
Abstract
A set of 1,2,4-triazine derivatives were designed as cyclooxygenase-2 (COX-2) inhibitors. These compounds were synthesized and screened for inhibition of cyclooxygenases (COX-1 and COX-2) based on a cellular assay using human whole blood (HWB) and lipoxygenase (LOX-15) that are key enzymes in inflammation. The results showed that 3-(2-(benzo[d][1,3]dioxol-5-ylmethylene)hydrazinyl)-5,6-bis(4-methoxyphenyl)-1,2,4-triazine (G11) was identified as the most potent COX-2 inhibitor (78%) relative to COX-1 (50%). Ferric reducing anti-oxidant power (FRAP) assay revealed that compound G10 possesses the highest anti-oxidant activity. The compound G3 with IC50 value of 124 μM was the most potent compound in LOX inhibitory assay. Molecular docking was performed and a good agreement was observed between computational and experimental results.
Introduction
Inflammation, a process whereby a huge number of mediators are released due to over-activation of different cascades, is associated with many diseases, such as rheumatoid arthritis (RA), osteoarthritis (OA), bronchospasm and psoriasisCitation1. Enzymatic action of phospholipases A2 (PLA2s) on phospholipid bilayer results in arachidonic acid release, which is a precursor in the production of eicosanoids including prostaglandins (PGs) and leukotrienes (LTs)Citation2. The transformation of arachidonic acid to PGs is mediated by cyclooxygenase (COX) enzyme family (COX-1, COX-2 and COX-3). The most important isoforms are COX-1 and COX-2, whereas, COX-3 is expressed in the brain and its biological role is still unknown.
COX-1 is the constitutive form of the enzyme and is present in the stomach, intestines, kidneys and platelets which causes gastrointestinal cytoprotection as well as renal effects. On the other hand, COX-2 is constitutively expressed only in low amounts in kidney, brain and ovaries, but its expression is considerably enhanced during inflammatory situations or mitogenic stimulationCitation3,Citation4.
Non-steroidal anti-inflammatory drugs (NSAIDs) are amongst the most widely used drugs worldwide. Traditional NSAIDs (classical NSAIDs or nonselective COX inhibitors), such as ibuprofen, indomethacin and naproxen inhibit both COX-1 and COX-2. Several serious side effects, such as gastrointestinal irritation and hemorrhage and kidney failure are caused by these agentsCitation5.
The discovery of molecules that selectively inhibit COX-2, named coxibs (celecoxib, rofecoxib and valdecoxib) has been a response to the necessity of lowering these side effects. Indeed, the selectivity of this class of drugs resulted in improved gastrointestinal tolerance. Consecutive clinical studies revealed that long term use of several coxibs is accompanied by increased risk level of cardiovascular problems, such as myocardial infarction, stroke, systemic and pulmonary hypertension, and congestive heart failureCitation6. Hence, finding effective and safe anti-inflammatory drugs seems to be an urgent necessity.
In previous investigations, we have reported some new compounds as anti-inflammatory agents. As a part of our ongoing effort to develop effective candidates for inhibition of inflammation, a set of 5,6-diaryl-1,2,4-triazine derivatives were designedCitation7.
In contrast with the traditional NSAIDs which belong to different categories of chemical structures, selective COX-2 inhibitors can be classified into three groups (): (1) diaryl ethers with methanesulfonamide at position-2 of one ring, Nemisulide and CGP-28237 can be considered as lead compounds in this classCitation8; (2) trans-stilbenoids, such as resveratrol, a natural product found in grapes and other food productsCitation9; (3) heterocyclic systems (such as 1,2,4 triazoleCitation10,Citation11, imidazoleCitation12, pyrazolCitation13, quinolineCitation14, indolCitation15, pyrimidineCitation16, 1,2,4-triazineCitation17,Citation18) with adjacent diaryl containing sulfamoyl (SO2NH2) or methylsulfonyl (SO2Me) substitution at position-4 of one ring which exist in celecoxib, SC-558 and etoricoxib.
Figure 1. Different classes of selective COX-2 inhibitors.
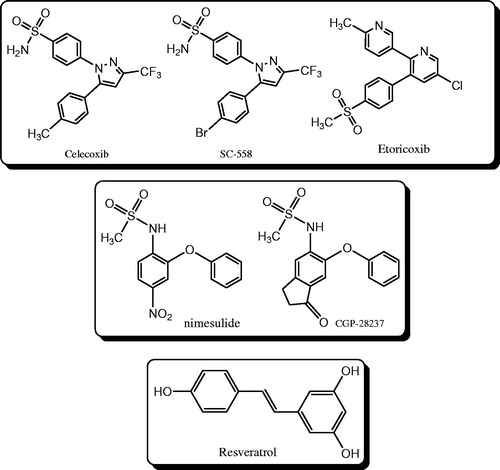
The compounds bearing 1,2,4-triazine core have been identified to exhibit a broad spectrum of biological effects, such as antifungalCitation19, anti-HIVCitation20, adenosine A2A receptor antagonistCitation21, kinase inhibitor antitumorCitation22,Citation23, analgesicCitation17, anti-anxiety and anti-inflammation effectsCitation24.
As a part of our research program toward the search of potent COX-2 inhibitors with lower side effects, improved affinity and efficacy and dual modes of action, molecular hybridization strategy was employed to design 1,2,4-triazine derivatives. After the synthesis of compounds, in vitro assays and molecular docking study were also performed.
Experimental protocols
Chemistry
NMR spectra were recorded on Brucker 500 spectrometer or Varian unity 400 spectrometer relative to tetramethylsilane (TMS) as an internal standard using CDCl3 or dimethyl sulfoxide (DMSO)-d6 as the solvents. The mass spectra were obtained on an Agilent 6410 instrument. The IR spectra were recorded on a FT-IR Perkin-Elmer spectrophotometer (KBr disks). Melting points were determined on a Kofler hot stage apparatus and are uncorrected. Elemental analyses were performed on a CHN–O– rapid elemental analyzer (Heraeus, Hanau, Germany).
General procedure
All 3-(2-arylidenehydrazinyl) 5,6-bis(4-methoxyphenyl)-1,2,4-triazine derivatives were prepared according to the procedure describedCitation7.
