Abstract
The relationship between the exposure to drug metabolites and overall drug safety has become an integral part of the drug-development process. In-depth discussions in the scientific community, as well as recent guidelines on Drug Safety Testing of Metabolites from the US FDA (often referred to as the MIST guidance and ICH M3(R2) from the International Conference on Harmonization (ICH), has brought clarity to the regulatory requirements of the sponsor company in providing documentation on circulating levels of qualifying metabolites. However, less attention has been given to the challenges now faced by the bioanalytical community in supporting these new guidance policies. In this paper, the European Bioanalysis Forum (EBF) is providing a recommendation on which quality standards to apply when assessing the (relative) abundance or absolute concentrations of metabolites. This paper is the result of both an intensive consultation within the EBF (through internal surveys amongst EBF member companies and discussions) and consultation of the broader bioanalytical community (through discussions at international conferences). These recommendations will provide an increased understanding of how to apply a tiered approach to metabolite quantification as part of the bioanalytical strategy. As such, it aims to provide support to the bioanalytical community on the appropriate level of validation required at each stage of the drug-development process.
Δ: Difference; PA: Peak area; RAD: Radioactivity detection.
PD: Pharmacodynamic; PK: Pharmacokinetic; UD: Unchanged drug.

ICH: International Conference on Harmonization.
FIH: First-in-human; M: Metabolite; MD: Molecular dynamics; PK: Pharmacokinetic; SD: Stochastic dynamics; Tox: Toxicology.
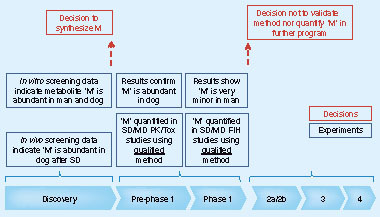
M: Metabolite; MD: Molecular dynamics; PK: Pharmacokinetic; SD: Stochastic dynamics; Tox: Toxicology.
In recent years, the need to provide quantitative data on drug metabolite exposures as part of the overall safety evaluation of drugs in development has been identified. Following the publication of a discussion paper in 2002 Citation[1], a robust scientific discussion on the relative merits, challenges and implications of metabolite quantification in drug development began, and this continued to be debated in peer-reviewed scientific literature Citation[2,3]. The result of this debate was a guidance document from the US FDA; a first draft for comments published in 2006, followed by the final guidance published in 2008, entitled ‘Guidance for Industry, Safety Testing of Drug Metabolites’ Citation[4]. Additionally, the discussion of metabolites being part of safety assessment prompted International Conference on Harmonization (ICH) to include this subject into the ICH M3(R2) guidance, ‘guidance on nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals’ which was finalized in 2009 Citation[5]. Both guidelines provide clear expectations in both the nature and timing of drug metabolite exposure data required to support drug registration.
Although the bioanalytical community is experienced in quantifying metabolites in clinical and preclinical studies, the rationale for metabolite quantification is typically prompted by a need to document concentrations of pharmacologically active metabolites that may be able to contribute to the overall pharmacology of the drug in patients. Bioanalysis is usually performed using validated methods to meet the requirements of the 2001 FDA Guidance on Bioanalytical Methods validation Citation[6]. As a consequence of the regulatory guidance on MIST, safety assessments involving metabolite quantification have assumed a greater degree of importance and are more commonly explored in earlier phases of the drug-development process. As a result, metabolite quantification has become an integral part of the bioanalytical support for the drug-development process, focusing both on quantification for pharmacokinetic and efficacy assessments, as well as on the determination of exposure margins in preclinical species versus human. There has been a significant increase in the effort expended on obtaining quantitative information on drug metabolite exposures in animals and in man. With this increased effort, came the additional challenges of quantifying metabolites that have diverse physicochemical properties, potentially having significant influence on the sample preparation behavior or stability of the metabolite in the biological matrix, as well as on the chromatographic and MS ionization properties. At the same time, limited supplies of reference standards may represent a significant challenge from a chemical synthesis perspective and in providing robust methods and exposure data.
The topic of metabolite quantification was also discussed at the 2006 Crystal City 3 meeting in Arlington (VA, USA) and proceedings from this conference were published in 2007 Citation[7]. In summary, both the FDA and the bioanalytical community jointly recognized that characterization of metabolites (referred to as UMMs, a terminology currently abandoned) should proceed using a flexible ‘tiered’ approach to bioanalytical method validation. This tiered approach, a process driven by a priori established scientifically appropriate criteria, would allow metabolite screening studies in early drug development using bioanalytical methods with limited validation and, subsequently, increasing validation criteria as products move into clinical trials.
