
Abstract
Novel bisindolyl-cycloalkane indoles resulted from the reaction of aliphatic dialdehydes and indole. As bisindolyl-natural alkaloid compounds have recently been reported as inhibitors of the methicillin-resistant Staphylococcus aureus (MRSA)-pyruvate kinase (PK), we tested our novel compounds as MRSA PK inhibitors and now report first inhibiting activities. We discuss structure–activity relationships of structurally varied compounds. Activity influencing substituents have been characterized and relations to antibacterial activities of the most active compounds have been proved.
Introduction
As novel antibiotic drugs have been developed during the last century decades the treatment of infectious diseases seemed to be a manageable problemCitation1. Occurring resistances were fought with antibiotics which showed β-lactamase stability in the case of the β-lactam antibiotics or which interacted with different but limited target structures within the bacterial metabolism like RNA or the protein biosynthesisCitation1–4. One of the early discovered bacteria with a critical antibiotic resistance was the methicillin-resistant Staphylococcus aureus (MRSA), but also formerly noncritical bacteria like E. coli variants developed resistances against used antibioticsCitation5–9.
Sources for novel antibiotics are presently seen in natural products like marine sponges or marine invertebratesCitation10–12. Extracts of them are screened for an antibacterial activity and partly novel structures have been discovered. However, such natural compounds mostly own complicated structures with chiral atoms so that an access to such compounds is difficult by the use of synthetic chemistry. Extraction procedures often yield poor amounts of compounds for a perspective therapeutical use.
We discovered novel bisindolyl-cycloalkane indoles 1–4 which are easily accessible in a simple one-pot reaction as will be discussed below. Their bisindolyl-partial structures have been found as structural elements in recently reported bisindolyl alkaloids from marine sources which showed inhibiting activities of the MRSA-pyruvate kinase (PK) as a novel antibacterial target structure and are shown in Citation12.
Figure 1. Bisindolyl alkaloidts from marine source with MRSA PK-inhibiting activies.
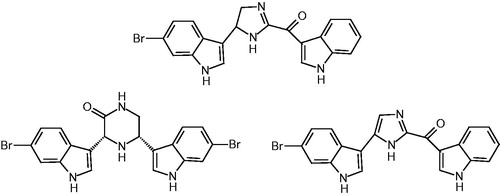
We will present the synthesis of our novel MRSA PK inhibitor class and discuss first structure–activity relationships of both MRSA inhibition and first antibacterial data of profiled compounds.
Experimental
General
Commercial reagents were used without further purification. Succinaldehyde was given by the treatment of 2,5-dimethoxytetrahydrofuran with hydrochloric acid in THF after stirring under reflux for 2 h. Adipaldehyde resulted from the sodium metaperiodate oxidation of cyclohexane epoxide in THF at room temperature. The 1H-NMR spectra (400 MHz) were measured using tetramethylsilane as internal standard. Thin layer chromatography (TLC) was performed on E. Merck 5554 silica gel plates. The EI mass spectra were measured with an AMD 402 mass spectrometer, the ESI spectra were recorded on a Finnigan LCQ Classic mass spectrometer. IR spectra were recorded on a FT-IR spectrometer. Elemental analysis indicated by the symbols of the elements was within ±0.4% of the theoretical values and was performed using a Leco CHNS-932 apparatus.
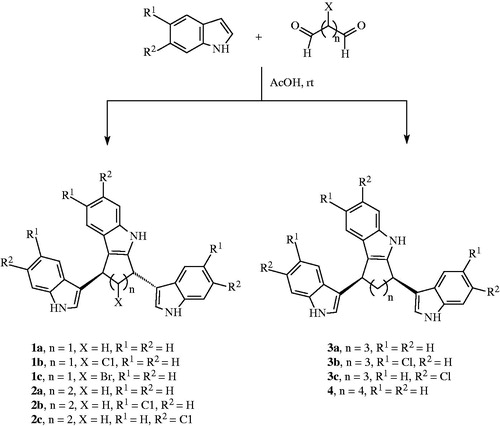
Formation of the bis(indolyl) cycloalkane indoles 1a–c, 2a–c, 3a–c, and of 4
The respective dialdehyde (2 mmol) was dissolved in glacial acetic acid (15 mL). Then the corresponding indole (5 mmol) was added to the solution. The mixture was left stirring overnight and the product was detected by TLC in CH2Cl2 (100%). The reaction mixture was worked up by neutralizing the acid with NaOH (10%) and then extracted with CH2Cl2 for three times. The organic layer was washed with water and brine each for three times. Then it was dried over sodium sulfate, filtered and concentrated in vacuum. The reaction products were given after column chromatography using CH2Cl2 as eluent (Scheme 1).
