
Abstract
Background: Current therapeutic options for advanced pancreatic cancer have been largely disappointing with modest results at best, and though adjuvant therapy remains controversial, most remain in agreement that Gemcitabine should stand as part of any combination study. The inhibitor of apoptosis (IAP) protein Survivin is a key factor in maintaining apoptosis resistance, and its dominant-negative mutant (Survivin-T34A) has been shown to block Survivin, inducing caspase activation and apoptosis.
Methods: In this study, exosomes, collected from a melanoma cell line built to harbor a tetracycline-regulated Survivin-T34A, were plated on the pancreatic adenocarcinoma (MIA PaCa-2) cell line. Evaluation of the presence of Survivin-T34A in these exosomes followed by their ability to induce Gemcitabine-potentiative cell killing was the objective of this work.
Results: Here we show that exosomes collected in the absence of tetracycline (tet-off) from the engineered melanoma cell do contain Survivin-T34A and when used alone or in combination with Gemcitabine, induced a significant increase in apoptotic cell death when compared to Gemcitabine alone on a variety of pancreatic cancer cell lines.
Conclusion: This exosomes/Survivin-T34A study shows that a new delivery method for anticancer proteins within the cancer microenvironment may prove useful in targeting cancers of the pancreas.
To access the supplementary material to this article, please see Supplementary files under Article Tools online.
Pancreatic cancer is the fourth leading cause of cancer death in the United States, with an average 5-year survival rate of 5% for all stages of the disease (Citation1). Pancreatic cancer has an annual mortality rate of approximately 95% with over 250,000 patients dying worldwide (Citation2). Pancreatic cancer exhibits no clear early warning signs or symptoms and it is often detected after it is too late for pancreatic resection. Currently, if diagnosed at early stages, surgical resection remains the most efficacious treatment and offers the best patient outcome. However, only 20% of pancreatic cancer patients meet these criteria (Citation3). There is a need to discover and implement new therapies or therapeutic combinations that increase the survival rate of those afflicted with this pancreatic cancer. Gemcitabine remains the gold-standard for chemotherapy (Citation4). However, while Gemcitabine has shown significant benefit in clinical applications, its ability to effectively impact pancreatic cancer is limited. Currently, combinatory treatments using Gemcitabine and other therapeutics have shown no significant improvements in survival rates (Citation5–Citation7). However, the cancer research field is moving rapidly towards combinatorial therapies, including combined multiple chemotherapy drugs (Citation8), radiation with chemotherapy (Citation7) and virotherapy with chemotherapy (Citation9). The inhibitor of apoptosis (IAP) Survivin seems to be one of the key players in resistance to many of these cancer therapies (Citation10–Citation12) and therefore a strategy to inhibit its action, when combined with standard treatment options may prove beneficial.
Survivin is a possible prognostic marker for pancreatic cancer patients (Citation13–Citation15). Though expressed in most human cancers and present during embryonic and foetal development (Citation16), its aberrantly high protein expression in cancer cells and low level of expression in most normal tissues makes Survivin an important anti-cancer target (Citation17). Survivin overexpression in cancer has been described as a predictive factor in determining response to chemotherapy and radiotherapy (Citation7, Citation18) (Citation19). Survivin reduces cell death induced by several anti-cancer agents including paclitaxel, etoposide and tumour necrosis factor alpha. Conversely, inhibition of Survivin reduces tumour growth potential and sensitizes tumour cells to many of the same chemotherapeutic agents (Citation20).
We have reported a marked enhancement of Survivin's role in therapeutic resistance to both chemo- and radiotherapy in pancreatic cancer (Citation7). Reduction of Survivin levels and/or inhibition of the protein's anti-apoptotic properties may assist in making cancer cells more susceptible to existing (and future) therapeutic regimens. Successful strategies against intracellular Survivin include molecular antagonists such as antisense oligos, RNA inhibition, Survivin-specific cytolytic T cells, the non-phosphorylatable dominant-negative Survivin mutant Thr34→Ala (T34A) and, most recently, binding interface mimetics (Citation21–Citation28).
The loss of phosphorylation at the Survivin Thr34 site is significant as it results in the dissociation of the caspase-9/Survivin protein complex, leading to anti-tumour effects (Citation22, Citation29–Citation32). Mesri et al. employed Survivin-T34A treatment in vivo via adenoviral vectors, with their results yielding only modest levels of success (Citation23). A better delivery method will need to be used if this Survivin inhibitor is to prove efficacious in vivo.