Understanding the challenges of metabolite quantification
Following the publication of the MIST guideline and the Crystal City 3 conference report, bioanalytical laboratories worldwide were challenged with an increased demand for metabolite quantification. In a significant number of laboratories, existing processes were not sufficiently robust or flexible to respond to this changing requirement. One approach to addressing these new challenges was a tiered approach to the bioanalysis of metabolites as described by Leclercq et al.Citation[8].
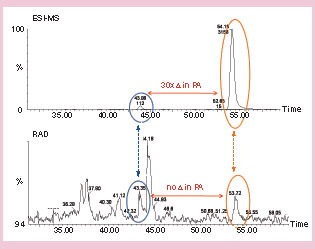
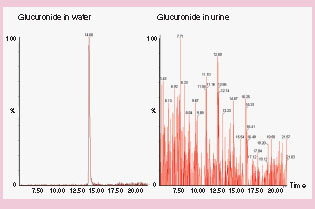
In the early phase of characterizing drug disposition, data arose from multiple sources using a variety of detection techniques. Care needs to be taken in reaching conclusions on the relative importance of metabolites as illustrated in . In , the example shows where the difference in instrument response between LC–ESI–MS/MS and LC–radioactivity detection (RAD) can lead to confusion, if the data are interpreted without the appropriate calibration standards. In , glucuronide metabolites may go undetected if the limitations caused by matrix suppression are not recognized, or if no reference standards or radioactive tracers are available to document susceptibility of the analyte towards matrix suppression.
Metabolites & EBF
In 2007, following the publication of the Crystal City conference report Citation[7], the European Bioanalysis Forum (EBF) agreed that the challenges related to metabolite quantification needed increased attention by the bioanalytical community. Initially, the EBF used a member-based survey to investigate the current laboratory procedures, the frequency at which metabolite quantification was performed and the timing when metabolite quantification was requested. The results highlighted the complexity of metabolite quantification for the bioanalytical laboratory and the multiple levels of quality applied to quantify metabolites in the different stages of development. The results were shared at the first EBF open symposium in Barcelona in 2008 Citation[9].
This survey reinforced activities within the EBF to further focus on providing interpretation and harmonization on the multiple approaches applied in metabolite qualification and quantification. A sub-team was formed within the EBF to propose optimal processes for metabolite quantification in relation to MIST.
The initial goal was to provide guidance and insight on the different tiers of validation required for metabolite quantification during drug development. However, it was soon recognized that the concept of a tiered approach has a broader area of application than in metabolite quantification alone. An array of studies were identified where the use of a validated method as required by the FDA Guidance on Bioanalytical Methods Validation Citation[6,7]) is either not needed or, owing to limitations such as lack of an appropriate reference standard, not possible. As a consequence, the scope of the sub-team was broadened to also prepare recommendations on the application of a tiered approach in other studies besides the studies related to quantification of metabolites.
Whilst this article is limited to providing recommendations on applying best practice for a tiered approach to metabolite quantification, a separate article is being prepared aimed at providing recommendations on the overall implementation of tiered approaches in bioanalytical laboratories. In summary, the proposal from the EBF is to classify the tiered approach into three levels and to provide clarity about the levels and associated requirements for validation and data quality.
Screening methods
These are methods or procedures applied to samples for which no a priori acceptance criteria are available. The outcome may range from qualitative, relative response or estimated abundance and can be used for early decision making. Typically, these assays are applied in studies where absolute quantification is not a prerequisite (i.e., discovery phase or pre-GLP studies, and in vitro studies allowing a relative output or initial metabolite studies).
Qualified methods
These are methods with appropriate levels of scientific validation (e.g., accuracy, precision and stability). When applied to study samples, they generate absolute concentration data, which allow documented and reproducible decision making. Typically these assays are applied in discovery phase, pharmacokinetic studies and in early/late development mechanistic toxicokinetic, pharmacokinetic or pharmacodynamic studies, and in studies where the application of validated assays is either not needed or not possible (i.e., rare matrices and tissue homogenates).
Validated methods
These are methods that follow the procedure around method development and validation as described in the FDA Guidance on Bioanalytical Methods Validation, supplemented with the recommendations from the recently published conference report Citation[6,7]. Guidelines from other regulatory authorities (i.e., European Medicines Agency [EMA]) may apply as they become relevant.