Scheme 1. Formation of target compounds.
(1R/S, 3R/S)-1,3-Di(1H-indol-3-yl)-1,2,3,4-tetrahydrocyclopenta[b]indole 1a. Yield 0.200 g (20%); light-brownish powder; mp 110–115 °C; 1H NMR (acetone-D6) δ10.19 (s, 1H, NH), 9.99 (s, 1H, NH), 9.97 (s, 1H, NH), 7.58-7.56 (m, 1H, aromat. H), 7.39-7.37 (m, 2H, aromat. H), 7.28-7.26 (m, 4H, aromat. H), 7.09-7.06 (m, 1H, aromat. H), 7.03–7.01 (m, 2H, aromat. H), 6.92-6.90 (m, 2H, aromat. H), 6.83-6.82 (m, 1H, aromat. H), 6.47-6.45 (m, 1H, aromat. H), 5.09 (d, J = 7.9 Hz, 2H, alkyl CH), 1.89 (t, J = 7.9 Hz, 2H, CH2); MS (EI), m/z = 387 [M+]; IR (ATR): 3404 (NH), 2923 (CH2) cm−1. Anal. (C27H21N3) Calc. C 83.69, H 5.46, N 10.85; Found C 83.35, H 5.24, N 10.66.
(1R/S, 3R/S)-2-Chloro-1,3-di(1H-indol-3-yl)-1,2,3,4-tetrahydrocyclopenta[b]indole 1b. Yield 0.236 g (28%); light-brownish powder; mp 168–171 °C; 1H NMR (acetone-D6) δ10.46 (s, 1H, NH), 10.10 (s, 1H, NH), 9.88 (s, 1H, NH), 7.63 (d, J = 8.2 Hz, 2H, aromat. H), 7.43-7.35 (m, 2H, aromat. H), 7.19 (d, J = 7.7 Hz, 2H, aromat. H), 7.11-7.07 (m, 4H, aromat. H), 6.97-6.92 (m, 4H, aromat. H), 4.92 (t, J = 7.9 Hz, 1H, CHCl), 3.30 (d, J = 7.9 Hz, 2H, alkyl CH); MS (ESI), m/z = 423 [M + H+]; IR (ATR): 3403 (NH), 1455 (CHCl) cm−1. Anal. (C27H20ClN3) Calc. C 76.86, H 4.78, N 9.96; Found C 76.58, H 4.65, N 9.94.
(1R/S, 3R/S)-2-Bromo-1,3-di(1H-indol-3-yl)-1,2,3,4-tetrahydrocyclopenta[b]indole 1c. Yield 0.215 g (23%); light-brownish powder; mp 130–132 °C; 1H NMR (acetone-D6) δ11.35 (s, 1H, NH), 10.91 (s, 1H, NH), 10.67 (s, 1H, NH), 7.56 (d, J = 7.7 Hz, 1H, aromat. H), 7.44 (d, J = 8.0 Hz, 3H, aromat. H), 7.42-7.35 (m, 2H, aromat. H), 7.28 (d, J = 7.5 Hz, 2H, aromat. H), 7.20 (“t”, J = 8.4 Hz, 3H, aromat. H), 7.03 (d, J = 7.2 Hz, 1H, aromat. H), 6.98-6.96 (m, 2H, aromat. H), 4.85 (t, J = 7.8 Hz, 1H, CHBr), 3.58 (d, J = 7.8 Hz, 2H, alkyl CH); MS (EI), m/z = 466 [M+]; IR (ATR): 3401 (NH), 1454 (CHBr) cm−1. Anal. (C27H20 BrN3) Calc. C 69.54, H 4.32, N 9.01; Found C 69.18, H 4.05, N 8.86.