3-(2-Benzylidenehydrazinyl)-5,6-bis(4-methoxyphenyl)1,2,4-triazine (G1)Citation7
Compound G1 was prepared by the described procedure using 106 mg (1 mmol) of benzaldehyde (yield 83%), m.p.: 245–247 °C; 1H NMR (DMSO-d6, 500 MHz), δH: 10.29 (s, 1H, NH), 8.24 (s, 1H, N=CH), 7.81 (d, J = 7.1 Hz, 2H, ArH), 7.66 (d, J = 8.8 Hz, 2H, ArH), 7.50 (d, J = 8.7 Hz, 2H, ArH), 7.43–7.38 (m, 3H, ArH), 6.93 (d, J = 8.7 Hz, 2H, ArH), 6.88 (d, J = 8.7 Hz, 2H, ArH), 3.86 ppm (s, 6H, 2 × O–CH3H); IR (KBr, ν): 3440, 3211, 1605, 1571, 831 cm−1; MS (EI) m/z (%): 411.2 (M+, 29), 334 (20), 308 (14), 238 (100), 223 (49), 195 (12), 152 (11). Anal. Calcd. for C24H21N5O: C, 70.06; H, 5.14; N, 17.02; found: C, 70.21; H, 4.86; N, 16.95.
3-(2-(4-Nitrobenzylidene)hydrazinyl) 5,6-bis(4-methoxyphenyl)-1,2,4-triazine (G2)Citation7
The titled compound was synthesized by the described procedure from 4-nitrobenzaldehyde (151 mg, 1 mmol) as yellow solid (yield 88%), m.p.: 265–267 °C; 1H NMR (DMSO-d6, 500 MHz), δH: 12.23 (s, 1H, NH), 8.36 (s, 1H, N=CH), 8.31 (d, J = 8.6 Hz, 2H, ArH), 7.99 (d, J = 8.0 Hz, 2H, ArH), 7.52 (d, J = 8.3 Hz, 2H, ArH), 7.39 (d, J = 8.6 Hz, 2H, ArH), 6.98–6.96 (m, 4H, ArH), 3.79 ppm (s, 6H, 2 × O–CH3); IR (KBr, ν): 3421, 3205, 1607, 1585, 838 cm−1; MS (EI) m/z (%): 456 (M+, 21), 308 (16), 238 (100), 223 (25), 195(11). Anal. Calcd. for C24H20N6: C, 63.15; H, 4.42; N, 18.41; found: C, 62.78; H, 4.12; N, 18.51.
3-(2-(4-Fluorobenzylidene)hydrazinyl)-5,6-bis(4methoxyphenyl)-1,2,4-triazine (G3)Citation7
The titled compound G3 was synthesized by the general procedure using 4-fluorobenzaldehyde (124 mg, 1 mmol) (yield 72%), m.p.: 257–259 °C; 1H NMR (DMSO-d6, 500 MHz), δH: 11.64 (s, 1H, NH), 8.25 (s, 1H, N=CH), 8.06–8.02 (m, 2H, ArH), 7.49 (d, J = 8.5 Hz, 2H, ArH), 7.35 (d, J = 8.5 Hz, 2H, ArH), 7.30–7.26 (m, 2H, ArH), 6.96–6.93 (m, 4H, ArH), 3.78 ppm (s, 6H, 2 × O–CH3); 13C NMR (DMSO-d6, 125 MHz), δC = 163.39, 161.45, 160.99, 159.39, 158.21, 155.45, 150.64, 141.87, 137.48, 131.19, 130.86, 130.80, 130.21, 128.63, 127.87, 122.86, 116.03, 115.86, 113.83, 112.42, 112.24, 55.27, 55.11 ppm; IR (KBr, ν): 3444, 3223, 2913, 1607, 1589, 1514, 1256, 836 cm−1; MS (EI) m/z (%): 429 (M, 36), 334 (29), 308 (10), 238 (100), 223 (53), 195 (12), 152 (11). Anal. Calcd. for C24H20FN5O2: C, 67.12; H, 4.69; N, 16.31; found: C, 66.85; H, 4.81; N, 15.98.
5,6-Bis(4-methoxyphenyl)-3-(2-(thiophen-2-ylmethylene)hydrazinyl)-1,2,4-triazine (G4)Citation7
The yellow solid product G4 was prepared from thiophene-2-carbaldehyde (112 mg, 1 mmol) in similar manner described in general procedure (yield: 91%), m.p.: 250–253 °C; 1H NMR (DMSO-d6, 500 MHz), δH: 11.79 (s, 1H, NH), 8.45 (s, 1H, N=CH), 7.61 (s, 1H, ArH), 7.44–7.46 (m, 2H, ArH), 7.39–7.35 (m, 3H, ArH), 7.12 (brs, 1H, ArH), 6.96–6.95 (m, 4H, ArH), 3.78 ppm (s, 6H, 2 × O–CH3); 13C NMR (DMSO-d6, 125 MHz), δC = 160.94, 159.34, 158.03, 155.37, 151.23, 150.33, 139.65, 138.66, 131.13, 130.17, 129.28, 128.67, 127.90, 127.76, 55.27, 55.10 ppm. IR (KBr, ν): 3350, 3220, 1608, 1514, 1360, 831 cm−1; MS (EI) m/z (%): 417(M+, 61), 308 (10), 283 (10), 252 (11), 238 (100), 223 (57), 195(20), 180 (13), 152 (23), 39 (16). Anal. Calcd. for C22H19N5O4S: C, 66.23; H, 4.59; N, 16.78; found: C, 65.94; H, 4.74; N, 16.94.
3-(2-(4-Hydroxy-3-methoxybenzylidene)hydrazinyl)-5,6-bis(4-mthoxyphenyl)-1,2,4-triazine (G5)Citation7
Compound G5 was obtained according to the described general procedure using 4-hydroxy-3-methoxybenzaldehyde (152 mg, 1 mmol) as yellow solid (yield 88%): m.p.: 245–248 °C; 1H NMR (DMSO-d6, 500 MHz), δH: 11.62 (s, 1H, NH), 9.46 (s, 1H, N=CH), 8.15 (s, 1H, ArOH), 7.49 (d, J = 8.5 Hz, 2H, ArH), 7.36 (d, J = 8.5 Hz, 2H, ArH), 7.31 (s, 1H, ArH), 7.11 (d, J = 8.1 Hz, 1H, ArH), 6.96–6.94 (m, 4H, ArH), 6.84 (d, J = 8.1 Hz, 1H, ArH), 3.84 (s, 3H, OCH3), 3.78 ppm (s, 6H, 2 × O–CH3); 13C NMR (DMSO-d6, 125 MHz), δC = 161.48, 159.29, 158.22, 155.28, 149.99, 148.27, 147.91, 144.08, 131.12, 130.93, 130.72, 130.14, 129.16, 128.78, 128.01, 126.29, 120.87, 115.51, 113.79, 113.70, 113.57, 109.18, 55.50, 55.25, 55.09 ppm; IR (KBr, ν): 3485, 3225, 1607, 1508, 1257, 843 cm−1; MS (EI) m/z (%): 417 (M+, 61), 323 (10), 308 (37), 252 (14), 238 (100), 223 (53), 195 (14), 152 (12). Anal. Calcd. for C25H23N5O4: C, 65.63; H, 5.07; N, 15.31; found: C, 65.93; H, 5.36; N, 15.51.