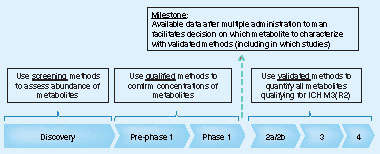
visualizes the bioanalytical support given for preclinical and clinical studies through a typical development process relative to the quality level proposed for these studies in support of assessing the exposure of drugs (and metabolites).
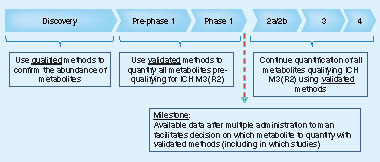
A separate article focuses on the definition and application of qualified assays in support of PK or related studies and will provide a recommendation to the bioanalytical community on how to make best use of the laboratory resources while safeguarding scientific quality, as well as data integrity and overall compliance. For biomarkers, the fit-for-purpose strategy, as described in Lee’s paper Citation[10] would of course still be the preferred route. In , a summary is provided on the level of validation or qualification experiments required in each of the three tiers. The results were discussed and shared at the Second EBF open symposium in Barcelona in 2008 Citation[11].
Application of tiered approach to MIST
Quantification of metabolites provides multiple bioanalytical challenges, which may be distinctly different from the analysis of the parent compound. Availability of reference material of drug metabolites, increased incidence of biochemical instability and insufficient identification of stereochemical structure are only a few of the increased challenges of metabolite quantification. A joint acknowledgement by the FDA and the pharmaceutical industry of the principles of a tiered approach to metabolite quantification offers the possibility of focusing on the scientific aspects, as well as the regulatory needs in support of metabolite characterization. This article offers additional insights, including the provision of a template that may serve as a starting point for decisions on which the level of quality (screening, qualification or validation) is applicable at the various stages of development when quantifying metabolites in the context of MIST. It is important to note that the EBF recommendation is not intended to replace the ICH M3(R2) or MIST guidance, for example when deciding which metabolite qualifies for further characterization, but offers a means by which to address the FDA and ICH guidance with respect to the bioanalytical challenges encountered when quantifying metabolites in different stages of pharmaceutical development.
Any qualitative prior knowledge on the identity of the metabolites available from in silico, in vitro or in vivo metabolite identification work should be integrated in the bioanalytical strategy. Furthermore, when the bioanalytical strategy has been defined, newly identified metabolites can be included as part of the quantification strategy or the quantification strategy can be revised based on the interpretation of additional quantitative data at any point in its application.
From a unique bioanalytical perspective, and prior to embarking on any quantification in the laboratory, the EBF identified only two types of metabolites in connection to the MIST guideline.
Type 1
Metabolites with anticipated low or unknown contribution to the pharmacological activity or toxicity of parent drug in humans. Prior to having data on the abundance of a metabolite, all metabolites without structural alerts would qualify for this category. Most common examples are conjugates resulting from phase 2 metabolism (e.g., glucuronides). For the quantification of these metabolites, the EBF proposes to use process A, in which all three tiers of quality are defined: screening, qualified and validated.
Type 2
Metabolites with known and anticipated contribution to the pharmacological activity, or known or anticipated contribution to the toxicity (i.e., structural alerts) of parent drug in humans. For the quantification of these metabolites, the EBF proposes to apply process B, in which only two tiers of quality are defined: qualified and validated.
At any stage in the drug development, and based on new information that becomes available on the pharmacological activity or toxicity, a metabolite can move from type 1 to type 2 or vice versa.