(1R/S, 4R/S)-1,4-Di(1H-indol-3-yl)-2,3,4,9-tetrahydro-1H-carbazole 2a. Yield 0.683 g (85%); light-brownish powder; mp 139–145 °C; 1H NMR (DMSO-D6) δ10.88 (s, 1H, NH), 10.67 (s, 1H, NH), 10.42 (s, 1H, NH), 7.59 (d, J = 7.9 Hz, 1H, aromat. H), 7.45 (d, J = 7.9 Hz, 1H, aromat. H), 7.39-7.28 (m, 3H, aromat. H), 7.18 (“t”, J = 6.5 Hz, 1H, aromat. H), 7.05 (“t”, J = 7.0 Hz, 2H, aromat. H), 6.94-6.81 (m, 4H, aromat. H), 6.67-6.60 (m, 2H, aromat. H), 4.58-4.49 (m, 2H, alkyl CH), 2.23-2.05 (m, 4H, CH2); MS (ESI), m/z = 402 [M + H+]; IR (ATR): 3398 (NH), 2923 (CH2) cm−1. Anal. (C28H23N3) Calc. C 83.76, H 5.77, N 10.47; Found C 83.80, H 5.73, N 10.51.
(1R/S,4R/S)-6-Chloro-1,4-bis(5-chloro-1H-indol-3-yl)-2,3,4,9-tetrahydro-1H-carbazole 2b. Yield 0.777 g (77%); white powder; mp 210–214 °C; 1H NMR (DMSO-D6) δ11.16 (s, 1H, NH), 11.01 (s, 1H, NH), 10.75 (s, 1H, NH), 7.60-7.56 (m, 1H, aromat. H), 7.42-7.36 (m, 3H, aromat. H), 7.22 (d, J = 8.5 Hz, 1H, aromat. H), 7.16-7.15 (m, 1H, aromat. H), 7.07 (d, J = 8.7 Hz, 2H, aromat. H), 6.92 (dd, J = 8.7, 1.9 Hz, 2H, aromat. H), 6.84 (s, 1H, aromat. H), 4.54-4.50 (m, 2H, alkyl CH), 2.17-1.99 (m, 4H, CH2); MS (EI), m/z = 504 [M+]; IR (ATR): 3411 (NH), 3009 (CH2), 1689 (CCl) cm−1. Anal. (C28H20Cl3N3) Calc. C 66.62, H 3.99, N 8.32; Found C 66.45, H 3.75, N 8.17.
(1R/S,4R/S)-7-Chloro-1,4-bis(6-chloro-1H-indol-3-yl)-2,3,4,9-tetrahydro-1H-carbazole 2c. Yield 0.788 g (78%); light green powder; mp 154–158 °C; 1H NMR (acetone-D6) δ10.62 (s, 1H, NH), 10.19 (s, 1H, NH), 9.70 (s, 1H, NH), 8.10-7.96 (m, 1H, aromat. H), 7.51 (d, J = 8.6 Hz, 1H, aromat. H), 7.47-7.43 (m, 3H, aromat. H), 7.34-7.31 (m, 2H, aromat. H), 7.20 (“t”, J = 7.6 Hz, 1H, aromat. H), 7.00–6.98 (m, 2H, aromat. H), 6.97-6.87 (m, 1H, aromat. H), 2.69-2.43 (m, 2H, alkyl CH), 1.41-1.38 (m, 2H, CH2), 1.11-0.91 (m, 2H, CH2); MS (ESI), m/z = 503 [M - H+]; IR (ATR): 3418 (NH), 2923 (CH2), 1613 (CCl) cm−1. Anal. (C28H20Cl3N3) Calc. C 66.62, H 3.99, N 8.32; Found C 66.58, H 3.83, N 8.29.
(6R/S,10S/R)-6,10-Di(1H-indol-3-yl)-5,6,7,8,9,10-hexahydrocyclohepta[b]indole 3a. Yield 0.208 g (25%); brown powder; mp 152–155 °C; 1H NMR (DMSO-D6) δ10.97 (s, 1H, NH), 10.66 (s, 1H, NH), 9.66 (s, 1H, NH), 7.62 (d, J = 7.7 Hz, 1H, aromat. H), 7.43-7.33 (m, 4H, aromat. H), 7.19 (dd, J = 7.1, 2.3 Hz, 1H, aromat. H), 7.10-7.05 (m, 3H, aromat. H), 6.99-6.69 (m, 2H, aromat. H), 6.85 (“t”, J = 6.9 Hz, 1H, aromat. H), 6.74 (“t”, J = 6.9 Hz, 1H, aromat. H), 6.66 (dd, J = 7.5, 1.2 Hz, 1H, aromat. H), 4.91 (t, J = 8.5 Hz, 1H, alkyl CH), 4.66 (dd, J = 9.5, 3.10 Hz, 1H, alkyl CH), 2.19-2.09 (m, 4H, CH2), 1.74-1.62 (m, 2H, CH2); MS (ESI), m/z = 414 [M - H+]; IR (ATR): 3416 (NH), 2925, 2825 (CH2) cm−1. Anal. (C29H25N3) Calc. C 83.82, H 6.06, N 10.11; Found C 83.89, H 6.05, N 10.24.