Conditioned media (CM) collected from cervical, pancreatic, prostate, breast cancer, osteosarcomas, leukaemia cell lines (Citation33) and CM collected from a Survivin-T34A overexpressing HeLa cell line provided evidence that functional Survivin can be found extracellularly in the CM. We have recently shown extracellular Survivin to reside in small 50–100 nm vesicles called exosomes (Citation34). Exosomes have been described as a pivotal mechanism in the multicellular organism for cell-to-cell communication as they allow for cells to exchange information through transferal of soluble factors such as proteins, RNAs and miRNA (Citation35). More recently, exosomes have been exploited for cancer immunotherapy as there may be an opportunity to adapt them as drug delivery vehicles for therapeutic intervention (Citation36). The typical exosome is 50–100 nm in size and originates intracellularly, displaying MHC class I and class II, heat shock proteins, tetraspanin proteins and in our hands IAP proteins (Citation34, Citation37).
We showed in this undertaking Survivin-T34A CM eliciting anti-cancer effects such as induced apoptosis with loss of mitochondrial potential. The goal of the present study was to determine if the cells engineered to overexpress the Survivin-T34A dominant-negative mutant would produce a functional, exosomally packaged, Survivin-T34A which when used in combination with Gemcitabine might significantly enhance the death of pancreatic cancer cells, in vitro.
Materials and methods
Cell cultures
Pancreatic adenocarcinoma (MIA PaCa-2) cell line was obtained from the American Type Culture Collection (ATCC, Manassas, VA) and maintained in DMEM, supplemented with 100 units of penicillin, 100 µg/mL of streptomycin, 300 µg of L-glutamine, 10% heat-inactivated FBS (ATCC) and 2.5% horse serum. Exosomes were isolated from YUSAC 2, a melanoma cell line obtained from Dr. Doug Grossman at the Huntsman Cancer Institute in Salt Lake City, Utah. The two cell line derivatives from YUSAC 2 were designed to overexpress either Survivin-WT (4C7 cells) or Survivin-T34A (F5C4 cells) in the absence of tetracycline (tet), otherwise only normal endogenous levels of Survivin are produced. YUSAC 2 cells were maintained in DMEM (CellGro, Manassas, VA) supplemented with 100 units of penicillin, 100 µg/mL of streptomycin, 300 µg of L-glutamine, 5% newborn calf serum (Thermo Scientific HyClone, Rockford, IL), 0.5 µg/mL tetracycline (tet-off system), 1.5 mg/mL Geneticin G418 (Teknova, Holister, CA) and 2 mM NaOH. YUSAC 2 cells were grown to 60% confluency in the presence of tetracycline in order to establish a healthy monolayer culture. After which, cells were washed carefully twice in PBS followed by the addition of media in the absence of tetracycline. All cells were grown at 37°C in a humidified atmosphere containing 5% CO2.
Gemcitabine treatment
Gemcitabine (Sigma-Aldrich, St. Louis, MO) was dissolved in DMSO and added to cells simultaneously with exosome treatment. Final DMSO concentration was 0.03%. Final Gemcitabine concentration used was 10 M. The cells were returned to the incubator and harvested after 24, 48 or 72 hr for apoptosis analysis by flow cytometry.
Exosome isolation and quantification
Exosomes were isolated from CM as we have previously reported (Citation34). In brief, the culture media was collected from cells in culture after 24 hr. The CM was centrifuged at 10,000×g for 10 min at 4°C to pellet the minor amounts of cells and cell debris. CM was filtered through 0.22 µm syringe followed by centrifugation at 100,000×g for 18 hr at 4°C on a 30% sucrose cushion (Citation38). The sucrose cushion containing exosomes was collected and washed with PBS followed by additional centrifugation at 100,000×g for 18 hr at 4°C. Exosome pellets were collected and stored at −80°C. Exosome quantification was accomplished using acetylcholinesterase assay, according to the protocol of Lancaster and Febbraio (Citation39). Briefly, acetylcholinesterase activity was employed to determine the presence of exosome vesicles. 40 µL of the exosome fraction was suspended in 110 µL of PBS. This PBS-diluted exosome fraction (37.5 µL) was then added to individual wells on a 96-well flat bottom plate. 112.5 µL of 1.25 mM acetylthiocholine and 150 µL of 0.1 mM 5,5’-dithio-bis(2-nitrobenzoic acid) were added to PBS-diluted exosomes. The change in absorbance at 412 nm was monitored every 5 min for 30 min. The data presented represent acetylcholinesterase enzymatic activity after 30 min incubation. Exosome and whole cell lysates (WCL) protein was quantified using the BCA assay on a µQuant microplate spectrophotometer (Bio-Tek, Winooski, VT) and analyzed using KC Junior Software (Bio-Tek).
Apoptosis and cell cycle analysis
Sub-confluent cultures of the pancreatic cancer cells were incubated with vehicle (DMSO), Gemcitabine (0–500 µM) and/or exposed to exosomes treatment (0–1,500 µg/mL total protein, exosomes were sterilized using 0.22 µm syringe filter) for 0, 24, 48 and 72 hr at 37°C. Cells were harvested, prepared and analyzed for DNA content using a Becton Dickinson FACScan flow cytometer (Becton Dickinson, San Jose, CA) as described previously (Citation34, Citation40). The distribution of cells in the different phases of the cell cycle was analyzed from DNA histograms using BD CellQuest software (Becton Dickinson and Company, San Jose, CA).
