
Abstract
Sulfocoumarins behave as interesting inhibitors of the metalloenzyme carbonic anhydrase (CA, EC 4.2.1.1). Here, we report a new series of 7-substituted derivatives which were obtained by the click chemistry approach from 7-propargyloxy-sulfocoumarin and aryl azides incorporating halogens, hydroxy, methoxy and carboxyl moieties in their molecules. The new compounds were screened for the inhibition on four physiologically relevant human CA (hCA) isoforms, the cytosolic hCA I and II and the transmembrane tumor-associated hCA IX and XII. The new compounds did not inhibit the cytosolic isoforms but were low nanomolar inhibitors of the tumor-associated ones hCA IX and XII.
Introduction
1,2-Benzoxathiines-2,2-dioxides, also referred as sulfocoumarins, were recently validated as a novel class of inhibitors of the zinc enzyme carbonic anhydrase (CA, EC 4.2.1.1)Citation1. The design of sulfocoumarins as CA inhibitors (CAIs) was inspired by their corresponding bioisosters coumarinsCitation2–9, whose inhibition mechanism relies on the CA-mediated esterase activity. Indeed the sulfocoumarins, as well as coumarins, undergo a CA-mediated hydrolysis within the active site cavity with the generation of the inhibitory species. The inhibition mechanism of such hydrolysis products is different at the molecular level when compared to the classical sulfonamide-based CAIsCitation10,Citation11, that coordinate in the deprotonated form to the zinc metal ion, thus substituting the fourth ligand represented by a water molecule/hydroxide ion. As demonstrated by means of X-ray crystallographic and kinetic studies, the hydrolyzed coumarine derivative (i.e. cinnamic acid) is placed at the rim of the enzymatic cavity thus occluding it. The resulting ligand-enzyme complex is stabilized by interactions with the amino acids present therein that constitute the most variable region within CA isoforms known to date (15 human CA (hCA) isoforms are reported)Citation2–8,10. Conversely, 2-hydroxyphenyl-ω-ethenyl sulfonic acids which are formed from the original sulfocoumarins, tightly bind to the zinc-coordinated water molecule by means of hydrogen bonding, whereas the scaffold of the inhibitor establishes additional favorable interactions within the cavity ()Citation1.
The highly selective CA inhibition profiles shown by the coumarins and sulfocoumarins rely on this particular inhibition mechanism, which in turn depends from different binding modes of the hydrolysis products within the CAs catalytic cavitiesCitation10. Such a feature is highly desired in the current CAIs research as the main problem associated to the sulfonamide-type clinically used CAIs is represented by their indiscriminate inhibition of the hCAs leading thus to a plethora of side effectsCitation10,12–17. Since many CA isoforms are involved in diverse physio/pathological conditions such as glaucoma (hCA I, II, IV and XII), edema (hCA II, IV, XIV as the most important), central nervous system (CNS)-related pathologies (hCAVII and XIV are particularly involved in the epilepsy) and tumors (hCA IX and hCA XII are strictly associated with hypoxic tumors), it is not surprising that CAIs are used in the clinic for some applications for almost 70 yearsCitation18–24. Many synthetic efforts have been made for the development of specific CAIs: in the last 15 years, in addition to the classical “tail approach”Citation10–17,25–28 which is mainly applied to classical sulfonamide inhibitors and their isosters, novel CAIs scaffolds have been also identified such as the polyaminesCitation29, phenolsCitation30–32, dithiocarbamatesCitation33–36, xanthatesCitation37, coumarins, thiocoumarins, 2-thioxocoumarins, coumarine oximesCitation4,Citation38,Citation39. Sulfocoumarins are the latest CAI class identified and similarly to the coumarins showed the most selective inhibition profiles against the pathologically valuable CA isoformsCitation1,9,40–44.
Figure 1. CA inhibition mechanism of sulfocoumarins. (A) The sulfocoumarin undergoes an enzyme-mediated hydrolysis with the formation of the trans-2-hydroxy-phenyl-ω-ethenylsulfonic acid 3. (B) The sulfonic acid 3 binds to the hCA II active site, by anchoring to the zinc-coordinated water molecule. The Zn(II) ion (central larger sphere), its three His ligands (His94, 96 and 119), the water molecule coordinated to the zinc (small sphere) as well as active site residues Thr200 and Pro201 involved in the binding of the hydrolyzed sulfocoumarin are shown, as determined by X-ray crystallography (PDB file 4BCW)Citation1.
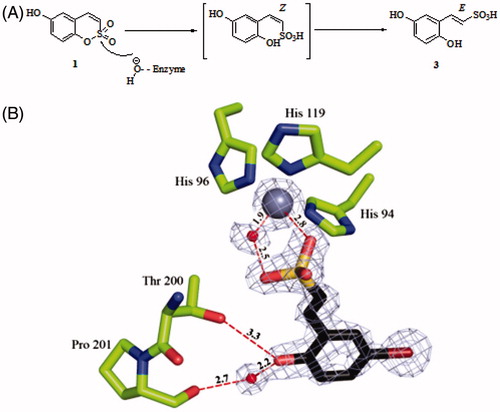
In analogy to coumarins, the substitution pattern at the sulfocoumarin scaffolds strongly influences the potency and selectivity profile against different hCA isoformsCitation1,40–44. Indeed, in our previous reportsCitation40–43, we investigated large series of sulfocoumarins bearing the tetrazolyl, triazolyl or aryl/alkyl moieties at 6 position, which showed low nanomolar hCAIX/XII inhibitory potencies, without particular effects on the cytosolic hCAI/II. On the contrary, derivatives bearing small substituents as well as benzyl esters at the 7 position act as low nanomolar hCAII inhibitorsCitation40.
To date, sulfocoumarins incorporating bulky and flexible moieties at 7 position were not reported, thus herein we report the synthesis, characterization and in vitro inhibition profiles against four relevant hCA isoforms (hCAI, II, IX and XII) of a small series of 7-substituted sulfocoumarins bearing the aryl-triazolyl moieties linked through an oxymethylene group, which were synthesized by means of the click chemistry approach.