Process A: metabolites with anticipated low or unknown contribution to the pharmacological activity or toxicity
For compounds qualifying for process A, the EBF recommends the following steps:
For studies spanning the discovery phase of development, screening methods should suffice to assess the presence of metabolites in a limited but well-selected number of samples originating from a limited number of in vivo and/or in vitro studies. Data generated during this phase should give an indication on the relative abundance of the most important metabolites and will guide the responsible scientist to decide whether metabolites qualify for quantification using qualified assays during the preclinical phase and the first-in-human studies. In most cases, this decision is made by a team where the bioanalyst works together with the pharmacokineticist (nonclinical and clinical), toxicologist and/or pharmacologist to give the right interpretation of the metabolite concentration data;
From the start of the GLP studies until a definitive assessment of the exposure in samples from human volunteers is possible, the EBF recommends quantifying metabolites that were flagged for further characterization in the discovery phase using qualified methods. The quantification can be bundled into discrete bioanalytical campaigns and limited to a selection of samples from a selected number of studies. Typically, single and multiple dose studies in the toxicological species, as well as single and multiple ascending dose studies in human volunteers/patients, qualify as studies from which samples should be selected. With respect to the timing of the actual analysis, the bioanalyst must be mindful of the sampling and storage conditions and their potential impact on the stability of the metabolites. This may be a driver of the timing of the bioanalytical phase and may be a reason to perform the analysis soon after the sampling. On the other hand, it may be beneficial to postpone the quantification until the samples from all species/selected studies, including human, are available. The analysis can then be performed in one or two well-designed bioanalytical experiments (e.g., including matrix matching to cover for species and/or matrix differences). If the anticipated stability of the metabolites allows, the latter strategy is preferred because potential effects of interbatch variability will be limited and cross-species comparison is optimally warranted. Limitations in availability of appropriate reference standards of certain metabolites will influence bioanalytical strategy which can be applied. For some metabolites, it will be impossible to generate absolute quantitative data using qualified assays. For these cases, screening assays can be applied to generate relative abundance data, as these screening assays are suited to provide insight if a certain metabolite is disproportionally present in samples from human volunteers or not. In cases where a reference standard is not available, it may even be a prerequisite that samples from different species are analyzed in one analytical experiment to safeguard comparison across the species. Also, the lack of appropriate standards at this stage will impact the decision making for the next phase or call for increased efforts to synthesize appropriate reference standards;
All data available allowing the first inter-species comparison between man and the preclinical species, should enable strategic guidance on which metabolites need to be quantified in future preclinical and/or clinical studies. In line with the MIST and ICH M3(R2) guidance documents, some metabolites will require further characterization using validated methods. Depending on the level of complexity or the physicochemical difference of the metabolites and the unchanged drug, the bioanalytical scientist may decide to validate combined assays for one or more metabolites or continue with separate assays. Again, for metabolites that are extremely difficult to synthesize, the bioanalytical scientist may need to deviate from the desired strategy and continue characterizing the metabolite using alternative methods until an appropriate reference standard is available. Last but not least, special consideration should be given to the number of studies and how long the quantification of the metabolite(s) needs to continue. For some metabolites, such as those with high abundance or with a significant contribution to the activity, it can be unequivocally necessary to continue quantification in all future studies in the development process or even after registration of the drug. At the same time, for some metabolites, the opposite may be the case and their quantification should be limited in time and discontinued when full characterization is achieved. The fact that a metabolite ‘can’ be quantified does not mean it ‘should’ be quantified in each and every sample.
Process B: metabolites with known or anticipated contribution to the pharmacological activity or toxicity
For metabolites with a known or anticipated contribution to the pharmacological activity or toxicity, the EBF recommends applying process B. Prior knowledge of the potential contribution of some metabolites on the toxicological and/or pharmacological effect can lead to a decision to already apply higher quality standards earlier on in the drug-development process, to assess the relative and absolute exposure of certain metabolites:
In the discovery stage, an understanding of the potential contribution of metabolites to the pharmacological activity/toxicity of a compound represents valuable information. As a consequence, investment in providing absolute concentration data allowing early decision making to optimize the compound selection or deselection is desirable. Hence, quantifying the drug and the metabolite with a qualified method should already be considered. Prior to the decision to set up a qualified assay, other screening assays may have provided sufficient and reliable information to decide on the deselection of a compound (e.g., metabolites with structural alerts) or redirection of the design-make-test cycle towards a more favorable chemical space. The real need to set up qualified assays for metabolites in this early stage can therefore be limited but should nevertheless be considered;
Moving forward, as early as in the first GLP studies, some metabolites may qualify for quantification with a validated assay as part of the validation strategy for that particular project. There should be sufficient consideration that at this stage of development, compliance with the MIST or ICH M3 guidance may not be the reason for the decision to set up a validated method (the impact of human exposure cannot be assessed at this stage) and other considerations may influence the bioanalytical strategy. In order to understand the total exposure of all analytes contributing to pharmacological or toxicological effect, exposure data may be needed prior to first dosing in humans. Consequently, a more accurate assessment of the risk/benefit and/or safety margin towards first into human dosing can be more relevant and a driver for the bioanalytical strategy. Similarly, this strategy may continue into the first-in-human studies in order to generate comparable exposure data in humans;
Interpretation of the available concentration data from preclinical and/or clinical studies after the multiple ascending dose to humans should enable strategic guidance as to which metabolites qualify for further quantification in preclinical and/or clinical studies. Typically at that stage in drug development, all the data from validated, qualified or screening methods are available to relate the exposure data to the MIST and ICH M3(R2) guidance document. Similarly, as for process A, there may be metabolites that are extremely difficult to synthesize, which results in a change from the desired strategy and the continuing characterization of these metabolites using alternative levels of quality until a reference standard of appropriate quality is available.