Western blot analysis
WCLs were prepared as previously described (Citation7) and quantified as described above. Proteins (30 µg) were separated using 12 or 15% Bis-Tris polyacrylamide gels and then transferred onto nitrocellulose membranes (Bio-Rad) and probed using 1–5 µg/mL of the following antibodies: rabbit polyclonal anti-Survivin (Novus, Littleton, CO), rabbit anti-ppSurvivin-Thr34 (Novus), rabbit polyclonal anti--actin (BioLegend, San Diego, CA) and mouse polyclonal anti-LAMP-1 (Cell Signaling Technologies, Beverly, MA). Secondary antibodies (IR-Dye-conjugated) were goat anti-rabbit and goat anti-mouse immunoglobulin (LI-COR, Lincoln, Nebraska) used at a 1:5,000 dilution. Membranes were blocked for 1 hr using blocking reagent purchased from LI-COR. Membranes were incubated overnight in primary antibody followed by three 15-min PBS-Tween wash steps and a final 1-hr secondary antibody incubation followed again by three 15-min PBS-Tween washings. Immunoreactive bands were detected using the Odyssey imaging system (LI-COR). β-actin or LAMP-1 was used as Western blot loading controls for either cell lysates or exosomal protein, respectively.
Statistical analysis
Multiple comparisons among different groups were calculated by using Multiple Analysis of Variance (MANOVA) as we have done in the past (Citation41). Student t-test (two-tailed) was used to evaluate the significance of changes between control groups and experimental groups. Probability values p<0.05 were considered statistically significant.
Results
The presence of Survivin-T34A induces time-dependent apoptosis
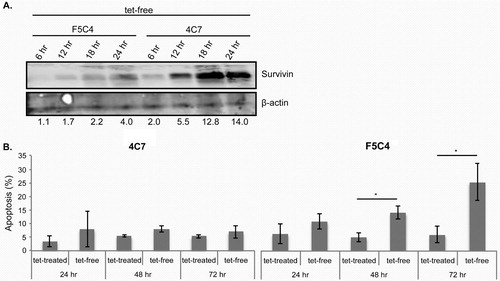
The melanoma cell line, YUSAC 2, was engineered to overexpress WT Survivin (4C7) or mutant T34A (F5C4), as previously reported (Citation22). The YUSAC 2 cells were continuously treated with 0.5 µg/mL of tetracycline (tet) to prevent overexpression of Survivin-WT (4C7 cells) or Survivin-T34A (F5C4 cells) in this tet-off system. Cells were harvested 6, 12, 18 and 24 hr after removal of tet for analysis by Western blot and flow cytometry (). Following tet removal, Survivin protein was concomitantly increased in both cell lines in a time-dependent manner (A), as previously reported (Citation22). Removal of tet from F5C4 cells resulted in increased apoptosis in a time-dependent manner, maximizing at 72 hr, while no increase in apoptosis was recorded in the 4C7 cells treated under the same conditions (B).
Fig. 1. A. Western blots of whole cell lysates of YUSAC 2 cell line derivatives. 4C7 or F5C4 cells were incubated in the presence or absence of tetracycline for 6, 12, 18 and 24 hr, which in the absence of tetracycline will overexpress Survivin-WT or Survivin-T34A, respectively. Survivin is increased in a time-dependent manner when incubated in the absence of tetracycline. Beta-actin was used as the loading control, and molecular weights (kDa) are shown on the left. Densitometry was undertaken to show the degree of Survivin increase. B. Histogram representing the percentage of apoptosis using propidium iodide (PI) analysis by flow cytometry, 4C7 show similar apoptosis levels for both tet-treated and tet-free conditions. F5C4 cells have increasing apoptosis in a time-dependent manner when incubated in tet-free media. Data are the mean±SD of three independent experiments (*p<0.05).
Survivin-T34A overexpressing cells showed decreased phosphorylated-Survivin in a time-dependent manner
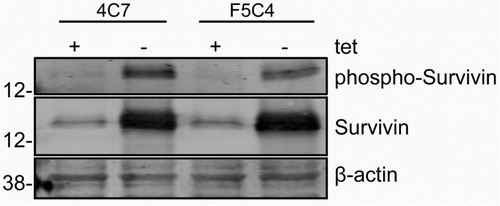
4C7 and F5C4 cells were incubated in either the presence or absence of tet for 24 hr, and levels of Survivin, phospho-Survivin and β-actin were analyzed by Western blotting. Both 4C7 and F5C4 cells expressed increased Survivin levels in the absence of tet compared to tet-treated control cells (). Survivin phosphorylation at Thr34 was confirmed by Western blotting using a phospho-specific Survivin Thr34 antibody as has been previously shown (Citation22). Tet-free F5C4 cells showed a decreased phospho-Survivin band compared to tet-free 4C7 cells.