3-(2-(2,4-Dichlorobenzylidene)hydrazinyl)-5,6-bis(4-methoxyphenyl)-1,2,4-triazine (G6)Citation7
The titled compound G6 was synthesized according to the general procedure using 2,4-dichlorobenzaldehyde (175 mg, 1 mmol) as pale yellow solid (yield 78%), m.p.: 248–251 °C; 1H NMR (DMSO-d6, 500 MHz), δH: 12.14 (s, 1H, NH), 8.61 (s, 1H, N=CH), 8.06 (d, J = 8.6 Hz, 1H, ArH), 7.72 (d, J = 2.0 Hz, 1H, ArH), 7.53 (dd, J = 2.0 Hz, J = 8.5 Hz, 1H, ArH), 7.50 (d, J = 8.7 Hz, 2H, ArH), 7.38 (d, J = 8.7 Hz, 2H, ArH), 6.97–6.95 (m, 4H, ArH), 3.79 ppm (s, 6H, 2 × O–CH3); IR (KBr, ν): 3444, 3183, 1607, 1511, 1369.82 (M+2, 5), 334 (16), 308 (15), 238 (100), 223 (45), 195 (13), 152 (10). Anal. Calcd. for C24H19Cl2N5O2: C, 60.01; H, 3.99; N, 14.58; found: C, 59.84; H, 4.16; N, 14.41.
5,6-Bis(4-methoxyphenyl)-3-(2-((5-nitrofuran-2-yl)methylene)hydrazinyl)-1,2,4-triazine (G7)
The titled compound G7 was synthesized according to the general procedure using 5-nitrofuran-2-carbaldehyde (141 mg, 1 mmol) as a pale yellow solid (yield 78%), m.p.: 248–251 °C 1H NMR (DMSO-d6, 500 MHz), δH: 12.34 (s, 1H, NH), 8.74 (s, 1H, N=CH), 7.82 (d, J = 3.9 Hz, 1H, ArH), 7.49–7.47 (m, 2H, ArH), 7.39–7.37 (m, 2H, ArH), 7.21 (d, J = 3.9 Hz, 1H, ArH), 6.97–6.95 (m, 4H, ArH), 3.79 ppm (s, 6H, 2 × O–CH3) IR (KBr, ν): 3443, 2839, 1608, 1514, 1353, 1254, 1071, 834 cm−1 MS (EI) m/z (%): 446 (M+, 17), 400 (100), 267 (24), 238 (79), 223 (86), 195 (23), 180 (22), 152 (21), 134 (17), 119 (16), 79 (12). Anal. Calcd. for C22H18N6O5: C, 59.19; H, 4.06; N, 18.83; found: C, 59.64; H, 3.94; N, 18.11.
5,6-Bis(4-methoxyphenyl)-3-(2-[(1H-indol-3-yl)methylene]hydrazinyl)-1,2,4-triazine (G8)Citation7
The titled compound G8 was synthesized according to the general described procedure using 1H-indole-3-carbaldehyde (145 mg, 1 mmol) as yellow solid (yield: 74%), m.p.: 258–262 °C; 1H NMR (DMSO-d6, 500 MHz), δH: 11.48 (s, 1H, NH), 8.51 (d, J = 7.2 Hz, 1H, ArH), 8.45 (s, 1H, N=CH), 7.77 (brs, 1H, NH), 7.56 (d, J = 7.7 Hz, 2H, ArH), 7.44 (d, J = 7.7 Hz, 2H, ArH), 7.39 (d, J = 8.4 Hz, 2H, ArH), 7.21–7.18 (m, 2H, ArH), 6.99–6.96 (m, 4H, ArH), 3.80 ppm (s, 6H, 2 × O–CH3); 13C NMR (DMSO-d6, 125 MHz), δC = 160.95, 159.24, 158.44, 155.01, 149.39, 140.71, 136.98, 131.82, 131.12, 130.13, 129.01, 128.92, 128.26, 124.44, 122.50, 122.32, 120.23, 113.83, 113.74, 112.35, 111.68, 55.32, 55.12 ppm; IR (KBr, ν): 3414, 3010, 1609, 1510, 1249, 1065, 839 cm−1; MS (EI) m/z (%): 450 (M+, 4), 308 (14), 238 (100), 223(47), 195 (15), 142 (18), 116 (16). Anal. Calcd. for C26H22N6O2: C, 69.32; H, 4.92; N, 18.66; found: C, 68.97; H, 4.76; N, 18.34.
3-(2-(2-Chloro-6-nitrobenzylidene)hydrazinyl)-5,6-bis(4-methoxyphenyl)-1,2,4-triazine (G9)Citation7
The yellow solid product G9 was prepared from 2-chloro-6-nitrobenzaldehyde (185 mg, 1 mmol) according to the general procedure (yield: 71%), m.p.: 258–262 °C; 1H NMR (DMSO-d6, 500 MHz), δH: 12.33 (s, 1H, NH), 8.68 (s, 1H, N=CH), 8.14–8.13(m, 2H, ArH), 7.70 (dd, J = 2.45 Hz, J = 8.8 Hz, 1H, ArH), 7.51 (d, J = 8.8 Hz, 2H, ArH), 7.39 (d, J = 8.7 Hz, 2H, ArH), 6.98–6.96 (m, 4H, ArH), 3.79 ppm (s, 6H, 2 × O–CH3); IR (KBr, ν): 3424, 3121, 1602, 1519, 1125, 831 cm−1; MS (EI) m/z (%): 490 (M+, 21), 492 (M+2, 6), 334 (16), 308 (15), 238 (100), 223 (45), 195 (13), 152(10). Anal. Calcd. for C24H19ClN6O4: C, 58.72; H, 3.90; N, 17.12; found: C, 58.97; H, 3.72; N, 17.16.