Materials and methods
Chemistry
Anhydrous solvents and all reagents were purchased from Sigma-Aldrich (St. Louis, MO), Alfa Aesar (Haverhill, MA) and TCI (Eschborn, Germany). All reactions involving air- or moisture-sensitive compounds were performed under a nitrogen atmosphere using dried glassware and syringe techniques to transfer solutions. Nuclear magnetic resonance (1H-NMR, 13C-NMR) spectra were recorded using a Bruker Avance III 400 MHz spectrometer in deuterated dimethyl sulfoxide (DMSO-d6). Chemical shifts are reported in parts per million and the coupling constants (J) are expressed in Hertz (Hz). Splitting patterns are designated as follows: s, singlet; d, doublet; sept, septet; t, triplet; q, quartet; m, multiplet; brs, broad singlet; dd, doublet of doublets; appt, apparent triplet; appq, apparent quartet. The assignment of exchangeable protons (OH and NH) was confirmed by the addition of D2O. Analytical thin-layer chromatography (TLC) was carried out on Merck silica gel F-254 plates. Flash chromatography purifications were performed on Merck Silica gel 60 (230–400 mesh American Society for Testing and Materials (ASTM)) as the stationary phase and ethyl acetate/n-hexane were used as eluents. Melting points (m.p.) were measured in open capillary tubes with a Gallenkamp MPD350.BM3.5 apparatus and are uncorrected. All compounds were >95% pure by high performance liquid chromatography (HPLC).
General synthetic procedure of compounds 7–14Citation45
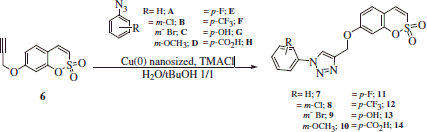
7-Prop-2-ynyloxy-benzo-[e][1,2]-oxathiine 2,2-dioxide 6 (1.0 eq) was added to a suspension of aryl azide A–H (1.1 eq) in H2O/tBuOH 1/1 (3.5 ml) at room temperature (r.t.), followed by addition of copper (0) nanosized (0.1 eq) and tetramethylammoniun chloride (TMACl) (1.0 eq). The suspension was stirred at 60 °C until starting materials were consumed (TLC monitoring), then quenched with H2O (20 ml) and the formed precipitate was filtered off and washed with H2O. The solid was dissolved in a minimal amount of acetone; the obtained solution was filtered through Celite 521® and then concentrated under vacuo to give a residue that was triturated with Et2O or dichloromethane (DCM) to afford the titled compounds 7–14.
Synthesis of 4-(2,2-dioxo-2H-2λ6-benzo-[e][1,2]-oxathiin-7-yloxymethyl)-1-phenyl-1H-[1,2,3]-triazole 7
4-(2,2-Dioxo-2H-2λ6-benzo-[e][1,2]-oxathiin-7-yloxymethyl)-1-phenyl-1H-[1,2,3]-triazole 7 was obtained according to the general procedure earlier reported using phenylazide A (1.1 eq), 7-prop-2-ynyloxy-benzo-[e][1,2]-oxathiine 2,2-dioxide 6 (0.05 g, 1.0 eq) in tBuOH/H2O (1/1, 3.5 ml), TMACl (1.0 eq) and copper nanosize (0.1 eq). The reaction mixture was stirred for 7 h to give the titled compound 7 as a white solid.
4-(2,2-dioxo-2H-2λ6-benzo-[e][1,2]-oxathiin-7-yloxymethyl)-1-phenyl-1H-[1,2,3]triazole 7
Seventy-two percent yield; m.p. 141–143 °C; silica gel TLC Rf 0.39 (EtOAc/n-hexane 50% v/v); δH (400 MHz, DMSO-d6): 5.41 (s, 2H, CH2), 7.16 (dd, J = 2.4, 8.8, 1H), 7.30 (d, J = 2.4, 1H), 7.37 (d, J = 10.4, 1H), 7.55 (t, J = 7.6, 1H), 7.67 (m, 4H), 7.95 (d, J = 7.6, 2H), 9.05 (s, 1H); δC (100 MHz, DMSO-d6): 62.7, 105.6, 113.2, 114.4, 120.3, 121.2, 124.2, 129.8, 130.7, 132.2, 137.4, 137.4, 143.9, 153.2, 161.9; m/z (ESI positive) 356.0 [M + H]+.
Synthesis of 1-(3-chloro-phenyl)-4-(2,2-dioxo-2H-2λ6-benzo-[e][1,2]-oxathiin-7-yloxymethyl)-1H-[1,2,3]-triazole 8
1-azido-3-chlorobenzene B (1.1 eq) and 7-prop-2-ynyloxy-benzo-[e][1,2]-oxathiine 2,2-dioxide 6 (0.05 g, 1.0 eq) were dissolved in tBuOH/H2O 1/1 (3.5 ml) and then TMACl (1.0 eq) and copper (0) nanosize (10% mol) were added. The mixture was stirred at 60 °C for 3.5 h, and then treated as described in general procedure earlier reported to afford 8 as a white solid.
1-(3-Chloro-phenyl)-4-(2,2-dioxo-2H-2λ6-benzo-[e][1,2]-oxathiin-7-yloxymethyl)-1H-[1,2,3]-triazole 8
Seventy-one percent yield; m.p. 174–176 °C; silica gel TLC Rf 0.26 (EtOAc/n-hexane 50% v/v); δH (400 MHz, DMSO-d6): 5.41 (s, 2H, CH2), 7.16 (dd, J = 2.4, 8.8, 1H), 7.28 (d, J = 2.4, 1H), 7.37 (d, J = 10.4, 1H), 7.66 (m, 4H), 7.97 (d, J = 8.8, 1H), 8.10 (s, 1H), 9.11 (s, 1H); δC (100 MHz, DMSO-d6): 62.6, 105.6, 113.2, 114.4, 119.7, 120.3, 121.0, 124.4, 129.6, 132.2, 132.6, 135.2, 137.4, 138.5, 144.1, 153.2, 161.8; m/z (ESI positive) 390.0 [M + H]+.
