondrial dysfunction. Free Radic Biol Med 2009;47:644–53
- Batista DGJ, Pacheco MGO, Kumar A, et al. Biological, ultrastructural effect and subcellular localization of aromatic diamidines in Trypanosoma cruzi. Parasitology 2010;137:251–9
- Sajid M, Robertson SA, Brinen LS, McKerrow JH. Cruzain: the path from target validation to the clinic. Adv Exp Med Biol 2011;712:100–15