Fig. 2. Western blot of whole cell lysates of YUSAC 2 cell line derivatives. 4C7 and F5C4 cells were incubated in the presence or absence of tetracycline for 24 hr, which in the absence of tetracycline will overexpress wild-type or Survivin-T34A, respectively. 4C7 has increased Survivin in the absence of tetracycline, which is concurrent with phosphorylated-Survivin-Thr34. However, in the absence of tetracycline, F5C4 also overexpresses Survivin, but it is not phosphorylatable at the Thr34 site.
YUSAC 2 cells release Survivin-containing exosomes
In order to confirm that F5C4 cells release exosomes, we isolated exosomes by ultracentrifugation using a sucrose cushion, as previously described (Citation34, Citation38). Following ultracentrifugation, exosome levels were analyzed using the acetylcholinesterase enzyme (AChE) activity assay (A). No significant difference in exosome levels between tet-treated and tet-free cells was measured at 24 hr, signifying that the number of exosomes released is independent of the tet-system.
Fig. 3. A. Histogram representing an acetylcholinesterase enzyme activity assay to quantify exosome numbers relative to fresh complete media. Exosomes are present as represented by the graph when compared to control. There is no difference within the same cells when treated with tetracycline or without. Data are the mean±SD of 3 independent experiments. B. Western blots of both YUSAC 2–derived cell lines overexpress Survivin in the absence of tetracycline. C. Western blots of proteins isolated from exosomes collected from F5C4 cells after incubation in the presence or absence of tetracycline for 24 hr. LAMP-1 was used as the loading control with molecular weights (kDa) shown on the left.
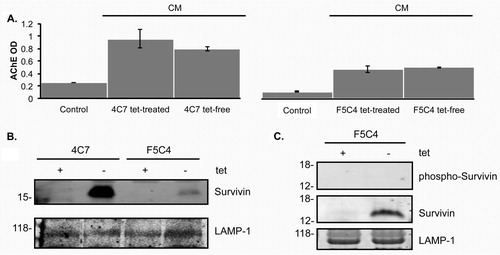
Exosomal Survivin content was evaluated by Western blotting. Survivin expression was elevated after tet removal in exosomes from both 4C7 and F5C4 cells (B). This is similar to observed levels of Survivin expression in WCL from YUSAC 2 cells. In order to evaluate Survivin-T34A presence in the exosomes released from F5C4 cells, we employed the phospho-specific Survivin Thr34 antibody for Western blots of these exosomes. Survivin was increased in the exosomes from tet-free F5C4 as was recorded in the immunoblots from the parent cells. However, there were no phosphorylated-Survivin bands from either exosomes of tet-treated or tet-free F5C4 cells (C). The presence of tet-removal-enhanced Survivin, coupled with the absence of phosphorylated-Survivin, provides strong evidence that these exosomes contain the dominant-negative Survivin-T34A.
Exosomes containing Survivin-T34A induce apoptosis in MIA PaCa-2 pancreatic cancer cells
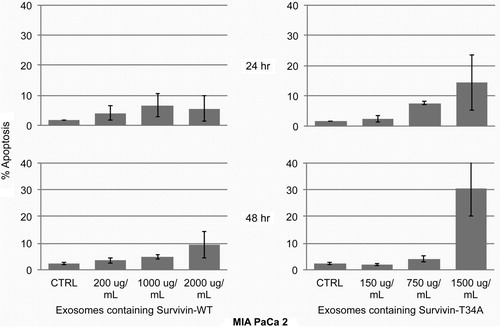
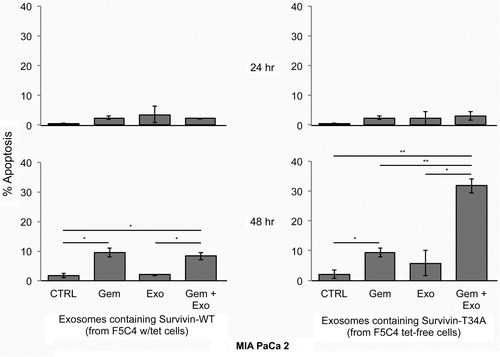
Exosomes isolated from tet-treated F5C4 (endogenous WT Survivin) cells were employed as the control for exosomes isolated from tet-free F5C4 cells (Survivin-T34A). There was no significant apoptotic induction (≤10%) after MIA PaCa 2 cells were treated with 200, 1,000 or 2,000 µg/mL of exosomes purified from tet-treated F5C4 cells after 24 and 48 hr (). MIA PaCa 2 cells were then treated using exosomes containing Survivin-T34A (150 and 750 µg/mL based on total exosomal protein). Results showed little apoptosis (~5%) versus treatment by control exosomes (from tet-treated F5C4 cells) at 24 hr (). However, using Survivin-T34A exosomes (1,500 µg/mL), a marked 48 hr increase in apoptosis (30.5%) was measured.