Synthesis of 1-(3-bromo-phenyl)-4-(2,2-dioxo-2H-2λ6-benzo-[e][1,2]-oxathiin-7-yloxymethyl)-1H-[1,2,3]-triazole 9
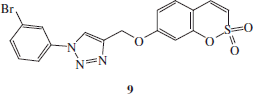
1-Azido-3-bromobenzene C (1.1 eq) and 7-prop-2-ynyloxy-benzo-[e][1,2]-oxathiine 2,2-dioxide 6 (0.05 g, 1.0 eq) were dissolved in tBuOH/H2O 1/1 (3.5 ml) and then TMACl (1.0 eq) and copper (0) nanosize (10% mol) were added. The mixture was stirred at 60 °C for 3.5 h and then treated as described in general procedure earlier reported to afford 9 as a white solid.
1-(3-Bromo-phenyl)-4-(2,2-dioxo-2H-2λ6-benzo-[e][1,2]-oxathiin-7-yloxymethyl)-1H-[1,2,3]-triazole 9
Seventy-nine percent yield; m.p. 171–173 °C; silica gel TLC Rf 0.49 (EtOAc/n-hexane 50% v/v); δH (400 MHz, DMSO-d6): 5.41 (s, 2H, CH2), 7.16 (dd, J = 2.4, 8.8, 1H), 7.30 (d, J = 2.4, 1H), 7.37 (d, J = 10.4, 1H), 7.61 (t, J = 8.4, 1H), 7.69 (m, 2H), 7.76 (d, J = 8.4, 1H), 8.01 (d, J = 8.4, 1H), 8.23 (t, J = 2.0, 1H), 9.11 (s, 1H); δC (100 MHz, DMSO-d6): 62.7, 105.6, 113.3, 114.4, 120.2, 120.4, 123.4, 123.8, 124.4, 132.2, 132.6, 132.8, 137.4, 138.6, 144.1, 153.2, 161.9; m/z (ESI positive) 434.0 [M + H]+.
Synthesis of 4-(2,2-Dioxo-2H-2λ6-benzo-[e][1,2]-oxathiin-7-yloxymethyl)-1-(3-methoxy-phenyl)-1H-[1,2,3]-triazole 10
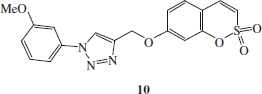
4-(2,2-dioxo-2H-2λ6-benzo-[e][1,2]-oxathiin-7-yloxymethyl)-1-(3-methoxy-phenyl)-1H-[1,2,3]-triazole 10 was obtained according the general procedure earlier reported using 1-azido-3-methoxybenzene D (1.1eq), 7-prop-2-ynyloxy-benzo-[e][1,2]-oxathiine 2,2-dioxide 6 (0.05 g, 1.0 eq) in tBuOH/H2O (1/1, 3.5 ml), TMACl (1.0 eq) and copper nanosize (0.1 eq). The reaction mixture was stirred for 6.5 h to give the titled compound 10 as a white solid.
4-(2,2-Dioxo-2H-2λ6-benzo-[e][1,2]-oxathiin-7-yloxymethyl)-1-(3-methoxy-phenyl)-1H-[1,2,3]-triazole 10
Sixty-eight percent yield; m.p. 129–131 °C; silica gel TLC Rf 0.40 (EtOAc/n-hexane 50% v/v); δH (400 MHz, DMSO-d6): 3.90 (s, 3H, CH3), 5.41 (s, 2H, CH2), 7.11 (d, J = 7.2, 1H), 7.16 (dd, J = 2.4, 8.8, 1H), 7.30 (d, J = 2.4, 1H), 7.37 (d, J = 10.4, 1H), 7.54 (m, 3H), 7.69 (m, 2H), 9.06 (s, 1H); δC (100 MHz, DMSO-d6): 56.6, 62.7, 105.6, 106.8, 113.1, 113.2, 114.3, 115.5, 120.3, 124.3, 131.8, 132.1, 137.4, 138.5, 143.9, 153.2, 161.1, 161.9; m/z (ESI positive) 386.0 [M + H]+.
Synthesis of 4-(2,2-dioxo-2H-2λ6-benzo-[e][1,2]-oxathiin-7-yloxymethyl)-1-(4-fluoro-phenyl)-1H-[1,2,3]-triazole 11
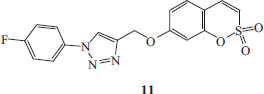
1-Azido-4-fluorobenzene E (1.1 eq) and 7-prop-2-ynyloxy-benzo-[e][1,2]-oxathiine 2,2-dioxide 6 (0.05 g, 1.0 eq) were dissolved in tBuOH/H2O 1/1 (3.5 ml) and then TMACl (1.0 eq) and copper (0) nanosize (10% mol) were added. The mixture was stirred at 60 °C for 2.5 h and then treated as described in general procedure earlier reported to afford 11 as a white solid.
4-(2,2-Dioxo-2H-2λ6-benzo-[e][1,2]-oxathiin-7-yloxymethyl)-1-(4-fluoro-phenyl)-1H-[1,2,3]-triazole 11
Sixty-three percent yield; m.p. 168–170 °C; silica gel TLC Rf 0.33 (EtOAc/n-hexane 50% v/v); δH (400 MHz, DMSO-d6): 5.41 (s, 2H, CH2), 7.16 (dd, J = 2.4, 8.8, 1H), 7.29 (d, J = 2.4, 1H), 7.37 (d, J = 10.4, 1H), 7.51 (t, J = 8.8, 2H), 7.69 (m, 2H), 8.00 (m, 2H), 9.01 (s, 1H); δF(376 MHz, DMSO-d6): −112.92 (s, 1F); δC (100 MHz, DMSO-d6): 62.7, 105.6, 113.2, 114.4, 117.7 (d, J2CF = 23.0), 120.3, 123.6 (d, J1CF = 8.9), 124.5, 132.2, 134.0, 137.4, 144.0, 153.2, 161.9, 162.7 (d, J1CF = 245.2); m/z (ESI positive) 374.0 [M + H]+.
