
Abstract
X-linked inhibitor of apoptosis (XIAP) and Chk1 are potential molecular targets in radiotherapy. However, their molecular association in the regulation of radiation sensitivity has been rarely studied. Here, we show that XIAP modulates radiation sensitivity by regulating stability of Chk1 in lung cancer cells. Both Chk1 and XIAP are highly expressed in various lung cancer cells. Overexpression of XIAP increased cell survival following genotoxic treatments by preventing downregulation of Chk1. However, XIAP reversed Chk1-protective activity in the presence of XIAP-associated factor 1 (XAF1) by degrading Chk1 via ubiquitination-dependent proteasomal proteolysis. The XIAP-XAF1 complex-mediated Chk1 degradation also required CUL4A and DDB1. Chk1 or XIAP was associated with DDB1 and CUL4A. Depletion of CUL4A or DDB1 prevented the XIAP-XAF1-mediated Chk1 degradation suggesting involvement of a CUL4A/DDB1-based E3 ubiquitin ligase in the process or its collaboration with XIAP E3 ligase activity. Taken together, our findings show that XIAP plays a dual role in modulation of Chk1 stability and cell viability following IR. In the absence of XAF1, XIAP stabilizes Chk1 under IR with corresponding increase of cell viability. By contrast, when XAF1 is overexpressed, XIAP facilitates Chk1 degradation, which leads to enhancement of radiation sensitivity. This selective regulation of Chk1 stability by XIAP and XAF1 could be harnessed to devise a strategy to modulate radiation sensitivity in lung cancer cells.
Abbreviations
| IR | = | ionizing radiation |
| DDR | = | DNA damage responses |
| XIAP | = | X-linked inhibitor of apoptosis |
| Chk1 | = | Checkpoint kinase 1 |
| XAF1 | = | XIAP-associated factor 1 |
| IFN-γ | = | interferon-gamma |
Introduction
The use of ionizing radiation (IR) in radiotherapy induces DNA damage, which in turn leads to various cellular responses including tumor cell death. Diverse cellular pathways have evolved to respond to the damage. The DNA damage response (DDR) involves the sensing of DNA injury as well as transduction of DDR signaling to various effector molecules to induce cell cycle checkpoints, DNA repair and cell death.Citation1
For instance, ATM (ataxia telangiectasia mutated) and ATR (ATM-Rad3-related) serve as initiating kinases following IR-induced DNA damage, and trigger cascade of DNA damage responses. Activated ATR and ATM phosphorylate and activate Chk1 and Chk2 respectively, and these in turn transduce signals to effector molecules such as CDC25A to cause cell cycle checkpoint.Citation2-4
Since the DDR coordinates cellular responses following genotoxic treatments, upregulation of the DDR activity protects cells from DNA injury by enhanced DNA repair, whereas attenuation or loss of DDR factors sensitizes cells to the treatments.Citation5,6 Therefore, disruption of the DDR may increase the impact of DNA injury following IR and enhance radiation sensitivity of cancer cells.
Chk1 is an essential factor in DDR and plays a crucial role in the S- and G2 checkpoints as well as in mitotic spindle checkpoint.Citation7 Genotoxic treatments induce G1 arrest mainly in a p53-dependent manner and G2 arrest in a Chk1-dependent manner. Since many cancer cells are defective in p53, these cells require Chk1-induced G2 checkpoint for their survival. As such, Chk1 is considered as a promising target for cancer therapy. Combined treatment of cancer cells with DNA-damaging agents and Chk1 inhibitors enhances efficacy of DNA damaging agents.Citation8 Or knockdown of Chk1 increases the sensitivity of p53-defective cancer cells to IR.Citation9 As the Chk1-mediated checkpoints are crucial for the DDR, the protein is tightly regulated. Activation of Chk1 by ATR-mediated phosphorylation on serine 317 and serine 345 leads to its ubiquitin-mediated degradation by E3 ubiquitin ligases that employ CUL1 and CUL4A.Citation10-12 Therefore modulation of Chk1 stability could be a key factor for cancer therapy.
X-linked inhibitor of apoptosis (XIAP), a member of the inhibitor of apoptosis (IAP) family, inhibits caspases and blocks the apoptotic pathway.Citation13 As such, overexpression of XIAP confers resistance to chemotherapy or to radiation,Citation14-16 whereas downregulation of XIAP restores chemo- or radiosensitivity to various cancer cell lines.Citation17-19 Domain structure of XIAP protein and binding of the domain with various cellular proteins determine the various roles of XIAP. For instance, the baculovirus IAP repeat (BIR) domains of XIAP bind and inhibit various caspases, whereas the RING domain of XIAP has E3 ubiquitin ligase activity and ubiquitylates a variety of substrates including caspases.Citation20 XIAP-associated factor 1 (XAF1) binds the RING domain of XIAP and activates E3 ubiquitin ligase activity of XIAP that targets Survivin for proteasomal degradation.Citation21,22 Moreover, XAF1 shows proapoptotic effects by reversing the anti-caspase activity of XIAP.Citation23
XIAP and XAF1 have been implicated in modulation of Chk1 and cell cycle. XIAP binds Chk1 as XAF1 does.Citation24,25 XIAP interacts with Chk1 during mitosis with implication in regulation of cell cycle and apoptosis.Citation24 XAF1 also interacts with and activates Chk1, which in turn results in G2/M cell cycle arrest through inactivation of CDC2-cyclinB complex.Citation25 Overexpression of XAF1 suppresses cancer cell growth and induces mitotic catastrophe.Citation25
XIAP and XAF1 have been also implicated in modulation of cellular radiation responses. Upregulation of XIAP following low dose ionizing irradiation increases resistance to radiation in non-small cell lung carcinoma,Citation16 whereas downregulation of XIAP by antisense approach in lung cancer cells enhances radiation sensitivity.Citation19 XAF1 overexpression by transient transfection or XAF1-inducing interferon-gamma (IFN-γ) treatment enhances radiation sensitivity in HeLa cells.Citation26 These results of XIAP, XAF1 and Chk1 imply that physical and functional association of the proteins might operate to modulate cellular processes such as radiation responses.