Fig. 4. Histograms representing the percent apoptosis in MIA PaCa2 cells following exosome treatment for 24 and 48 hr. MIA PaCa 2 cells treated for 24 hr showed little to no increase in apoptosis after exosomes treatment. After 48 hr treatment, only 1,500 µg/mL of exosomes containing Survivin-T34A induced apoptosis (30.5%). Percent apoptosis was determined from sub-G1 DNA content analyzing propidium iodide staining by flow cytometry. Data are the mean±SD of 2 independent experiments.
Survivin-T34A exosomes enhance Gemcitabine treatment on pancreatic cancer cells
In order to investigate whether exosomes containing Survivin-T34A could enhance Gemcitabine-killing, MIA PaCa 2 cells were treated with either exosomes containing Survivin-WT (200 µg/mL) or Survivin-T34A (150 µg/mL) or 10 µM Gemcitabine. Gemcitabine dosage levels conformed to the protocols previously established in our laboratory (Citation7) and Supplementaryfile. Exosome concentrations were chosen that did not induce a measurable level of apoptosis () at 24 and 48 hr, as an induced additive or potentiative response from the combination was the goal. We also measured apoptotic cell death after pancreatic cancer cells were treated with a combination of exosomes and Gemcitabine. At 24 hr, no marked differences between the various treatments (24 hr, <4%) were recorded (). As expected, Gemcitabine induced modest apoptosis vs. control (10 and 2%, respectively) after 48 hr. However, the combination of exosomes containing Survivin-T34A with 10 µM Gemcitabine treatment displayed the greatest enhanced apoptosis compared to control and the single modality treatments (32% apoptosis vs. less than 10% apoptosis) after 48 hr. The combination treatment using exosomes containing Survivin-T34A yielded the same levels of apoptosis as did treatment using 1,500 µg/mL Survivin-T34A exosomes.
Fig. 5. MIA PaCa 2 cells were treated with either 10 µM Gemcitabine, 150 or 200 µg/mL (total exosomal protein from F5C4) or the combination of both. MIA PaCa 2 cells treated for 24 hr showed little to no marked increase in apoptosis (<5%) after exosomes treatment. Gemcitabine (10 µM) had modest apoptosis induction over control (~11% vs. 2%, respectively). The combination of 10 µM Gemcitabine with 150 µg/mL exosomes containing Survivin-T34A yielded 32% apoptosis (~30% greater than control) after 48 hr. Percent apoptosis was determined from sub-G1 DNA content analyzing propidium iodide staining by flow cytometry. Data are the mean±SD of 2 independent experiments (*p<0.05, *p<0.01).
Discussion
We have previously described Survivin's (WT and T34A) presence in the extracellular CM (Citation33, Citation34) (Citation42, Citation43), and that extracellular Survivin can be taken up by cancer cells resulting in functional effects such as proliferation, invasion and resistance (Citation33). We have also shown that Survivin is released via small membrane-bound vesicles called exosomes (Citation34). Building upon these findings, we have sought to test whether or not exosomes containing the apoptosis-inducing Survivin mutant, T34A, could be produced and if so, used either alone or in combination with other treatment methods as a cancer therapeutic.
To this end, the YUSAC 2 tet-off system (Citation22) was employed. Exosomes must be collected early so that the vesicles being harvested are intact and functional with the production of apoptotic bodies kept at a minimum. The F5C4 cell lines provide a good system in which the tet-off regulatory mechanisms allow for Survivin-T34A to be controlled systematically in all cells. The released quantity of exosomes was independent of the tet-system, which is important because any changes in protein levels within the exosomes are the result of the tet-system and not the quantity of exosomes. Our results demonstrate for the first time that Survivin-T34A is released from the cells via exosomes.
Using exosomes for anti-cancer therapy is a relatively new idea, but a rapidly growing field (Citation44). Dendritic exosomes (DEX) are the most commonly used exosomes for such therapy (Citation37, Citation45–Citation48). However, DEXs are typically used as a form of immunotherapy – a way to prime the immune system against tumour cells (Citation44). Our aim in this study was to directly attack tumour cells with exosomes containing Survivin-T34A.