Synthesis of 4-(2,2-dioxo-2H-2λ6-benzo-[e][1,2]-oxathiin-7-yloxymethyl)-1-(4-trifluoromethyl-phenyl)-1H-[1,2,3]-triazole 12
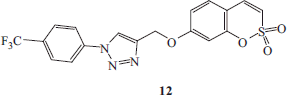
1-Azido-4-trifluoromethylbenzene F (1.1 eq) and 7-prop-2-ynyloxy-benzo-[e][1,2]-oxathiine 2,2-dioxide 6 (0.05 g, 1.0 eq) were dissolved in tBuOH/H2O 1/1 (3.5 ml) and then TMACl (1.0 eq) and copper (0) nanosize (10% mol) were added. The mixture was stirred at 60 °C for 5 h and then treated as described in general procedure earlier reported to afford 12 as a white solid.
4-(2,2-Dioxo-2H-2λ6-benzo-[e][1,2]-oxathiin-7-yloxymethyl)-1-(4-trifluoromethyl-phenyl)-1H-[1,2,3]-triazole 12
Seventy-four percent yield; m.p. 210–212 °C; silica gel TLC Rf0.40 (EtOAc/n-hexane 50% v/v); δH (400 MHz, DMSO-d6): 5.43 (s, 2H, CH2), 7.16 (dd, J = 2.4, 8.8, 1H), 7.29 (d, J = 2.4, 1H), 7.37 (d, J = 10.4, 1H), 7.69 (m, 2H), 8.04 (d, J = 8.4, 2H), 8.22 (d, J = 8.4, 2H), 9.19 (s, 1H); δC (100 MHz, DMSO-d6): 62.7, 105.6, 113.3, 114.4, 120.4, 121.7, 124.5, 124.8 (d, J1CF = 270.4), 128.2, 129.8 (d, J2CF = 32.0), 132.2, 137.4, 140.3, 144.4, 153.3, 161.9; m/z (ESI positive) 324.0 [M + H]+.
Synthesis of 4-(2,2-dioxo-2H-2λ6-benzo-[e][1,2]-oxathiin-7-yloxymethyl)-1-(4-hydroxy-phenyl)-1H-[1,2,3]-triazole 13
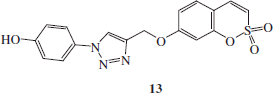
1-Azido-4-hydroxybenzene G (1.1 eq) and 7-prop-2-ynyloxy-benzo-[e][1,2]-oxathiine 2,2-dioxide 6 (0.05 g, 1.0 eq) were dissolved in tBuOH/H2O 1/1 (3.5 ml) and then TMACl (1.0 eq) and copper (0) nanosize (10% mol) were added. The mixture was stirred at 60 °C for 4 h and then treated as described in general procedure earlier reported to afford 13 as a white solid.
4-(2,2-Dioxo-2H-2λ6-benzo-[e][1,2]-oxathiin-7-yloxymethyl)-1-(4-hydroxy-phenyl)-1H-[1,2,3]-triazole 13
Eighty-two percent yield; m.p. 244–246 °C; silica gel TLC Rf 0.35 (EtOAc/n-hexane 50% v/v); δH (400 MHz, DMSO-d6): 5.38 (s, 2H, CH2), 6.98 (d, J = 8.8, 2H), 7.15 (dd, J = 2.4, 8.8, 1H), 7.28 (d, J = 2.4, 1H), 7.37 (d, J = 10.4, 1H), 7.69 (m, 4H), 8.84 (s, 1H), 10.00 (s, 1H, exchange with D2O, OH); δC (100 MHz, DMSO-d6): 62.7, 105.6, 113.2, 114.3, 117.0, 120.3, 123.0, 124.1, 129.6, 132.1, 137.42, 143.5, 153.2, 158.8, 161.9; m/z (ESI positive) 372.0 [M + H]+.
Synthesis of 1-(4-carboxy-phenyl)-4-(2,2-dioxo-2H-2λ6-benzo-[e][1,2]-oxathiin-7-yloxymethyl)-1H-[1,2,3]-triazole 14
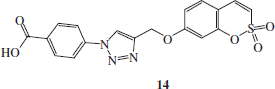
1-azido-carboxybenzene H (1.1 eq) and 7-prop-2-ynyloxy-benzo-[e][1,2]-oxathiine 2,2-dioxide 6 (0.05 g, 1.0 eq) were dissolved in tBuOH/H2O 1/1 (3.5 ml) and then TMACl (1.0 eq) and copper (0) nanosize (10% mol) were added. The mixture was stirred at 60 °C for 12.5 h and then treated as described in general procedure earlier reported to afford 14 as a white solid.
1-(4-Carboxy-phenyl)-4-(2,2-dioxo-2H-2λ6-benzo-[e][1,2]-oxathiin-7-yloxymethyl)-1H-[1,2,3]-triazole 14
Twenty-two percent yield; m.p. >300 °C; silica gel TLC Rf 0.42 (MeOH/DCM 10% v/v); δH (400 MHz, DMSO-d6): 5.43 (s, 2H, CH2), 7.17 (dd, J = 2.4, 8.8, 1H), 7.30 (d, J = 2.4, 1H), 7.38 (d, J = 10.4, 1H), 7.69 (m, 2H), 8.11 (d, J = 8.4, 2H), 8.19 (d, J = 8.4, 2H), 9.16 (s, 1H), 13.31 (bs, 1H, exchange with D2O, COOH); m/z (ESI positive) 400.0 [M + H]+.