(6R/S,10S/R)2-Chloro-6,10-bis(5-chloro-1H-indol-3-yl)-5,6,7,8,9,10-hexahydrocyclohepta[b]indole 3b. Yield 0.208 g (20%); brown powder; mp 130–133 °C; 1H NMR (CDCl3) δ8.27 (s, 1H, NH), 7.92 (s, 1H, NH), 7,23 (s, 1H, NH), 7.52 (s, 1H, aromat. H), 7.45-7.44 (m, 1H, aromat. H), 7.41-7.34 (m, 1H, aromat. H), 7.25-7.17 (m, 1H, aromat. H), 7.14-7.04 (m, 4H, aromat. H), 6.99-6.93 (m, 2H, aromat. H), 6.57 (s, 1H, aromat. H), 4.88 (t, J = 7.9 Hz, 1H, alkyl CH), 4.51–4.53 (m, 1H, alkyl CH), 2.70-2.61 (m, 2H, CH2), 2.21-2.11 (m, 2H, CH2), 1.96-1.72 (m, 2H, CH2); MS (ESI), m/z = 517 [M - H+]; IR (ATR): 3423 (NH), 2921, 1460 (CCl) cm−1. Anal. (C29H22Cl3N3) Calc. C 67.13, H 4.27, N 8.10; Found C 67.18, H 4.30, N 8.13.
(6R/S,10S/R)3-Chloro-6,10-bis(6-chloro-1H-indol-3-yl)-5,6,7,8,9,10-hexahydrocyclohepta[b]indole 3c. Yield 0.187 g (18%); brown powder; mp 107–110 °C; 1H NMR (CDCl3) δ8.25 (s, 1H, NH), 7.89 (s, 1H, NH), 7.62 (d, J = 8.5 Hz, 1H, aromat. H), 7,50 (s, 1H, NH), 7.44-7.37 (m 1H, aromat. H), 7.37-7.34 (m, 2H, aromat. H), 7.32-7.27 (m, 1H, aromat. H), 7.23 (d, J = 7.5 Hz, 1H, aromat. H), 7.16-7.11 (m 1H, aromat. H), 7.04–6.95 (m, 2H, aromat. H), 6.89 (dd, J = 8.5, 1.9 Hz, 1H, aromat. H), 6.53-6.52 (m, 1H, aromat. H), 4.93-4.91 (m, 1H, alkyl CH), 4.53 (dd, J = 11.3, 7.3 Hz, 1H, alkyl CH), 2.70-2.61 (m, 2H, CH2), 1.99-1.78 (m, 2H, CH2), 1.78-1.59 (m, 2H, CH2); MS (ESI), m/z = 517 [M - H+]; IR (ATR): 3432 (NH), 2919, 1464 (CCl) cm−1. Anal. (C29H22Cl3N3) Calc. C 67.13, H 4.27, N 8.10; Found C 67.05, H 4.14, N 8.03.