Interpretation of exposure data & examples for the different processes
The interpretation of exposure data can lead to multiple strategic analytical approaches required from the bioanalyst. Some illustrative examples are given below, (clearly, other scenarios are possible):
Exposure of humans to metabolite(s) is higher than in preclinical species. Consequently, metabolite(s) will (continue to) qualify for quantification with validated method(s) until additional safety data become available on the metabolites in preclinical species (e.g., higher exposure data in those species compared with humans);
Exposure of humans to metabolite(s) is lower than in preclinical species. Consequently, metabolite quantification may be discontinued;
Exposure of humans to metabolite(s) is lower than in preclinical species, but still exceeds the exposure limits mentioned in the ICH3 M3(R2) guidelines (being different from the MIST guidance). In this case, metabolite quantification may continue;
Exposure of humans to metabolite(s) is lower than in preclinical species, but metabolite quantification may be continued for other reasons than stipulated in the ICH M3(R2) guidance, such as to document metabolite exposure in support of a back-up program or other team or company related strategies.
From the above examples, these potential outcomes will contribute to the bioanalytical strategy for metabolite quantification, and careful deliberation is needed to build a strategy that fulfills compliance to all existing guidelines. The proposed bioanalytical strategy is summarized in . In , some actual examples are given how the above processes can be applied.
describes an example of process A, where screening data led to the decision to synthesize a mono hydroxylated metabolite. Data from in vivo and in vitro screening assays suggested that this metabolite may be important in humans. As such, more data needed to be gathered prior to Phase I and at the occasion of the first-in-human studies. Exposure data, generated with qualified methods in pre-Phase I, confirmed the high abundance of this hydroxylated metabolite in dogs. Nevertheless, and in contrast to in vitro data, the hydroxylated metabolite appeared to be minor after both single and multiple ascending dose in humans. In humans, unlike in dogs, hydroxylation of the parent drug followed by metabolic phase 2 conjugation was more abundant. If not already carried out as part of the initial validation of the bioanalytical method for the unchanged drug, the observation made on the conjugated metabolite may trigger additional stability and validation work. More importantly, and unless there are no other reasons (i.e., significant contribution to the pharmacological activity or susceptibility for drug–drug interactions), the data generated on the metabolite in humans and dogs should invite the team to abandon any further quantification of this metabolite in the continuation of the program.
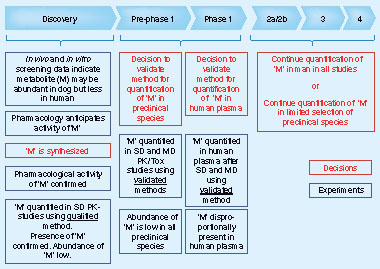
describes an example of a pharmacologically active metabolite. In addition, compounds from earlier development programs within the same chemical space have suggested that this metabolic pathway gave rise to pharmacologically active metabolites with longer half-lives in humans, as compared with preclinical species (i.e., the preclinical species may not be fully predictive for human PK). Upon availability of in vitro and in vivo screening data for this predicted metabolite, synthesis is initiated and the pharmacological activity confirmed. Therefore, absolute concentration data are requested to confirm the identity and abundance of the metabolite in vivo. Although in vivo and in vitro data agree on the qualitative presence of the active metabolite, quantitative data do not correspond and in vitro data potentially overestimate the importance of the metabolite. Nevertheless, and in view of the importance of the potential contribution of the metabolite to the overall pharmacology of drug-related dosed material, it is decided to set up validated assays for the metabolite in preclinical (toxicology) species and human. Preclinical data show that the abundance of the metabolite is relatively low. However, PK data in humans confirm that the metabolite has a significantly longer half-life in humans compared with preclinical species, confirming the prediction that the metabolite might be disproportionally present in humans compared with the preclinical species. Whilst potential actions are outside the scope of the recommendations given in this paper, it is evident that experiences in drug development suggest a number of actions may be undertaken, most usually following discussions with various subject matter experts. Most commonly, the generation of additional safety and exposure data on the metabolite in existing or additional preclinical species is undertaken. From a bioanalytical perspective, this example calls for a decision to continue the measurement in both preclinical and clinical studies using validated methods until additional exposure data in preclinical species have become available. At that point in time, the quantification of the metabolite in preclinical species may be abandoned, but the quantification may still continue in a selection or all human studies in the continuation of the program.