Synthesis 7-prop-2-ynyloxy-benzo-[e][1,2]-oxathiine 2, 2-dioxide 6
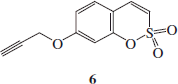
Propargyl bromide (1.2 eq) was added to a suspension of compound 5 (0.5 g, 1.0 eq) and K2CO3 (2.0 eq) in dry DMF (4 ml) under a nitrogen atmosphere and the mixture was stirred at r.t. for 2 h. The reaction mixture was quenched with H2O (20 ml) and extracted with EtOAc (3 × 15 ml). The organic layer was washed with brine (4 × 15 ml), dried over Na2SO4, filtered off and concentrated under vacuo to give a residue that was triturated with Et2O to afford the titled compound 6 as a white powder.
7-Prop-2-ynyloxy-benzo-[e][1,2]-oxathiine 2,2-dioxide 6
Fifty-four percent yield; m.p. 150–151 °C; silica gel TLC Rf 0.83 (EtOAc/n-hexane 50% v/v); δH (400 MHz, DMSO-d6): 3.70 (t, J = 2.4, 1H), 4.98 (d, J = 2.4, 2H), 7.07 (dd, J = 2.4, 8.4, 1H), 7.15 (d, J = 2.4, 1H), 7.37 (d, J = 10.4, 1H), 7.68 (m, 2H); δC (100 MHz, DMSO-d6): 57.21, 79.28, 80.03, 105.70, 113.43, 114.42, 120.51, 132.11, 137.40, 153.07, 161.03.
Synthesis of phenylazides A–HCitation46
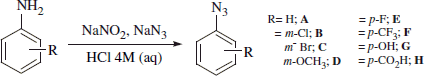
The proper aniline (0.5 g, 1.0eq) was dissolved in a 4 M HCl aqueous solution (5 ml) at 0 °C. NaNO2 (1.2 eq) was slowly added and the resulting solution was stirred at the same temperature for 0.5 h. Then NaN3 (1.5 eq) was added portion-wise and the mixture was stirred at r.t. for 0.5 h. The reaction mixture was filtered off or extracted with Et2O (2 × 15 ml) and the combined organic layers were dried over Na2SO4, filtered off and the solvent evaporated in vacuo to afford the corresponding phenylazide which was used without further purification.
Synthesis of phenylazide A
![]()
Phenylazide A was obtained according to the general procedure reported earlier.
Phenylazide A
Sixty percent yield; silica gel TLC Rf 0.76 (EtOAc/n-hexane 50% v/v); δH (400 MHz, DMSO-d6):7.12 (d, J = 7.6, 2H), 7.20 (t, J = 7.6, 1H), 7.42 (t, J = 7.6, 2H); δC (100 MHz, DMSO-d6): 120.0, 126.1, 131.0, 140.3.
Experimental in agreement with reported dataCitation46.
Synthesis of 3-chlorophenylazide B
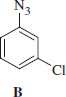
3-Chlorophenylazide B was obtained according to the general procedure reported earlier.
3-Chlorophenylazide B
Eighty-six percent yield; silica gel TLC Rf 0.84 (EtOAc/n-hexane 50% v/v); δH (400 MHz, DMSO-d6):7.14 (dd, J = 2.2, 8.0, 1H), 7.24 (t, J = 2.2, 1H), 7.30 (d, J = 2.2, 1H), 7.46 (t, J = 8.0, 1H); δC (100 MHz, DMSO-d6): 118.9, 120.1, 126.0, 132.4, 135.1, 142.2.
Experimental in agreement with reported dataCitation47.
Synthesis of 3-bromophenylazide C
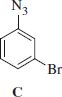
3-Bromophenylazide C was obtained according to the general procedure reported earlier.
3-Bromophenylazide C
Eighty-eight percent yield; silica gel TLC Rf 0.83 (EtOAc/n-hexane 50% v/v); δH (400 MHz, DMSO-d6):7.18 (dt, J = 1.6, 7,2, 1H), 7.38 (m, 3H); δC (100 MHz, DMSO-d6): 119.3, 122.8, 123.5, 128.9, 132.7, 142.3.
Experimental in agreement with reported dataCitation47.
Synthesis of 3-methoxyphenylazide D
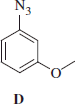
3-Methoxyphenylazide D was obtained according to the general procedure reported earlier.
3-Methoxyphenylazide D
Seventy-four percent yield; silica gel TLC Rf 0.78 (EtOAc/n-hexane 50% v/v); δH (400 MHz, DMSO-d6): 3.80 (s, 3H, CH3), 6.66 (t, J = 2.4, 1H), 6.74 (ddd, J = 0.8, 2.4, 8.2, 1H), 6.81 (ddd, J = 0.8, 2.4, 8.2, 1H), 7.36 (t, J= 8.2, 1H); δC (100 MHz, DMSO-d6): 56.3, 105.7, 112.0, 112.1, 131.7, 141.5, 161.5.
Experimental in agreement with reported dataCitation46.
Synthesis of 4-fluorophenylazide E
![]()
4-Fluorophenylazide E was obtained according to the general procedure reported earlier.
4-Fluorophenylazide E
Eighty-nine percent yield; silica gel TLC Rf 0.79 (EtOAc/n-hexane 50% v/v); δH (400 MHz, DMSO-d6): 67.19 (m, 2H), 7.29 (t, J = 8.8, 2H); δF (376 MHz, DMSO-d6): −117.77 (s, 1F); δC (100 MHz, DMSO-d6): 117.7 (d, J2CF = 23), 121.8 (d, J3CF = 9.0), 136.4, 160.3 (d, J1CF = 241.0). Experimental in agreement with reported dataCitation47.
Synthesis of 4-trifluoromethyl-phenylazide F
![]()
4-Trifluoromethylphenylazide F was obtained according to the general procedure reported earlier.
4-Trifluoromethylphenylazide F
Sixty-seven percent yield; silica gel TLC Rf 0.84 (EtOAc/n-hexane 50% v/v); δH (400 MHz, DMSO-d6): 7.35 (d, J = 8.8, 2H), 7.78 (d, J = 8.8, 2H); δF(376 MHz, DMSO-d6): −56.18 (s, 3F); δC (100 MHz, DMSO-d6): 120.8, 125.0 (d, J1CF = 269.6), 126.2 (q, J2CF = 32.0), 130.0, 144.7.