3-(2-(1,3-Dihydroxybenzylidene)hydrazinyl)-5,6-bis(4-methoxyphenyl)-1,2,4-triazine (G10)Citation7
The titled compound G10 was synthesized according to the general procedure from 2,4-dihydroxybenzaldehyde (138 mg, 1 mmol) as yellow solid (yield: 86%), m.p.: 262–266 °C; 1H NMR (DMSO-d6, 500 MHz), δH: 11.93 (s, 1H, NH), 11.71 (s, 1H, ArOH), 9.89 (s, 1H, ArOH) 8.31 (s, 1H, N=CH), 7.77 (s, 1H, ArH), 7.50 (d, J = 8.7 Hz, 2H, ArH), 7.38 (d, J = 8.7 Hz, 2H, ArH), 7.25 (d, J = 8.4 Hz, 1H, ArH), 6.97–6.95 (m, 4H, ArH), 6.37–6.35 (dd, J = 2.2 Hz, J = 8.3 Hz, 1H, ArH), 3.79 ppm (s, 6H, 2 × O–CH3); IR (KBr, ν): 3352, 2837, 1608, 1517, 1253, 834 cm−1; MS (m/z, %): 443 (M+, 22), 308 (11), 238 (100), 195 (11). Anal. Calcd. for C24H21N5O4: C, 65.01; H, 4.77; N, 15.79; found: C, 64.91; H, 4.83; N, 16.01.
3-(2-(Benzo[d][1,3]dioxol-5-ylmethylene)hydrazinyl)-5,6-bis(4-methoxyphenyl)-1,2,4-triazine (G11)Citation7
The titled compound G11 was synthesized by the described general procedure using 1,3-benzodioxole-5-carbaldehyde (piperonal) (150 mg, 1 mmol) (yield 88%), m.p.: 268–272 °C; 1H NMR (DMSO-d6, 400 MHz), δH: 11.62 (s, 1H, NH), 8.16 (s, 1H, N=CH), 7.49 (d, J = 7.6 Hz, 2H, ArH), 7.40–7.34 (m, 2H, ArH), 7.31 (s, 1H, ArH), 14 (d, J = 7.6 Hz, 2H, ArH), 6.98–6.94 (m, 4H, ArH), 6.08 (s, 2H, ArH), 3.78 ppm (s, 6H, 2 × O–CH3); IR (KBr, ν): 3429, 2962, 1606, 1515, 1175, 836 cm−1; MS (EI) m/z (%): 455 (M+, 18), 334 (10), 308 (16), 238 (100), 223 (38), 195 (11). Anal. Calcd. for C25H21N5O4: C, 65.93; H, 4.65; N, 15.38; found: C, 65.71; H, 5.01; N, 15.65.
3-(2-(Furan-2-ylmethylene)hydrazinyl)-5,6-bis(4-methoxyphenyl)-1,2,4-triazine (G12)Citation7
The yellow solid product G12 was prepared from furan-2-carbaldehyde (96 mg, 1 mmol) according to the general procedure (yield: 71%), m.p.: 258–262 °C; 1H NMR (DMSO-d6, 500 MHz), δH: 11.79 (s, 1H, NH), 8.15 (s, 1H, N=CH), 7.82 (s, 1H, ArH), 7.47 (d, J = 8.6 Hz, 2H, ArH), 7.36 (d, J = 8.6 Hz, 2H, ArH), 6.96–6.94 (m, 4H, ArH), 6.85 (d, J = 3.2, 1H, ArH), 6.63 (brs, 1H, ArH), 3.78 ppm (s, 6H, 2 × O–CH3); 13C NMR (DMSO-d6, 125 MHz), δC = 161.57, 161.42, 159.99, 158.72, 156.17, 151.82, 150.92, 150.33, 145.73, 144.97, 134.10, 131.66, 130.71, 129.16, 128.44, 116.15, 114.30, 114.22, 112.57, 55.77, 55.61 ppm; IR (KBr, ν): 3443, 2839, 1608, 1254, 834 cm−1; MS (EI) m/z (%): 401 (M+, 66), 372 (23), 308 (15), 267 (47), 252 (14), 238 (100), 223 (89), 195 (22), 152 (21), 135 (37). Anal. Calcd. for C22H19N5O3: C, 65.83; H, 4.77; N, 17.45; found: C, 65.51; H, 5.03; N, 17.12.
3-(2-(4-Methoxybenzylidene)hydrazinyl)-5,6-bis(4-methoxyphenyl)-1,2,4-triazine (G13)Citation7
The titled compound G13 was synthesized according to the general procedure from 4-methoxybenzaldehyde (136 mg, 1 mmol) as yellow solid (yield: 91%), m.p.: 254–256 °C; 1H NMR (DMSO-d6, 500 MHz), δH: 11.72 (s, 1H, NH), 8.21 (s, 1H, N=CH), 7.67 (d, J = 8.0 Hz, 2H, ArH), 7.49 (d, J = 8.0 Hz, 2H, ArH), 7.36 (d, J = 8.0 Hz, 2H, ArH), 7.02–6.94 (m, 6H, ArH), 3.80 (s, 3H, OCH3), 3.78 ppm (s, 6H, 2 × O–CH3); IR (KBr, ν): 3444, 3215, 1608, 1577, 1248, 834 cm−1; MS (EI) m/z (%): 441 (M+, 14), 308 (31), 238 (100), 223 (49), 195 (12), 152 (10). Anal. Calcd. for C25H23N5O3: C, 68.01; H, 5.25; N, 15.86; found: C, 68.28; H, 4.93; N, 15.67.
5,6-Bis(4-methoxyphenyl)-3-(2-(pyridin-4-ylmethylene)-hydrazinyl)-1,2,4-triazine (G14)Citation7
The yellow solid product G14 was prepared from pyridine-4-carbaldehyde (107 mg, 1 mmol) according to the general procedure (yield: 81%), m.p.: 255–258 °C; 1H NMR (DMSO-d6, 500 MHz), δH: 12.17 (s, 1H, NH), 8.63 (d, J = 4.4 Hz, 2H, ArH), 8.23 (s, 1H, N=CH), 7.67 (d, J = 5.7 Hz, 2H, ArH), 7.52 (d, J = 8.8 Hz, 2H, ArH), 7.39 (d, J = 8.6 Hz, 2H, ArH), 6.98–6.96 (m, 4H, ArH), 3.79 ppm (s, 6H, 2 × O–CH3); IR (KBr, ν): 3434, 3146, 1609, 1569, 1507, 1237, 841 cm−1; MS (EI) m/z (%): 412 (M+, 32), 334 (16), 308 (12), 238 (100), 223 (25), 195 (10). Anal. Calcd. for C23H20N6O2: C, 66.98; H, 4.89; N, 20.38; found: C, 66.75; H, 5.71;N, 19.93.