Conclusion
In conclusion, the EBF recommends using only three tiers of quality to assess the (relative) abundance or absolute concentrations of circulating metabolites: screening, qualifed or validated. For metabolites with anticipated low or unknown contribution to pharmacological activity or toxicity, we propose to use a combination of screening and qualified methods until sufficient data in human volunteers are available. At that moment, the bioanalytical strategy should be aligned with the ICH M3(R2) or MIST guidelines, resulting in the establishment of validated methods for those metabolites that qualify for continued documenting, or, as the situation dictates, the potential discontinuation of the quantication of some (or all) metabolites. For metabolites with known or anticipated contribution to activity or toxicity, we propose to already consider using a combination of qualified or validated methods earlier in the development process, potentially even prior to dosing to human volunteers. Again, when metabolite exposures in experimental animals can be correlated with exposure of these metabolites in human volunteers/patients, the bioanalytical strategy can be aligned to the ICH M3(R2) or MIST guidelines.
Table 1. Key elements of screening, qualified assays and validated assays as compared with screening and validated methods: ‘fit-for-purpose criteria’ applicable for biomarkers analysis may differ†.
Disclaimer
The views expressed in this article are those of the European Bioanalysis Forum and do not necessarily reflect the respective company’s position on the subject.
Contributors from other EBF member companies:
Stephanie Fischmann (Abbot)
Susanne Globig (Actelion Pharmaceuticals Ltd)
Carl-Johan Sennbro (Active Biotech)
Josep M Jansat (Laboratorios Almirall SA)
Hans Mulder (Astellas)
Elizabeth Thomas (AstraZeneca)
Margrete Brundy-Kloeppel (Bayer Schering Pharma AG)
Silke Luedtke (Boehringer-Ingelheim)
Magnus Knutsson (Ferring Pharmaceuticals
Klaus Pusecker (Grünenthal GmbH)
Stephen A White (GlaxoSmithKline)
Berthold Lausecker (F.Hoffman-La Roche)
Nathalie Mokrzycki-Issartel (MSD)
Fernando Romero (Novartis Pharma AG)
Michael Pilgård Anderson (Novo Nordisk)
Norbert Knebel (Nycomed)
Marcel de Zwart (MSD)
Dietmar Schmidt (Sanofi-Aventis)
Peter van Amsterdam (Abbot)
Richard Abbott (Shire Pharmaceuticals)
Pierre Boulanger (UCB Pharma)
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties No writing assistance was utilized in the production of this manuscript.
Bibliography
- Baillie T , CayenMN, FoudaHet al. Drug metabolites in safety testing. Toxicol. Appl. Pharmacol. 182, 188–196 (2002).
- Smith D , ObachRS. Seeing through the MIST: abundance versus percentage. Commentary on metabolites in safety testing. Drug Metab. Dispos. 33, 1409–1417 (2005).
- Robison T , JacobsA. Metabolites in safety testing. Bioanalysis1(7), 1193–1200 (2009).
- US FDA Guidance for Industry, Safety Testing of Drug Metabolites. US Department of Health and Human Services Food and Drug Administration. Center for drug evaluation and research (CDER). Pharmacology Toxicology (2008).
- CPMP/ICH/286/95 - ICH Topic M 3 (R2) Non-Clinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals (2009).
- Guidance for Industry Bioanalytical Method Validation, US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CVM) (2001).
- Viswanathan C , BansalS, BoothBet al. Workshop/Conference Report – quantitative bioanalytical methods validation and implementation: best practices for chromatographic and ligand binding assays. AAPS J. 9(1), E30–E42 (2007).
- Leclercq L Cuyckens F , de VriesRet al. Which human metabolites have we MIST? Retrospective analysis, practical aspects, and perspectives for metabolite identification and quantification in pharmaceutical development. Chem. Res. Toxicol. 22(2), 280–293 (2009).
- Abbott R , Brudny-KloeppelM. Conference report from 1st open symposium – burning issues in bioanalysis. Bioanalysis1(2), 273–276 (2009).
- Lee J , DevanarayanV, BarrettYCet al. Fit-for-purpose method development and validation for successful biomarker measurement. Pharm. Res. 23(2), 312–328 (2006).
- Abbott R . Conference report from 2nd Open Symposium – the broadening scope of validation. Bioanalysis2(4), 703–708 (2010).