Experimental in agreement with reported dataCitation48.
Synthesis of 4-hydroxy-phenylazide G
![]()
4-hydroxyphenylazide G was obtained according to the general procedure reported earlier.
4-Hydroxyphenylazide G
Forty-six percent yield; silica gel TLC Rf 0.84 (EtOAc/n-hexane 50% v/v); δH (400 MHz, DMSO-d6): 6.84 (d, J = 8.8, 2H), 6.97 (d, J = 8.8, 2H), 9.56 (bs, 1H, exchange with D2O, OH); δC (100 MHz, DMSO-d6): 117.5, 121.1, 130.6, 156.0.
Experimental in agreement with reported dataCitation49.
Synthesis of 4-azidobenzoic acid H
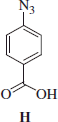
4-azidobenzoic acid H was obtained according to the general procedure reported earlier. 4-aminobenzoic acid was treated with NaNO2 and NaN3 in a 4 M HCl aqueous solution and the formed precipitate was filtered-off to afford the title compound H as a yellow solid.
4-Azidobenzoic acid H
Seventy-three percent yield; m.p. 188–190 °C dec; silica gel TLC Rf 0.71 (EtOAc/n-hexane 50% v/v); δH (400 MHz, DMSO-d6): 7.25 (d, J = 8.4, 2H), 7.99 (d, J = 8.4, 2H), 13.02 (bs, 1H, exchange with D2O, COOH); δC (100 MHz, DMSO-d6): 120.2, 128.3, 132.2, 144.9, 167.6. Experimental in agreement with reported dataCitation50.
CA inhibition
An applied photophysics stopped-flow instrument has been used for assaying the CA catalyzed CO2 hydration activityCitation51. Phenol red (at a concentration of 0.2 mm) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mm Hepes (pH 7.5) as buffer, and 20 mm Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mm for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mm) were prepared in distilled-deionized water and dilutions up to 0.01 nm were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 15 min to 6 h at room temperature (15 min) or 4 °C (6 h) prior to assay, in order to allow for the formation of the E-I complex. Data from were obtained after 6 h incubation of enzyme and inhibitor, as for the sulfocoumarins and coumarins reported earlierCitation1–4,7. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3 and the Cheng–Prusoff equation, as reported earlierCitation2,Citation5, and represent the mean from at least three different determinations. All CA isoforms were recombinant ones obtained in-house as reported earlierCitation52–56.
Table 1. Inhibition data of human CA isoforms hCA I, II, IX and XII with sulfocoumarins 7–14 reported here and the standard sulfonamide inhibitor acetazolamide (AAZ) by a stopped-flow CO2 hydrase assayCitation51.
Results and discussion
CA inhibition
Sulfocoumarins 7–14 were screened in vitro for the inhibition against four physiologically relevant hCA isoforms, the cytosolic hCAI and II and the trans-membrane tumor-associated hCA IX and XIICitation10,12,24,57–60. shows the inhibition data obtained and compared to the standard clinically used sulfonamide acetazolamide (AAZ) after a period of incubation of 6 h of the enzyme and inhibitors. Noteworthy the assay inhibition performed within the usual 15 min incubation period (as for the sulfonamides)Citation51 led to the very weak inhibition constants (data not shown). For this reason, herein is reported a 6 h incubation time instead.
The following structure-activity relationship (SAR) should be noted:
According to our previous reportsCitation40–44, isoform hCA I was not or was poorly inhibited by sulfocoumarins 7–14. Compounds 8 and 11 are high micromolar inhibitors, whereas the remaining ones did not significantly inhibit the enzyme below 10 μm inhibitor concentration.
The inhibition profile shown by sulfocoumarins 7–14 on the remaining three CA isoforms was quite surprising and unexpected. Indeed the 7-substituted sulfocoumarins earlierCitation40 reported were effective and selective inhibitors against the hCA II over the tumor-associated isoforms IX and XII. Surprisingly derivatives 7–14 herein reported did not exhibit any activity against the cytosolic isoform hCA II and were low nanomolar inhibitors against the transmembrane isoforms IX and XII (). Such a behavior might be ascribed that the previously synthesized derivatives incorporated small moieties or benzyl esters at the 7 position, whereas sulfocoumarins 7–14 bear flexible and bulky aryl-triazolyl moieties via a CH2O linker.
As mentioned, the transmembrane isoforms were strongly inhibited by the new sulfocoumarins 7–14 here reported, with KI values spanning between 19.0 and 36.5 nM for the hCA IX and 4.3–19.1 nM for the hCA XII, respectively. These values were comparable with the clinically used sulfonamide AAZ (KI: 2.5–5.7 nM). The definition of a proper SAR for hCA IX and hCA XII is not feasible, considering that all these compounds showed similar enzyme affinities. However the data we reported clearly showed that the nature of the substitution pattern -R on the phenyl moiety has a weak influence on the inhibitory properties, whereas the substitution itself within the sulfocoumarin scaffold at 7 position plays a pivotal role addressing the selectivity profiles.
Chemistry
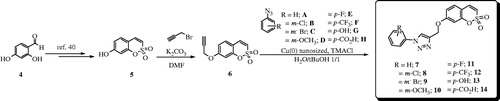
The general strategy of Zabulovski’s groupCitation1,Citation9 for the preparation of 6-substituted sulfocoumarins was recently validated by us for the synthesis of 7-substituted such derivatives. As reportedCitation40, the regioselective protection of the commercially available 2,4-dihydroxybenzaldehyde 4, followed by intramolecular cyclization and aromatization, afforded the desired 7-hydroxysulfocoumarin 5, which finally was converted to the derivative 6 through reaction with propargyl bromide (). Then, 6 was reacted with various freshly prepared aryl azides A–H in the presence of copper (0) nanosized as catalystCitation45 to afford the desired 1,2,3-triazolyl derivatives 7–14 ().