3-(2-(4-Bromobenzylidene)hydrazinyl)-5,6-bis(4-methoxyphenyl)-1,2,4-triazine (G15)
The yellow solid product G15 was prepared from 4-bromobenzaldehyde (185 mg, 1 mmol) according to the general procedure (yield: 82%), m.p.: 254–256 °C. 1H NMR (DMSO-d6, 500 MHz), δH: 11.92 (s, 1H, NH), 8.24 (s, 1H, N=CH), 7.69–7.64 (m, 4H, ArH), 7.50 (d, J = 8.2, 2H, ArH), 7.37 (d, J = 8.2 Hz, 2H, ArH), 6.97–6.95 (m, 4H, ArH), 3.79 ppm (s, 6H, 2 × O–CH3). IR (KBr, ν): 3422, 3223, 2907, 1607, 1592, 1510, 1254, 837 cm−1; MS (m/z, %): 489.1 (M+, 11.5), 491.1 (M+2, 11.4). Anal. Calcd. for C24H20BrN5O2: C, 58.79; H, 4.11; N, 14.28; found: C, 59.35; H, 4.91; N, 13.24.
Biological activities
In vitro COX inhibition assays
In vitro bioactivities of all the synthesized compounds were evaluated using celecoxib and indomethacin as the reference drugs. The HWB assay was employed to determine the activity of COX isoenzymesCitation36. Five-hundred microliters of fresh blood samples obtained from human volunteers were incubated with 1 μl of DMSO as a vehicle or 1 μl of 10 μM solution of synthesized compounds or indomethacin and celecoxib as the reference drugs. The plasma was separated after 1 h incubation at 37 °C. COX-1 level was measured by the production of thromboxane B2 (TxB2) using a commercially available enzyme immunoassay (EIA) kit.
In order to determine COX-2 activity, 500 μl of fresh blood samples obtained from human volunteers were collected in EDTA-containing tubes, incubated with 1 μl of DMSO as a vehicle or 1 μl of 10 μM solution of synthesized compounds or indomethacin and celecoxib as the reference drugs. The samples were incubated at 37 °C in the presence of 10 μg/ml LPS for 24 h, after which the plasma was separated. COX-2 level was measured by the production of PGE2 using a commercially available EIA kit.
Inhibition of 15-LOX
The assay was performed according to the previously reported methodCitation37. The compounds were dissolved in DMSO and phosphate buffer. The solution was added to the assay mixture that contained 9 ml of 0.1 M sodium phosphate buffer, pH 8 and enzyme (final concentration: 167 U/ml). The mixture was incubated for four minutes and then linoleic acid as substrate was added to reach the final concentration of 134 μM. Quercetin, a well-known inhibitor of 15-LOX was used as a positive control. Absorbance was recorded by spectrophotometer at 234 nm during the measurement period. Enzyme activity experiments were tested in triplicate and the values are expressed as means ± SD.
Determination of the anti-oxidant activity by FRAP assay
FRAP assayCitation33 was employed to evaluate the anti-oxidant activities of the target compounds. For this purpose, FRAP solution was prepared by adding 10 ml acetate buffer 300 mM at pH 3.6 to 1 ml of aqueous solution of FeCl3·6H2O and 1 ml (10 mM) acidic solution of 2,4,6-tris(2-pyridyl)-s-triazine (TPTZ). Twenty five microliters of 100 μM of compounds was added to FRAP solution. After incubation at 37 °C for 15 min, the absorbance at 585 nm was monitored relative to ferrous sulfate (FeSO4) as the standard. Results are reported as mmol Fe(II) per gram of dry mass of compounds. Ascorbic acid was selected as a reference. The data are mean of three independent experiments.
Molecular docking study
Molecular docking study was performed using Autodock 4.2 and Auto Dock Tools 1.5.4 (ADT). For this purpose, the X-ray crystal structure of COX-1 carrying diclofenac as cognate ligand in binding site (PDB code: 3N8Y) and X-ray crystal structure of COX-2 in complex with SC-558 (PDB code: 1CX2) were downloaded from protein data bank (http://www.rcsb.org). Water molecules and co-crystalized ligand were excluded. The PDB file of protein was regenerated by adding polar hydrogens and computing Gasteiger charges.
The 3D structures of ligands were created using Avogadro software. Each ligand structure was separately submitted to a molecular mechanics conformational search (MM+ force field) and the geometry of the most stable conformer was then optimized by semi-empirical method (AM1) in Hyperchem software. The Gasteiger charges were added and the degrees of torsions were defined to generate PDBQT format of the ligands.
The grid parameter file was generated using AutoGrid. Numbers of points in x, y, z dimensions were set to 60 × 60 × 60 with 0.375 Å grid spacing. Rigid macromolecule was chosen to define the docking parameter file using Lamarckian Genetic Algorithm (LGA), with 200 GA runs and 25 000 000 energy evaluations while the other parameters were left as default. The results revealed that all tested compounds fit much well in the COX-2 cavity rather than COX-1 cavity.
Results and discussion
Fragment-based design
The target 1,2,4-triazine derivatives were designed by the molecular hybridization approach based on selective COX-2 inhibitor etoricoxib (1) and a number of potent anti-inflammatory agents that are reported in the literature (compounds 2–7) (). Etoricoxib (1), commonly used for pain relief after surgery, is approved in more than 80 countries worldwide which is a selective COX-2 inhibitor, containing pyridine with bearing adjacent diaryls.
Figure 2. Design of COX-2 inhibitors using molecular hybridization method.