(6R/S,11S/R)-6,11-Di(1H-indol-3-yl)-6,7,8,9,10,11-hexahydro-5H-cycloocta[b]indole 4. Yield 0.232 g (27%); brown powder; mp 105–108 °C; 1H NMR (DMSO-D6) δ11.86 (s, 1H, NH), 10.85 (s, 1H, NH), 9.05 (s, 1H, NH), 7.65 (d, J = 7.8 Hz, 1H, aromat. H), 7.59-7.49 (m, 4H, aromat. H), 7.37 (d, J = 8.0 Hz, 1H, aromat. H), 7.32-7.18 (m, 4H, aromat. H), 7.18-7.15 (m, 1H, aromat. H), 7.07-7.00 (m, 2H, aromat. H), 6.95 (“t”, J = 7.1 Hz, 1H, aromat. H), 5.61-5.57 (m, 1H, alkyl CH), 4.61 (dd, J = 10.2, 2.1 Hz, 1H, alkyl CH), 2.07-1.98 (m, 6H, CH2), 1.95-1.86 (m, 2H, CH2); MS (ESI), m/z = 428 [M - H+]; IR (ATR): 3375 (NH), 2936 (CH2) cm−1. Anal. (C30H27N3) Calc. C 83.88, H 6.34, N 9.78; Found C 83.89, H 6.36, N 9.81.
Measurement of pyruvate kinase inhibition
MRSA PK was expressed in pET-28a (+) as recombinant protein in E. coli BL-21(DE3) and then purfied by using Ni-nitrilotriacetic acid (NTA) agarose (Quiagen, Inc., Germantown, MD). The PK activity was measured in a continuous assay coupled to lactate dehydrogenase (LDH) in which the change in absorbance at 340 nm owing to the oxidation of NADH was measured by using a benchmark Plus microplate spectrophotometer (Bio-Rad, Inc., Hercules, CA). The reaction mixture contained 60 mM Na+-HEPES (pH = 7.5), 5% glycerol, 67 mM KCl, 6.7 mM MgCl2, 0.24 mM NADH, 5.5 units LDH from rabbit muscle (Sigma Aldrich), 2 mM ADP, and 10 mM PEP in a total volume of 200 μL. Reaction was initiated by addition of the PK (15 nM). The PK activity proportional to the rate of the change at 340 nm was expressed as specific activity (μmol/min/mg) which is defined as the amount of PK which catalyzes the formation of 1 μmol of either product per minute. Inhibitors were dissolved in dimethyl sulfoxide (DMSO) with the final concentration of the solvent never exceeding 1% of the whole assay volume. IC50 values were calculated by curve fitting on a four-parameter dose response model with a variable slope using Graphpad Prism 5.0 (GraphPad Software, Inc., La Jolla, CA). The reactions were performed in triplicate at 30 °C for 5 min. Inhibition curves of respective compounds are shown in the available Supplementary material.
Antimicrobial activity determination of the compounds
The compounds and the standards were dissolved in 12.5% DMSO at concentrations of 200 μg/mL. Further dilutions of the compounds and standard drugs in the test medium were prepared at the required quantities of 400, 200, 100, 50, 25, 12.5, 6.25, 3.12, 1.56, and 0.78 μg/mL concentrations with Mueller-Hinton broth. The minimum inhibitory concentrations (MIC) were determined using the 2-fold serial dilution technique. All the compounds were tested for their in vitro growth inhibitory activity against Staphylococcus aureus (S. aureus) ATCC 25923 and MRSA standard ATCC 43300. ATCC strains of the microorganisms used in this study were obtained from the culture collection of the Refik Saydam Health Institution of Health Ministry, Ankara. The cultures were obtained from Mueller-Hinton broth (Difco, Becton Dickson Inc., Sparks, MD) for all the bacterial strains after 24 h of incubation at 37 ± 1 °C. Testing was carried out in Mueller-Hinton broth at pH 7.4 and the 2-fold serial dilution technique was applied. The final inoculum size was 105 CFU/mL. A set of tubes containing only inoculated broth was used as controls. After incubation for 24 h at 37 ± 1 °C, the last tube with no growth of microorganism was recorded to represent the MIC (expressed in micrograms per milliliter).
Results and discussion
Chemistry
Indoles preferably react with electrophils at the electron-rich 3-positionCitation13,Citation14. Thus, the indole reaction with an aldehyde primarily leads to a hydroxyalkyl addition product which may undergo addition to a second indole under acidic conditions after elimination of a water molecule. Such reactions lead to bisindolyl-alkanes by the use of various protic acids or lewis acids used as catalysts to promote the reactionCitation15–18.
We first investigated the reaction between indole and a dialdehyde under variation of the aldehyde alkyl chain length. The reaction was carried out under mild conditions using glacial acetic acid at room temperature.
The yielded reaction products have been the bisindolyl-cycloalkane indoles 1–4. The mechanistic discussion of their formation suggests an addition of one of the aldehyde functions to each one indole reactant at the preferred electron-rich 3-position giving a bisindolyl-alkane substructure.