Scheme 1. Synthesis of 7-substituted sulfocoumarins 7–14.
Conclusion
Herein, we report a series of 7-substituted sulfocoumarins bearing the 1,2,3-triazolyl moieties via a CH2O linker obtained by the click chemistry approach. Unlike 7-substituted sulfocoumarins investigated earlier,Citation40 which were potent and selective hCA II inhibitors and ineffective as hCA I, IX and XII inhibitors, compounds from this new series exhibit effective and selective inhibitory properties for the tumor-associated isoforms over hCA IX and XII.
As hCA IX and hCA XII were recently validated as antitumor/antimetastatic drug targets, with one inhibitor in Phase I clinical trialsCitation61, this prodrug, isoform-selective CAIs, reported here might be considered of interest for such biomedical applications.
Declaration of interest
This work was financed in part by a Marie Curie ITN FP7 project (Dynano). The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Related Research Data
References
- Tars K, Vullo D, Kazaks A, et al. Sulfocoumarins (1,2-benzoxathiine-2,2-dioxides): a class of potent and isoform-selective inhibitors of tumor-associated carbonic anhydrases. J Med Chem 2013;56:293–300
- Maresca A, Temperini C, Vu H, et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–62
- Touisni N, Maresca A, McDonald PC, et al. Glycosyl coumarin carbonic anhydrase IX and XII inhibitors strongly attenuate the growth of primary breast tumors. J Med Chem 2011;54:8271–7
- Carta F, Maresca A, Scozzafava A, et al. 5- and 6-membered (thio)lactones are prodrug type carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2012;22:267–70
- Maresca A, Temperini C, Pochet L, et al. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem 2010;53:335–44
- Maresca A, Scozzafava A, Supuran CT. 7,8-Disubstituted but not 6,7-disubstituted coumarins selectively inhibit the transmembrane, tumor-associated carbonic anhydrase isoforms IX and XII over the cytosolic ones I and II in the low nanomolar/subnanomolar range. Bioorg Med Chem Lett 2010;20:7255–8
- Bonneau A, Maresca A, Winum JY. Metronidazole-coumarin conjugates and 3-cyano-7-hydroxy-coumarin act as isoform-selective carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2013;28:397–401
- Davis RA, Vullo D, Maresca A, et al. Natural product coumarins that inhibit human carbonic anhydrases. Bioorg Med Chem 2013;21:1539–43
- Grandane A, Belyakov S, Trapencieris P, et al. Facile synthesis of coumarin bioisosteres 1,2-benzoxathiine 2,2-dioxides. Tetrahedron 2012;68:5541–6
- Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68
- Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2010;20:3467–74
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81
- Aggarwal M, Kondeti B, McKenna R. Insights towards sulfonamide drug specificity in α-carbonic anhydrases. Bioorg Med Chem 2013;21:1526–33
- Aggarwal M, McKenna R. Update on carbonic anhydrase inhibitors: a patent review (2008 − 2011). Expert Opin Ther Pat 2012;22:903–15
- Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72
- Supuran CT, Scozzafava A, Casini A. Carbonic anhydrase inhibitors. Med Res Rev 2003;23:146–89
- Pastorekova S, Parkkila S, Pastorek J, et al. Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J Enzyme Inhib Med Chem 2004;19:199–229
- Carta F, Supuran CT, Scozzafava A. Sulfonamides and their isosters as carbonic anhydrase inhibitors. Future Med Chem 2014;6:1149–65
- Supuran CT. Carbonic anhydrases: from biomedical applications of the inhibitors and activators to biotechnological use for CO(2) capture. J Enzyme Inhib Med Chem 2013;28:229–30
- Svastová E, Hulíková A, Rafajová M, et al. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett 2004;577:439–45
- Thiry A, Dogné JM, Masereel B, et al. Targeting tumor-associated carbonic anhydrase IX in cancer therapy. Trends Pharmacol Sci 2006;27:566–73
- Alterio V, Hilvo M, Di Fiore A, et al. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc Natl Acad Sci U S A 2009;106:16233–8
- Ahlskog JK, Dumelin CE, Trüssel S, et al. In vivo targeting of tumor-associated carbonic anhydrases using acetazolamide derivatives. Bioorg Med Chem Lett 2009;19:4851–6
- Lou Y, McDonald PC, Oloumi A, et al. Targeting tumor hypoxia: suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res 2011;71:3364–76
- Supuran CT. Carbonic anhydrase inhibitors and activators for novel therapeutic applications. Future Med Chem 2011;3:1165–80
- Aggarwal M, Boone CD, Kondeti B, et al. Structural annotation of human carbonic anhydrases. J Enzyme Inhib Med Chem 2013;28:267–77
- Supuran CT, Ilies MA, Scozzafava A. Carbonic anhydrase inhibitors. Part 29. Interaction of isozymes I, II and IV with benzolamide-like derivatives. Eur J Med Chem 1998;33:739–52
- Supuran CT, Maresca A, Gregáň F, et al. Three new aromatic sulfonamide inhibitors of carbonic anhydrases I, II, IV and XII. J Enzyme Inhib Med Chem 2013;28:289–93
- Carta F, Temperini C, Innocenti A, et al. Polyamines inhibit carbonic anhydrases by anchoring to the zinc-coordinated water molecule. J Med Chem 2010;53:5511–22
- Innocenti A, Vullo D, Scozzafava A, et al. Carbonic anhydrase inhibitors: interactions of phenols with the 12 catalytically active mammalian isoforms (CA I-XIV). Bioorg Med Chem Lett 2008;18:1583–7