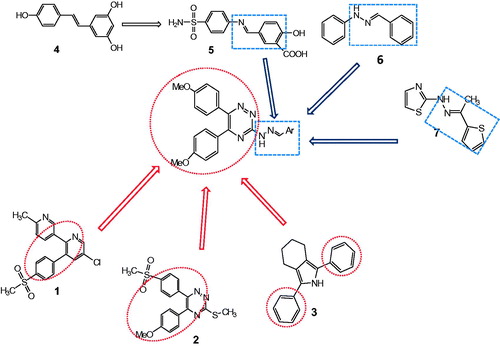
It has been reported that 1,2,4-triazine heterocyclic compounds with diaryl substituent (2) are effective COX-2 inhibitorsCitation17,Citation18. Some experiments have reported structures in which the sulfonyl moiety on the aromatic ring is missing (3)Citation25–27. Niedballa et al. showed that sulfonyl moiety can be replaced with alkoxy groupsCitation28.
The natural product (4) resveratrol, has been introduced as a COX-2 inhibitorCitation29. Lin et al. demonstrated that bioisosteric replacement of the central –N=N– linkage of (5) with –N=C–double bond leads to 114-fold more COX-2 inhibitory effect than COX-1Citation30.
2-Benzylidene-1-phenylhydrazine derivative (6) has been proposed as a COX-2 inhibitor and hydrazine derivative (7) is described as a dual COX/5-LOX inhibitorCitation31. With respect to these structural features, we designed a set of COX-2 inhibitors. The target compounds were synthesized via the same route that we had previously reportedCitation7.
Chemistry
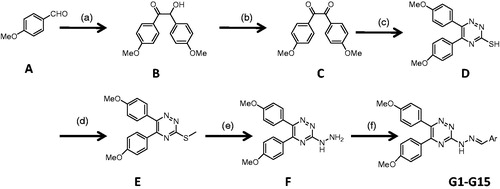
The synthetic reactions used for the synthesis of the designed fifteen 1,2,4-triazine derivatives (G1–G15) are outlined in Scheme 1. 2-Hydroxy-1,2-bis(4-methoxyphenyl) ethanone (B) was prepared by thiamine hydrochloride-mediated coupling of 4-methoxy benzaldehyde (A). Oxidation of compound B afforded the corresponding 1,2-bis(4-methoxyphenyl)ethane-1,2-dione (C). 5,6-Diaryl-1,2,4-triazine-3-thiol (D) was acquired through cyclization of compound (C) with thiosemicarbazide. Compound (D) was treated with methyl iodide to afford corresponding methylthio compound (E). In the presence of excess amount of hydrazine hydrate, compound (F) was prepared. The final compounds (G1–G15) were prepared by treatment of (F) with equimolar amounts of the appropriate aldehydes. All the steps were carried out according to our previously reported proceduresCitation7.
Scheme 1. Synthesis pathway for the preparation of triazine derivatives. Reagents and conditions: (a) thiamine hydrochloride, NaOH, EtOH, reflux, 3 h; (b) HNO3, 60 °C; (c) thiosemicarbazide, CH3COOH, reflux, 3 h; (d) CH3I, NaOH, EtOH, 2 h, rt; (e) NH2NH2·H2O, EtOH, reflux, 12 h; (f) ArCHO, EtOH, 1–10 h, rt.
Biological activity
In vitro COX inhibition assay
The human whole blood (HWB) assay, initially developed by Patrignani et al.Citation32, was performed to assess the ability of synthesized compounds to inhibit COX-2 and COX-1 isoenzymes. Celecoxib and indomethacin were used as the reference drugs. The results are summarized in
Table 1. COX-1 and COX-2 inhibitory activities of synthesized 1,2,4-triazine compounds.
The introduction of electron-withdrawing moieties, such as nitro or fluoro on phenyl ring resulted in compounds with the lowest activities against COX-2 in this set. This effect can be seen in compounds G2 and G3.
Para-substituted phenyl rings by both electron-donating and electron-withdrawing groups represented reduced activities. G13 and G15 showed a lower affinity toward COX-1 and hence represented higher COX-2/COX-1 ratios.
Compounds with five membered heterocyclic rings, such as G4 and G12 displayed COX-2 selectivity. G11 and G8 with heterocyclic fused ring aldehydes were the most selective ones among the tested compounds.
In vitro 15-lipoxygenase (LOX) inhibitory activity
In vitro 15-LOX inhibitory activities of synthesized compounds were tested. The results are reported as IC50 values in
Table 2. The 15-lipoxygenase inhibitory and anti-oxidant activities of the synthesized 1,2,4-triazine compounds.
Ferric reducing anti-oxidant power (FRAP) assay
The anti-oxidant potential of target compounds were determined using FRAP assayCitation33. This method is based on the ability of compounds in the reduction of ferric tripyridyl triazine complex to ferrous (Fe2+) form. The result is production of an intense blue color. The change in the absorbance at 585 nm is a tool for monitoring the reaction progress. Higher increase in the absorbance indicates that the potential of reduction is higher. The results are presented in . Ascorbic acid (vitamin C) was considered as a reference compound. Compound G10 showed the highest anti-oxidant activity.
Molecular modeling
In order to clarify the mechanism of COX inhibition and virtual interactions between synthetic compounds and active sites of COX-1 (PDB code: 3N8Y)Citation34 and COX-2 (PDB code: 1CX2)Citation35, the molecular docking was performed. Substitution of Ile523 in COX-1 relative to Val523 in COX-2 which is a less bulky amino acid and hence the conformational change at Tyr355 results in the presence of an accessible 2°-pocket in COX-2 active site. While the similar pocket in COX-1 is inaccessible. The other reason for effective COX-2 inhibition is the replacement of His513 in COX-1 by Arg513 in COX-2 which enables the ligands to form more H-bonds in the COX-2 active site. Therefore, because of the differences between the active sites of enzymes, it enables us to design selective COX-2 inhibitors.