The other aldehyde function will have reacted with a third indole unit at the 3-position. The resulting hydroxyalkane group will have been protonated accordingly. An addition at the 2-position of the neighbored indole will have proceeded to give the cycloalkane-indole substructure after a water elimination. In the case of the cyclopentane and cyclohexane derivatives 1a–c and 2a–c the stereochemistry of the indoyl-bearing cycloalkane CH ring atoms was the same being each (S/S) or (R/R), respectively, with both hydrogen atoms at the carbon atoms resulting in a signal for just one proton in the 1H NMR spectra. Consequently, both indole residues have a trans-orientation. In the case of the cycloheptane and cyclooctane derivatives the stereochemistry of each indolyl-bearing cycloalkane CH ring atom was different being either (S/R) or (R/S) with resulting in each one signal for the different hydrogen atoms at those cycloalkane ring atoms. That stereochemistry practically meant a cis-orientation of both attached indolyl residuesCitation19.
MRSA PK inhibition
PK has recently been discovered as an essential hub protein in the interactome of MRSACitation20,Citation21. PK plays a central role in the carbohydrate metabolism. It catalyzes the final rate-limiting step in the glycolysis which converts phospoenolpyruvate (PEP) to pyruvate under ATP formation from ADP in an irreversible processCitation22. Both metabolism products PEP as well as pyruvate are related to other biosynthetic pathways, so that a potential inhibition of PK may result in a hampered metabolism of MRSACitation12. So PK was identified as a hopeful target structure for perspective novel antibiotics. However, it remained uncertain whether such a PK inhibition may lead to an antibacterial activity of the respective PK inhibitor. First MRSA inhibitors have been bisindole alkaloid compounds from a marine natural libraryCitation12. Their antibacterial S. aureus activity was just moderate. That just moderate antibacterial activity reveals a discussed potential difference between enzyme inhibiting activity and antibacterial potencyCitation12.
The bisindolyl alkaloids show a structural similarity to our novel bisindolyl-cycloalkane indoles. The different heterocycles of those alkaloids with both indolyl substituents are practically replaced by our cycloalkane ring part with varying ring sizes. However, the attached indole part in our cycloalkane indoles increases this structural core.
We determined the PK-inhibiting activities of our unsubstituted cycloalkane indoles 1a, 2a, 3a, and 4 with an increasing size of the cycloalkane ring ().
Table 1. MRSA-pyruvate kinase inhibition as IC50 values of our target compounds 1a–c, 2a–c, 3a–c, and for 4.
The cyclohexane compound 2a showed the best activity with an IC50 value of 8.5 μM. The cycloheptane and the cyclooctane compounds 3a and 4 were less active and the cyclopentane compound 1a showed lowest activity with an IC50 value of 16.8 μM.
We then introduced halogen atoms into the cyclopentane scaffold. Such halogen atoms may show halogen bond donator functions and thus may potentially increase the inhibitory activity. The corresponding chloro- and bromo-substituted compounds 1b and 1c were easily accessible because their aldehyde precursors were commercially available.
The chloro substitution in derivative 1b led to increases in the PK-inhibiting activities with an IC50 value of 6.8 μM thus being lower than that IC50 value of the cyclohexane compound 2a as best one of the unsubstituted compound series.
The bromo substitution in derivative 1c led to best PK-inhibiting activities within the cyclopentane compounds thus suggesting a role in a potential halogen bonding.
We then introduced chloro substituents in the indole part of our compounds with varying positions in both the 5- and the 6-position of the indole scaffold. Such an indole substitution in one of the reported bisindolyl alkaloids in the 6-position led to a complete loss of activity of the respective derivativeCitation12. However, the substituent in that case was a hydroxyl group with a certain hydrogen bond donator function.
The 5-chloroindol substitution of cyclohexane compound 2a with best activity of the unsubstituted series led to main decreases in the PK inhibitory activity for 2b with an IC50 value of merely 50 μM. The position-varied 6-chloroindolyl compound 2c showed no more activity against PK.
These results may be comparable with those reported for the inactive 6-hydroxyindolyl substituted alkaloid compound.