MIA PaCa 2 cells treated in culture with exosomes containing Survivin-T34A experienced enhanced apoptosis. When we compared Survivin-T34A exosome-induced apoptosis levels to that induced by Gemcitabine alone, 100 µM Gemcitabine yielded 50% less apoptosis in the pancreatic cancer cell lines PANC1 and MIA PaCa-2 (Citation7) than did 1,500 µg/mL of Survivin-T34A-containing exosomes on the same cell lines. We also could not obtain similar levels of apoptosis as exosomal Survivin-T34A using our highest concentration of Gemcitabine (500 µM, data not shown). We therefore combined low doses of exosomal Survivin-T34A (150 µg/mL total protein) with Gemcitabine (10 µM) to investigate whether Survivin-T34A could enhance Gemcitabine's killing effects. Alone, these doses did not yield an increase in apoptosis over control. However, there was a marked enhancement of apoptosis when Survivin-T34A exosomes and Gemcitabine were combined. Use of 150 µg/mL of Survivin-T34A exosomes and 10 µM of Gemcitabine resulted in apoptosis levels of more than 30% after 48 hr of combined treatment. We believe that the enhancement from the combination treatments occurred because of the mechanism of Gemcitabine. Gemcitabine has been shown to reduce G2/M cell cycle arrest, which reduces cellular Survivin levels (Citation29). We also have found that a modest amount of stress induces wild-type Survivin in this model system (data not shown). This combined with Survivin-T34A exosomes, which target Survivin, enhance apoptosis with much lower dosages because it is believed that Survivin-T34A targets and disrupts Survivin-associated protection of the cancer cell (Citation29, Citation49) (Citation50).
In summary, delivery has been the confounding factor for using the novel Survivin-T34A as an effective therapeutic. This study not only adds to the importance of using Survivin-T34A as a cancer therapeutic for the treatment of pancreatic cancer, but that exosome delivery may provide a potential mechanism for effective tumour delivery. Cancer research and therapy have been rapidly moving towards combinatorial therapies but clinically, Gemcitabine remains the most prominent player in effective pancreatic cancer therapy. In closing, we would like to emphasize the importance of continued exploration of the potential of combining exosome delivery of Survivin-T34A with Gemcitabine and other anticancer therapeutic regimens. These studies may prove relevance for the discovery and implementation of novel pancreatic cancer therapeutic strategies to improve the efficacy of chemotherapy-induced apoptosis in patients (Citation51).
Financial Support
NCMHD Project EXPORT Program 5P20MD001632/Project 3 (NRW). Funding was also obtained as part of a start-up package from Loma Linda University's Center for Molecular Biology and Gene Therapy, now the Center for Health Disparities Research and Molecular Medicine (NRW) and a National Merit Test Bed (NMTB) award sponsored by the Department of the Army under Cooperative Agreement Number DAMD17-97-2-7016 (NRW). This work was also supported by The Hirshberg Foundation for Pancreatic Cancer Research (NRW).
Disclaimer
The views expressed in the submitted article are those of the authors and are not an official position of the institution or funder.
Conflict of interest and funding
The authors declare no conflict of interest, financial or otherwise.
Acknowledgements
This work would have been impossible without a generous grant from the Hirshberg Foundation for Pancreatic Cancer Research and the friendship, inspiration and mentoring of Agi Hirshberg. Funding for our laboratory has also come from grants for health disparity research: NIH-NCMHD Project EXPORT Program 5P20MD001631/Project 3 (NRW). Funding also came from a National Merit Test Bed (NMTB) award sponsored by the Department of the Army under Cooperative Agreement Number DAMD17-97-2-7016 (NRW). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. We thank Dr. Doug Grossman for the kind gift of the YUSAC cells. We also thank Mr. Ron Moyron and the entire NRW lab for a careful review of the manuscript.
Notes
To access the supplementary material to this article, please see Supplementary files under Article Tools online.
Related Research Data
References
- Kleeff J, Michalski C, Friess H, Buchler MW. Pancreatic cancer: from bench to 5-year survival. Pancreas. 2006; 33: 111–18.
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011; 61: 69–90.
- Muller MW, Friess H, Koninger J, Martin D, Wente MN, Hinz Uet al. Factors influencing survival after bypass procedures in patients with advanced pancreatic adenocarcinomas. Am J Surg. 2008; 195: 221–8.
- Ueno H, Kiyosawa K, Kaniwa N. Pharmacogenomics of gemcitabine: can genetic studies lead to tailor-made therapy?. Br J Cancer. 2007; 97: 145–51.
- Reni M, Cereda S, Galli L. PEFG (cisplatin, epirubicin, 5-fluorouracil, gemcitabine) for patients with advanced pancreatic cancer: the ghost regimen. Cancer Lett. 2007; 256: 25–8.
- Oettle H, Neuhaus P. Adjuvant therapy in pancreatic cancer: a critical appraisal. Drugs. 2007; 67: 2293–310.
- Galloway NR, Aspe JR, Sellers C, Wall NR. Enhanced antitumor effect of combined gemcitabine and proton radiation in the treatment of pancreatic cancer. Pancreas. 2009; 38: 782–90.