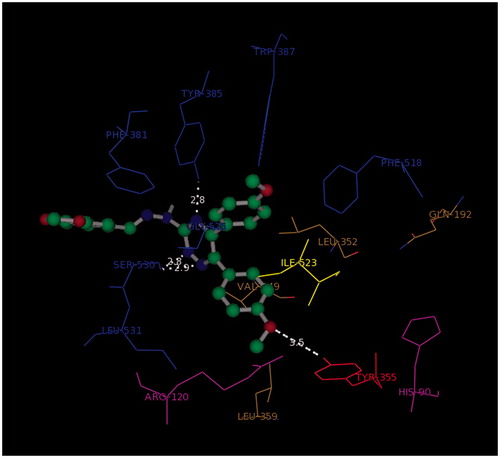
The docking study was performed using Autodock4.2. All the ligands showed a similar orientation in COX-2 active site (). Compound G11 docked in COX-2 is shown (). In general, it is observed that the oxygen atom of methoxy phenyl moiety is placed close to the side chain of Arg-120 (guanidine H2N…OCH3, 2.5 Å) while the other methoxy phenyl is accommodated in hydrophobic pocket formed by Tyr385, Gly526, Ser530, Ala527. 1,2,4-Triazine and hydrazine moieties were inserted in the selectivity pocket that was created as a result of Val523 presence in COX-2 by Ile523 in COX-1, then they got involved in H-bond with Leu352 (N=N…C=O, 2.2 Å), (triazine N…C=O, 2.9 Å), Ser353 (triazine N…C=O, 3.1 Å), Tyr355 (triazine N…O–H, 2.8 Å). The other hydrogen bond was formed by benzo[1,3]dioxole fragment (O…N, 3.2 Å).
Figure 3. Superimposition of ligands in the COX-2 binding pocket.
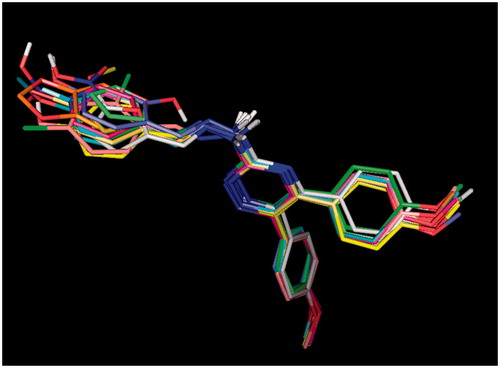
Figure 4. 3D orientation model of G11 (ball and stick) in COX-2 active pocket (blue: hydrophobic pocket, red: Tyr353, yellow: responsible for the COX-2 selectivity, purple: selectivity pocket).
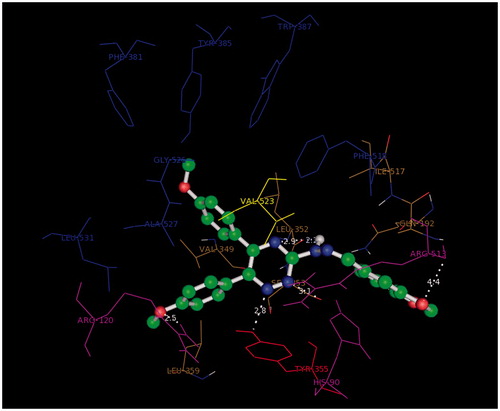
A similar method was used to compare ligand interactions in COX-1 (PDB code: 3N8Y) enzyme active site. All compounds showed a different conformation in COX-1 active site from their conformation in COX-2 active site.
The compound G11 as a representative example is shown in . As it was mentioned before, the presence of an inaccessible pocket in COX-1 resulted in different conformation and hence different H-bonds with Ser530 (triazine N…C=O, 2.8 Å, 2.9 Å), Tyr385 (triazine N…OH, 2.8 Å) and Tyr355 (CH3-O…OH, 3.5 Å).
Figure 5. 3D model of the orientation of G11 (ball and stick) in COX-1 active site (blue: hydrophobic pocket, red: Tyr353, yellow: responsible for the COX-2 selectivity, purple: selectivity pocket).
Conclusion
In summary, fragment based design strategy was utilized to design 1,2,4-triazine derivatives as selective COX-2 inhibitors. The designed compounds were synthesized and their biological activities were evaluated. The results demonstrated that compounds G9, G4, G13, G8 and G11 exhibited stronger effect against COX-2 in comparison with their activity against COX-1 at 10 μM. In vitro 15-LOX inhibitory activity of synthesized compounds was also tested. Compound G3 with an IC50 value of 124 μM was the most potent derivative. The anti-oxidant potential of synthesized compounds was determined by FRAP assay and compound G10 showed the highest anti-oxidant activity. The binding patterns of compounds inside the COX-2/COX-1 active sites were predicted using a docking technique. Considering the docking results, it can be suggested that 1,2,4-triazine and hydrazine moieties are positioned in the selectivity pocket in COX-2 active site, which provides a useful scaffold for novel compounds with improved inhibitory activities.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article. The authors acknowledge the financial support of Shiraz University of Medical Sciences, Vice Chancellor of Research (Grant: 7442).
Related Research Data
References
- Khanapure SP, Garvey DS, Janero DR, et al. Eicosanoids in inflammation: biosynthesis, pharmacology, and therapeutic frontiers. Curr Top Med Chem 2007;7:311–40
- Samuelsson B. The discovery of the leukotrienes. Am J Respir Crit Care Med 2000;161:S2–6
- Pairet M, Engelhardt G. Distinct isoforms (COX‐1 and COX‐2) of cyclooxygenase: possible physiological and therapeutic implications. Fundam Clin Pharmacol 1996;10:1–15
- Simmons DL, Xie W, Chipman JG, et al. Multiple cyclooxygenases: cloning of a mitogen-inducible form. In: Bailey JM, ed. Prostaglandins, leukotrienes, lipoxins, and PAF. Springer US; 1991:67–78
- Fosslien E. Adverse effects of nonsteroidal anti-inflammatory drugs on the gastrointestinal system. Ann Clin Lab Sci 1998;28:67–81
- McGettigan P, Henry D. Cardiovascular risk and inhibition of cyclooxygenase: a systematic review of the observational studies of selective and nonselective inhibitors of cyclooxygenase 2. JAMA 2006;296:1633–44
- Khoshneviszadeh M, Ghahremani MH, Foroumadi A, et al. Design, synthesis and biological evaluation of novel anti-cytokine 1,2,4-triazine derivatives. Bioorg Med Chem 2013;21:6708–17
- Cignarella G, Vianello P, Berti F, et al. Synthesis and pharmacological evaluation of derivatives structurally related to nimesulide. Eur J Med Chem 1996;31:359–64
- Jang M, Cai L, Udeani GO, et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 1997;275:218–20
- Bhardwaj A, Kaur J, Sharma SK, et al. Hybrid fluorescent conjugates of COX-2 inhibitors: search for a COX-2 isozyme imaging cancer biomarker. Bioorg Med Chem Lett 2013;23:163–8
- Jiang B, Huang X, Yao H, et al. Discovery of potential anti-inflammatory drugs: diaryl-1,2,4-triazoles bearing N-hydroxyurea moiety as dual inhibitors of cyclooxygenase-2 and 5-lipoxygenase. Org Biomol Chem 2014;12:2114–27