- Innocenti A, Vullo D, Scozzafava A, et al. Carbonic anhydrase inhibitors: inhibition of mammalian isoforms I-XIV with a series of substituted phenols including paracetamol and salicylic acid. Bioorg Med Chem 2008;16:7424–8
- Davis RA, Hofmann A, Osman A, et al. Natural product-based phenols as novel probes for mycobacterial and fungal carbonic anhydrases. J Med Chem 2011;54:1682–92
- Carta F, Aggarwal M, Maresca A, et al. Dithiocarbamates: a new class of carbonic anhydrase inhibitors. Crystallographic and kinetic investigations. Chem Commun (Camb) 2012;48:1868–70
- Maresca A, Carta F, Vullo D, et al. Dithiocarbamates strongly inhibit the β-class carbonic anhydrases from Mycobacterium tuberculosis. J Enzyme Inhib Med Chem 2013;28:407–11
- Monti SM, Maresca A, Viparelli F, et al. Dithiocarbamates are strong inhibitors of the beta-class fungal carbonic anhydrases from Cryptococcus neoformans, Candida albicans and Candida glabrata. Bioorg Med Chem Lett 2012;22:859–62
- Carta F, Aggarwal M, Maresca A, et al. Dithiocarbamates strongly inhibit carbonic anhydrases and show antiglaucoma action in vivo. J Med Chem 2012;55:1721–30
- Carta F, Akdemir A, Scozzafava A, et al. Xanthates and trithiocarbonates strongly inhibit carbonic anhydrases and show antiglaucoma effects in vivo. J Med Chem 2013;56:4691–700
- Carta F, Vullo D, Maresca A, et al. New chemotypes acting as isozyme-selective carbonic anhydrase inhibitors with low affinity for the offtarget cytosolic isoform II. Bioorg Med Chem Lett 2012;22:2182–5
- Carta F, Maresca A, Scozzafava A, et al. Novel coumarins and 2-thioxo-coumarins as inhibitors of the tumor-associated carbonic anhydrases IX and XII. Bioorg Med Chem 2012;20:2266–73
- Tanc M, Carta F, Bozdag M, et al. 7-Substituted-sulfocoumarins are isoform-selective, potent carbonic anhydrase II inhibitors. Bioorg Med Chem 2013;21:4502–10
- Grandane A, Tanc M, Zalubovskis R, et al. 6-Triazolylsubstituted sulfocoumarins are potent, selective inhibitors of the tumor-associated carbonic anhydrases IX and XII. Bioorg Med Chem Lett 2014;24:1256–60
- Grandane A, Tanc M, Zalubovskis R, et al. Synthesis of 6-tetrazolyl-substituted sulfocoumarins acting as highly potent and selective inhibitors of the tumor-associated carbonic anhydrase isoforms IX and XII. Bioorg Med Chem 2014;22:1522–8
- Grandane A, Tanc M, Di Cesare Mannelli L, et al. 6-substituted sulfocoumarins are selective carbonic anhdydrase IX and XII inhibitors with significant cytotoxicity against colorectal cancer cells. J Med Chem 2015;58:3975–83
- Tanc M, Carta F, Scozzafava A, et al. 6-substituted 1,2-benzoxathiine-2,2-dioxides are isoform-selective inhibitors of human carbonic anhydrases IX, XII and VA. Org Biomol Chem 2015;13:77–80
- Pala N, Micheletto L, Sechi M, et al. Carbonic anhydrase inhibition with benzenesulfonamides and tetrafluorobenzenesulfonamides obtained via click chemistry. ACS Med Chem Lett 2014;5:927–30
- Kwok S, Fotsing J, Fraser R, et al. Transition-metal-free catalytic synthesis of 1,5-diaryl-1,2,3-triazoles. Org Lett 2010;12:4217–19
- Dai Z, Chen Y, Zhang M, et al. Synthesis and antifungal activity of 1,2,3-triazole phenylhydrazone derivatives. Org Biomol Chem 2015;13:477–86
- Ciocoiu C, Nikolic N, Nguyen H, et al. Synthesis and dual PPARalpha/delta agonist effects of 1,4-disubstituted 1,2,3-triazole analogues of GW 501516. Eur J Med Chem 2010;45:3047–55
- Baron A, Herrero C, Quaranta A, et al. Click chemistry on a ruthenium polypyridine complex. An efficient and versatile synthetic route for the synthesis of photoactive modular assemblies. Inorg Chem 2012;51:5985–7
- Suwal S, Pflum M. Phosphorylation-dependent kinase-substrate cross-linking. Angew Chem Int Ed Engl 2010;49:1627–30
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73
- Liu F, Martin-Mingot A, Lecornué F, et al. Carbonic anhydrases inhibitory effects of new benzenesulfonamides synthesized by using superacid chemistry. J Enzyme Inhib Med Chem 2012;27:886–91
- Ekinci D, Kurbanoglu NI, Salamci E, et al. Carbonic anhydrase inhibitors: inhibition of human and bovine isoenzymes by benzenesulphonamides, cyclitols and phenolic compounds. J Enzyme Inhib Med Chem 2012;27:845–8
- Hilvo M, Baranauskiene L, Salzano A, et al. Biochemical characterization of CA IX, one of the most active carbonic anhydrase isozymes. J Biol Chem 2008;283:27799–809
- Nocentini A, Carta F, Ceruso M, et al. Click-tailed coumarins with potent and selective inhibitory action against the tumor-associated carbonic anhydrases IX and XII. Bioorg Med Chem 2015;23:6955–66
- Casini A, Scozzafava A, Mincione F, et al. Carbonic anhydrase inhibitors: water-soluble 4-sulfamoylphenylthioureas as topical intraocular pressure-lowering agents with long-lasting effects. J Med Chem 2000;43:4884–92
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77
- Pacchiano F, Carta F, McDonald PC, et al. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J Med Chem 2011;54:1896–902
- Dubois L, Lieuwes NG, Maresca A, et al. Imaging of CA IX with fluorescent labelled sulfonamides distinguishes hypoxic and (re)-oxygenated cells in a xenograft tumour model. Radiother Oncol 2009;92:423–8
- Dubois L, Peeters S, Lieuwes NG, et al. Specific inhibition of carbonic anhydrase IX activity enhances the in vivo thera-peutic effect of tumor irradiation. Radiother Oncol 2011;99: 424–31
- https://clinicaltrials.gov/ct2/show/study/NCT02215850?term=SLC_0111&rank=1