We then introduced the halogen function into the cycloheptane compound 3a which was the next best inhibitor of the unsubstituted series. The corresponding 5-chloroindolyl compound 3b surprisingly showed improved PK-inhibiting activities with an IC50 value of 2.6 μM. The position-varied 6-chloroindolyl compound 3c showed the best PK-inhibiting activity within this cycloheptane compound series.
So we can conclude that the cycloalkane ring size as well as various substitution patterns play important roles in the inhibiting activities of our discovered novel class of PK inhibitors.
Furthermore, we determined the mode of action of our best compound 3c toward PK. The reported bisindolyl alkaloids as PK inhibitors were demonstrated to act as allosteric inhibitors of PK by blocking a hydrophobic pocket at the interface of two PK monomers of the tetrameric kinase structureCitation12. So we carried out enzyme kinetic studies with our compound 3c to analyzes whether the mode of enzymatic action is also a noncompetitive one similar to the reported bisindolyl alkaloids (
Figure 2. Inhibition curves of the MRSA-pyruvate kinase for respective inhibitor concentrations of compound 3c upon phosphoenolpyruvate (PEP) initiation.
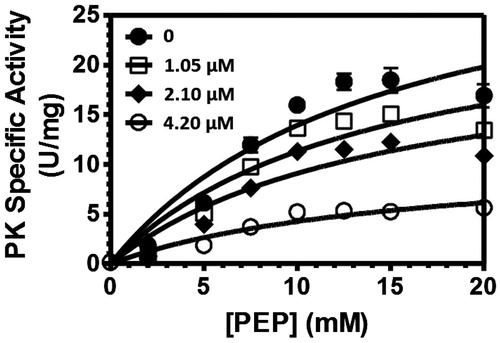
We determined the velocity of the PEP cleavage using increasing inhibitor concentrations of our compound 3c from 1.05 μM to 4.2 μM. We found a lowered velocity vmax of the compound cleavage under the increasing inhibitor concentrations with a vmax value of 34.33 μmol/min/mg for the uninhibited reaction to a vmax value of 11.04 μmol/min/mg for the inhibited reaction at the 4.2 μM concentration of compound 3c. The determined Michaelis Menten kinetics of the reaction remained nearly unchanged with values of 14.64 mM for the uninhibited reaction to 15.90 mM for the inhibited reaction at the 4.2 μM inhibitor concentration. These results proved a noncompetitive binding mode of our compound similar to the reported bisindolyl alkaloids.
Antibacterial activity
Best PK-inhibiting compounds have been further evaluated as inhibitors of both S. aureus and MRSA. In the antibacterial testing the MIC value was determined as the minimal inhibitory concentration which obviously stopped the bacterial growth. We used ampicillin as control antibiotic agent ().
Table 2. Antibacterial inhibition data of target compounds 1b and c, 2a, 3b, and c determined as minimal inhibitory concentration (MIC) values for total growth inhibition.
The most active PK-inhibiting cyclohexane compound 2a showed excellent antimicrobial activities with MIC values of 3.125 μg/mL against both S. aureus and MRSA if compared to the ampicillin standard with a MIC value of 50 μg/mL against MRSA.
The halogen substituted cyclopentane derivatives 1b and 1c as best ones of the cyclopentane series also showed good antimicrobial activities against S. aureus and MRSA with both MIC values making 3.125 μg/mL.
Finally, the chloro-substituted cycloheptane derivatives 3b and 3c were evaluated. Both compounds showed antibacterial activities with MIC values of 3.125 μg/mL against S. aureus and of 6.25 μg/mL against MRSA.
So it can be summarized that all compounds Mapping the protein interaction network in methicillin-resistant Staphylococcus aureus.with highest activities as PK inhibitors showed antibacterial activities against both S. aureus variants. The antibacterial activities were better than those reported for the first bisindolyl-alkaloid compounds. Thus, a first class of novel PK inhibitors was identified which gives perspectives for further developments as effective antimicrobial agents against both S. aureus and MRSA.
Supplementary material available online
IENZ_1118685_Supplementary_Material.pdf
Download PDF (97.2 KB)Declaration of interest
The authors acknowledge the financial support of their work by the Egyptian Government with a scholarship to Mardia Telep El-Sayed.