- Navidpour L, Shadnia H, Shafaroodi H, et al. Design, synthesis, and biological evaluation of substituted 2-alkylthio-1,5-diarylimidazoles as selective COX-2 inhibitors. Bioorg Med Chem 2007;15:1976–82
- Qiu K-M, Yan R, Xing M, et al. Synthesis, biological evaluation and molecular modeling of dihydro-pyrazolyl-thiazolinone derivatives as potential COX-2 inhibitors. Bioorg Med Chem 2012;20:6648–54
- Ghodsi R, Zarghi A, Daraei B, et al. Design, synthesis and biological evaluation of new 2,3-diarylquinoline derivatives as selective cyclooxygenase-2 inhibitors. Bioorg Med Chem 2010;18:1029–33
- Reddy M, Billa VK, Pallela VR, et al. Design, synthesis, and biological evaluation of 1-(4-sulfamylphenyl)-3-trifluoromethyl-5-indolyl pyrazolines as cyclooxygenase-2 (COX-2) and lipoxygenase (LOX) inhibitors. Bioorg Med Chem 2008;16:3907–16
- Sharma B, Singh P, Pilania P, et al. QSAR of 2-(4-methylsulphonylphenyl) pyrimidine derivatives as cyclooxygenase-2 inhibitors: simple structural fragments as potential modulators of activity. J Enzyme Inhibit Med Chem 2012;27:249–60
- Irannejad H, Kebriaieezadeh A, Zarghi A, et al. Synthesis, docking simulation, biological evaluations and 3D-QSAR study of 5-aryl-6-(4-methylsulfonyl)-3-(metylthio)-1, 2, 4-triazine as selective cyclooxygenase-2 inhibitors. Bioorg Med Chem 2014;22:865–73
- Dadashpour S, Tuylu Kucukkilinc T, Unsal Tan O, et al. Design, synthesis and in vitro study of 5,6‐diaryl‐1,2,4‐triazine‐3‐ylthioacetate derivatives as COX‐2 and β‐amyloid aggregation inhibitors. Archiv Pharm 2015;348:179–87
- Shivarama Holla B, Gonsalves R, Sooryanarayana Rao B, et al. Synthesis of some new biologically active bis-(thiadiazolotriazines) and bis-(thiadiazolotriazinyl) alkanes. Il Farmaco 2001;56:899–903
- Abdel-Rahman RM, Morsy J, Hanafy F, et al. Synthesis of heterobicyclic nitrogen systems bearing the 1,2,4-triazine moiety as anti-HIV and anticancer drugs: Part I. Die Pharm 1999;54:347–51
- Congreve M, Andrews SP, Doré AS, et al. Discovery of 1,2,4-triazine derivatives as adenosine A(2A) antagonists using structure based drug design. J Med Chem 2012;55:1898–903
- Hunt JT, Mitt T, Borzilleri R, et al. Discovery of the pyrrolo [2, 1-f][1, 2, 4] triazine nucleus as a new kinase inhibitor template. J Med Chem 2004;47:4054–9
- Walters T, Aur R, Hernandez K, et al. 6-Azauridine in combination chemotherapy of childhood acute myelocytic leukemia. Cancer 1972;29:1057–60
- Mullick P, Khan SA, Begum T, et al. Synthesis of 1,2,4-triazine derivatives as potential anti-anxiety and anti-inflammatory agents. Acta Pol Pharm Drug Res 2009;66:379–85
- Portevin B, Tordjman C, Pastoureau P, et al. 1,3-Diaryl-4,5,6,7-tetrahydro-2 H-isoindole derivatives: a new series of potent and selective COX-2 inhibitors in which a sulfonyl group is not a structural requisite. J Med Chem 2000;43:4582
- Black W, Bayly C, Belley M, et al. From indomethacin to a selective COX-2 inhibitor: development of indolalkanoic acids as potent and selective cyclooxygenase-2 inhibitors. Bioorg Med Chem Lett 1996;6:725–30
- Leblanc Y, Black W, Chan C, et al. Synthesis and biological evaluation of both enantiomers of L-761,000 as inhibitors of cyclooxygenase 1 and 2. Bioorg Med Chem Lett 1996;6:731–6
- Niedballa U, Bottcher I. eds. US Patent 4,440,776, 1984. Chem Abstr; 1980
- Zykova TA, Zhu F, Zhai X, et al. Resveratrol directly targets COX-2 to inhibit carcinogenesis. Mol Carcinogen 2008;47:797–805
- Lin S-J, Tsai W-J, Chiou W-F, et al. Selective COX-2 inhibitors. Part 2: Synthesis and biological evaluation of 4-benzylideneamino- and 4-phenyliminomethyl-benzenesulfonamides. Bioorg Med Chem 2008;16:2697–706
- Almasirad A, Hosseini R, Jalalizadeh H, et al. Synthesis and analgesic activity of 2-phenoxybenzoic acid and N-phenylanthranilic acid hydrazides. Biol Pharm Bull 2006;29:1180–5
- Patrignani P, Panara MR, Greco A, et al. Biochemical and pharmacological characterization of the cyclooxygenase activity of human blood prostaglandin endoperoxide synthases. J Pharmacol Exp Ther 1994;271:1705–12
- Benzie IF, Strain J. Ferric reducing/antioxidant power assay: direct measure of total antioxidant activity of biological fluids and modified version for simultaneous measurement of total antioxidant power and ascorbic acid concentration. Methods Enzymol 1999;299:15–27
- Sidhu RS, Lee JY, Yuan C, et al. Comparison of cyclooxygenase-1 crystal structures: cross-talk between monomers comprising cyclooxygenase-1 homodimers. Biochemistry 2010;49:7069–79
- Kurumbail RG, Stevens AM, Gierse JK, et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996;384:644–8
- Baytas S, Turan Dural N, Ozkan Y, et al. Synthesis, anti-inflammatory, antiplatelet and in silico evaluations of (E)-3-(3-(2, 3-dihydro-3-methyl-2-oxo-3H-benzoxazole-6-yl)-1-phenyl-1H-pyrazole-4-yl) acrylamides. Turk J Chem 2012;36:367–82
- Malterud KE, Rydland KM. Inhibitors of 15-lipoxygenase from orange peel. J Agric Food Chem 2000;48:5576–80