- Vishnu P, Colon-Otero G, Kennedy GT, Marlow LA, Kennedy WP, Wu KJet al. RhoB mediates antitumor synergy of combined ixabepilone and sunitinib in human ovarian serous cancer. Gynecol Oncol. 2012; 124: 589–97.
- Karapanagiotou EM, Roulstone V, Twigger K, Ball M, Tanay M, Nutting Cet al. Phase I/II trial of carboplatin and paclitaxel chemotherapy in combination with intravenous oncolytic reovirus in patients with advanced malignancies. Clin Cancer Res. 2012; 18: 2080–9.
- Li F. Survivin study: what is the next wave?. J Cell Physiol. 2003; 197: 8–29.
- Asanuma K, Kobayashi D, Furuya D, Tsuji N, Yagihashi A, Watanabe N. A role for Survivin in radioresistance of pancreatic cancer cells. Jpn J Cancer Res. 2002; 93: 1057–62.
- Kami K, Doi R, Koizumi M, Toyoda E, Mori T, Ito Det al. Downregulation of Survivin by siRNA diminishes radioresistance of pancreatic cancer cells. Surgery. 2005; 138: 299–305.
- Sarela AI, Verbeke CS, Ramsdale J, Davies CL, Markham AF, Guillou PJ. Expression of Survivin, a novel inhibitor of apoptosis and cell cycle regulatory protein, in pancreatic adenocarcinoma. Br J Cancer. 2002; 86: 886–92.
- Kami K, Doi R, Koizumi M, Toyoda E, Mori T, Ito Det al. Survivin expression is a prognostic marker in pancreatic cancer patients. Surgery. 2004; 136: 443–8.
- Satoh K, Kaneko K, Hirota M, Masamune A, Satoh A, Shimosegawa T. Expression of Survivin is correlated with cancer cell apoptosis and is involved in the development of human pancreatic duct cell tumors. Cancer. 2001; 92: 271–8. [PubMedAbstract].
- Adida C, Crotty PL, McGrath J, Berrebi D, Diebold J, Altieri DC. Developmentally regulated expression of the novel cancer anti-apoptosis gene Survivin in human and mouse differentiation. Am J Pathol. 1998; 152: 43–9. [PubMedAbstract].
- Fukuda S, Pelus LM. Survivin, a cancer target with an emerging role in normal adult tissues. Mol Cancer Ther. 2006; 5: 1087–98.
- Zhu H, Wang Q, Hu C, Zhang W, Quan L, Liu Met al. High expression of Survivin predicts poor prognosis in esophageal squamous cell carcinoma following radiotherapy. Tumour Biol. 2011; 32: 1147–53.
- Shen X, Zheng JY, Shi H, Zhang Z, Wang WZ. Survivin Knockdown enhances gastric cancer cell sensitivity to radiation and chemotherapy in vitro and in nude mice. Am J Med Sci. 2012; 344: 52–8.
- Mita AC, Mita MM, Nawrocki ST, Giles FJ. Survivin: key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin Cancer Res. 2008; 14(16): 5000–5.
- Andersen MH, Pedersen LO, Capeller B, Brocker EB, Becker JC, thor Straten P. Spontaneous cytotoxic T-cell responses against Survivin-derived MHC class I-restricted T-cell epitopes in situ as well as ex vivo in cancer patients. Cancer Res. 2001; 61: 5964–8. [PubMedAbstract].
- Grossman D, Kim PJ, Schechner JS, Altieri DC. Inhibition of melanoma tumor growth in vivo by Survivin targeting. Proc Natl Acad Sci U S A. 2001; 98: 635–40.
- Mesri M, Wall NR, Li J, Kim RW, Altieri DC. Cancer gene therapy using a Survivin mutant adenovirus. J Clin Invest. 2001; 108: 981–90.
- Wall NR, O'Connor DS, Plescia J, Pommier Y, Altieri DC. Suppression of Survivin phosphorylation on Thr34 by flavopiridol enhances tumor cell apoptosis. Cancer Res. 2003; 63: 230–5. [PubMedAbstract].
- Plescia J, Salz W, Xia F, Pennati M, Zaffaroni N, Daidone MGet al. Rational design of shepherdin, a novel anticancer agent. Cancer Cell. 2005; 7: 457–68.
- Olie RA, Simoes-Wust AP, Baumann B, Leech SH, Fabbro D, Stahel RAet al. A novel antisense oligonucleotide targeting Survivin expression induces apoptosis and sensitizes lung cancer cells to chemotherapy. Cancer Res. 2000; 60: 2805–9. [PubMedAbstract].
- Kanwar JR, Shen WP, Kanwar RK, Berg RW, Krissansen GW. Effects of Survivin antagonists on growth of established tumors and B7-1 immunogene therapy. J Natl Cancer Inst. 2001; 93: 1541–52.
- Andersen MH, Svane IM, Becker JC, Straten PT. The universal character of the tumor-associated antigen survivin. Clin Cancer Res. 2007; 13: 5991–4.