Related Research Data
References
- Kohanski MA, Dwyer DJ, Collins JJ. How antibiotics kill bacteria: from targets to networks. Nat Rev Microbiol 2010;8:423–35
- Floss HG, Yu TW. Rifamycin – mode of action, resistance, and biosynthesis. Chem Rev 2005;105:621
- Tomasz A. The mechanism of the irreversible antimicrobial effects of penicillins: how the beta-lactam antibiotics kill and lyse bacteria. Annu Rev Microbiol 1979;33:113–37
- Vakulenko SB, Mobashery S. Versatility of aminoglycosides and prospects for their future. Clin Microbiol Rev 2003;16:430–50
- Otto M. Methicillin-resistant Staphylococcus aureus infection is associated with increased mortality. Future Microbiol 2012;7:189–91
- Chalfine A, Kitzis M-D, Bezie Y, et al. Ten-year decrease of acquired methicillin-resistant Staphylococcus aureus (MRSA) bacteremia at a single institution: the result of a multifaceted program combining cross-transmission prevention and antimicrobial stewardship. Antimicrob Resist Infect Control 2012;1:18
- Deleo FR, Otto M, Kreiswirth BN, Chambers HF. Community-associated meticillin-resistant Staphylococcus aureus. Lancet 2010;375:1557–68
- Meir S, Weber R, Zbinden R, et al. Extended-spectrum β-lactamase-producing Gram-negative pathogens in community-acquired urinary tract infections: an increasing challenge for antimicrobial therapy. Infection 2011;39:333–40
- Kumarasamy KK, Toleman MA, Walsh TR. Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: a molecular, biological, and epidemiological study. Lancet Infect Dis 2010;10:597–602
- Motomasa K, Shunji A, Gato K, et al. Marine natural products. XXXIV. Trisindoline, a new antibiotic indole trimer, produced by a bacterium of Vibrio sp. separated from the marine sponge Hyrtios altum. Chem Pharm Bull 1994;42:2449–51
- Oka R, Wagner-Döbler V, Laatsch IIH. New indole alkaloids from the North Sea bacterium Vibrio parahaemolyticus Bio249. J Nat Prod 2003;66:1520–3
- Zoraghi R, Worrall L, See RH, et al. Methicillin-resistant Staphylococcus aureus (MRSA) pyruvate kinase as a target for bis-indole alkaloids with antibacterial activities. J Biol Chem 2011;286:44716–25
- Raju BC, Rao JM. An easy and efficient synthesis of bisindolylmethanes and tetraindolylmethane Tröger's base catalyzed by AgBF4. Ind J Chem 2008;47B:623
- Sujatha K, Perumal PT, Muralidharan D, Rajendran M. Synthesis and biological evaluation of some novel heterocyclic derivatives of paracetamole as potential analgesic and antipyretic agents. Ind J Che 2009;48B:267
- Pore D-M, Desai U-V, Thopate TS, Wadgaonkar P-P. A mild, expedient, solventless synthesis of bis(indolyl)alkanes using silica sulphuric acid as a reusable catalyst. Arkivoc 2006;12–75
- Karthik M, Tripathi AK, Gupta NM, et al. Zeolite catalyzed electrophilic substitution reaction of indoles with aldehydes: synthesis of bis(indolyl)methanes. Catal Commun 2004;5:371–5
- Kidwai M, Bura N, Mishra NK. Nioboum(V) pentachloride-catalyzed efficient and highly rapid synthesis of bis(indolyl)-methanes under mild conditions. Ind J Chem 2011;50B:229
- Ke BW, Qin Y, Wang Y, Wang FP. Synth Commun 2005;35:1209–12
- The compound stereochemistry has been additionally proved by X-ray structure analysis of trisacetylated derivative 2a with published crystallographic data under CCDC-939747 which can be obtained free of charge via http://www.ccdc.cam.ac.uk/data_request/cif
- Cherkasov A, Hsing M, Zoraghi R, et al. Mapping the protein interaction network in methicillin-resistant Staphylococcus aureus. J Proteome Res 2011;10:1139–50
- Zoraghi R, See RH, Axerio-Cilies P, et al. Identification of pyruvate kinase in methicillin-resistant Staphylococcus aureus as a novel antimicrobial drug target. Antimicrob Agents Chemother 2011;55:2042–53
- Valentini G, Chiarelli L, Fortin R, et al. The allosteric regulation of pyruvate kinase. J Biol Chem 2000;275:18145