- Aspe JR, Wall NR. Survivin-T34A: molecular mechanism and therapeutic potential. Onco Targets Ther. 2010; 3: 247–54. [PubMedAbstract] [PubMedCentralFull Text].
- Li HX, Zhao XY, Wang L, Wang YS, Kan B, Xu JRet al. Antitumor effect of mSurvivinThr34→Ala in murine colon carcinoma when administered intravenously. Med Oncol. 2010; 27: 1156–63.
- Peng XC, Yang L, Yang LP, Mao YQ, Yang HS, Liu JYet al. Efficient inhibition of murine breast cancer growth and metastasis by gene transferred mouse survivin Thr34→Ala mutant. J Exp Clin Cancer Res. 2008; 27: 46.
- Shen C, Liu W, Buck AK, Reske SN. Pro-apoptosis and anti-proliferation effects of a recombinant dominant-negative survivin-T34A in human cancer cells. Anticancer Res. 2009; 29: 1423–8. [PubMedAbstract].
- Khan S, Aspe JR, Asumen MG, Almaguel F, Odumosu O, Acevedo-Martinez Set al. Extracellular, cell-permeable survivin inhibits apoptosis while promoting proliferative and metastatic potential. Br J Cancer. 2009; 100: 1073–86.
- Khan S, Jutzy JM, Aspe JR, McGregor DW, Neidigh JW, Wall NR. Survivin is released from cancer cells via exosomes. Apoptosis. 2011; 16: 1–12.
- Bobrie A, Thery C. Exosomes and communication between tumours and the immune system: are all exosomes equal?. Biochem Soc Trans. 2013; 41: 263–7.
- EL Andaloussi S, Mager I, Breakefield XO, Wood MJ. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov. 2013; 12: 347–57.
- Tan A, De La Pena H, Seifalian AM. The application of exosomes as a nanoscale cancer vaccine. Int J Nanomedicine. 2010; 5: 889–900. [PubMedAbstract] [PubMedCentralFull Text].
- Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. ;Chapter :Unit 3. 2006; 3: 22. [PubMedAbstract] [PubMedCentralFullText].
- Lancaster GI, Febbraio MA. Exosome-dependent trafficking of HSP70: a novel secretory pathway for cellular stress proteins. J Biol Chem. 2005; 280: 23349–55.
- Li F, Ackermann EJ, Bennett CF, Rothermel AL, Plescia J, Tognin Set al. Pleiotropic cell-division defects and apoptosis induced by interference with survivin function. Nat Cell Biol. 1999; 1: 461–6.
- Khan S, Jutzy JM, Valenzuela MM, Turay D, Aspe JR, Ashok Aet al. Plasma-derived exosomal survivin, a plausible biomarker for early detection of prostate cancer. PloS One. 2012; 7: e46737.
- Bokarewa M, Lindblad S, Bokarew D, Tarkowski A. Balance between survivin, a key member of the apoptosis inhibitor family, and its specific antibodies determines erosivity in rheumatoid arthritis. Arthritis Res Ther. 2005; 7: R349–58.
- Mera S, Magnusson M, Tarkowski A, Bokarewa M. Extracellular survivin up-regulates adhesion molecules on the surface of leukocytes changing their reactivity pattern. J Leukoc Biol. 2008; 83: 149–55.
- Khan S, Jutzy JM, Aspe JR, Asuncion-Valenzuela MM, Park JS, Turay D, Gali-Muhtasib H. The application of membrane vesicles for cancer therapy. 2011; Advances in cancer therapy: intech.
- Zitvogel L, Regnault A, Lozier A, Wolfers J, Flament C, Tenza Det al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med. 1998; 4: 594–600.
- Wolfers J, Lozier A, Raposo G, Regnault A, Thery C, Masurier Cet al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med. 2001; 7: 297–303.
- Couzin J. Cell biology: the ins and outs of exosomes. Science. 2005; 308: 1862–3.
- Chaput N, Flament C, Viaud S, Taieb J, Roux S, Spatz Aet al. Dendritic cell derived-exosomes: biology and clinical implementations. J Leukoc Biol. 2006; 80: 471–8.
- Blanc-Brude OP, Mesri M, Wall NR, Plescia J, Dohi T, Altieri DC. Therapeutic targeting of the survivin pathway in cancer: initiation of mitochondrial apoptosis and suppression of tumor-associated angiogenesis. Clin Cancer Res. 2003; 9: 2683–92. [PubMedAbstract].
- Liu T, Brouha B, Grossman D. Rapid induction of mitochondrial events and caspase-independent apoptosis in Survivin-targeted melanoma cells. Oncogene. 2004; 23: 39–48.
- Nicholson DW. From bench to clinic with apoptosis-based therapeutic agents. Nature. 2000; 407: 810–16.