We explored the possibility whether physical association of XIAP, XAF1 and Chk1 contributes to modulation of Chk1 and to cellular radiation responses. We observed formation of a complex composed of XIAP, XAF1 and Chk1. In the absence of XAF1, XIAP protected Chk1 from downregulation following genotoxic stresses, whereas when XAF1 is present, XIAP reversed the activity by degrading Chk1 suggesting a dual function of XIAP. In addition, we elucidated mechanism of the XIAP-XAF1-mediated Chk1 degradation and identified E3 ubiquitin ligase components that are involved in the protein degradation. Chk1 downregulation by the XIAP-XAF1 complex or by XAF1-inducing IFN-γ treatment enhanced radiation sensitivity in lung cancer cells. These results might be utilized to develop a strategy in radiotherapy to enhance cancer cell killing through modulation of Chk1 protein stability.
Results
XIAP protects Chk1 from downregulation following genotoxic treatments
Both XIAP and Chk1 have been considered as potential therapeutic targets for cancer treatment in which downregulation or inhibition of the proteins in cancer cells enhances their sensitivity to chemo- or radiotherapy.Citation27-29
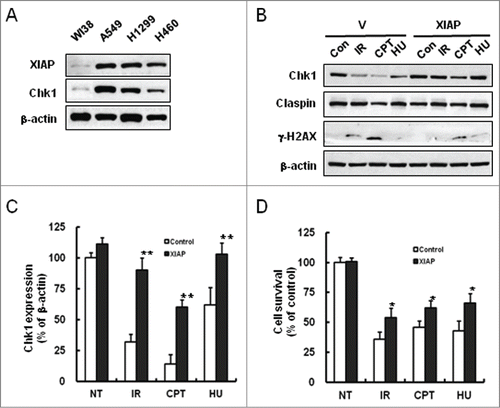
We sought to find relationship between XIAP and Chk1, and thus examined endogenous expression levels of the proteins in several lung cancer cell lines such as A549, H1299 and NCI-H460. Interestingly, expression levels of XIAP and Chk1 were correlated in the cells with elevated expression when compared to WI38 normal lung cells (). Since Chk1 protein is degraded following treatment with certain types of genotoxic agents,Citation12 we examined whether XIAP overexpression has any effect on Chk1 stability under those conditions. Treatment of empty vector-transfected A549 cells with various genotoxic agents such as comphthecin (CPT), hydroxyurea (HU), or IR decreased endogenous levels of Chk1, whereas XIAP-overexpressed cells by transfection with XIAP-encoding construct preserved significant amount of Chk1 protein under the same experimental conditions ( and C). In accordance with a role of XIAP in cellular protection under genotoxic treatments, transient overexpression of XIAP increased cell survival following treatments with IR, CPT or HU as shown by MTT assay (). These results suggest that overexpression of XIAP in the lung cancer cells might enhance cell survival following genotoxic treatments by preventing down-regulation of Chk1.
Figure 1. Prevention of Chk1 downregulation by XIAP under genotoxic treatments. (A) Whole cell extracts from WI38, A549, NCI-H1299, and NCI-H460 cells were immunoblotted with XIAP, Chk1, and β-actin antibodies. WI38 is a human diploid lung fibroblast cell line, whereas the others are human lung cancer cell lines. (B) A549 cells were transfected either with vector control or with plasmid construct encoding XIAP and incubated for 36 hr. The cells were harvested after 5 hr following treatments with 10 Gy IR, 500 nM camptothecin (CPT), or 5 mM hydroxyurea (HU). Whole cell extracts were prepared and immunoblotted with the specified antibodies. β-actin antibody was used as a protein loading control. γ-H2AX detection was used as a marker of DNA damage. (C) Graphical representation of the results in B. The relative Chk1 protein levels were normalized to β-actin expression as determined by densitometry. Values are means ± standard deviation (SD) of 3 experiments. **P < 0.01 versus control. (D) Cell survival was measured by MTT assay following genotoxic treatments. A549 cells were transfected either with vector control (control) or with plasmid construct encoding XIAP (XIAP) and treated as in B. The cell survival was normalized against untreated control (NT). Values are means ± SD. Statistical analysis of the results was carried out as in C.
XIAP determines radiation sensitivity by modulating Chk1 levels
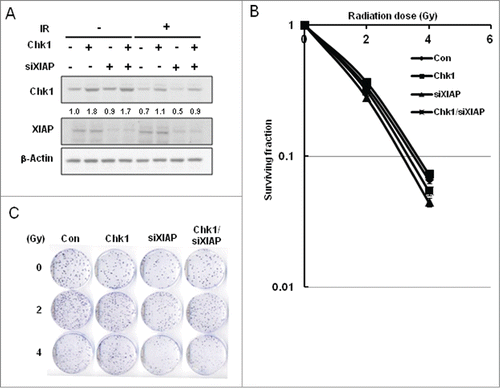
Since Chk1 is essential for cell survival following IR,Citation30 methods that are able to downregulate Chk1 levels in cancer cells might be combined with IR to enhance cancer cell killing. Thus we tested whether depletion of XIAP by its corresponding siRNA results in IR-induced Chk1 downregulation and consequent decrease of cell survival. Chk1 levels were affected little by XIAP depletion under normal condition (non-IR), whereas Chk1 levels decreased by IR with further decrease by XIAP depletion (). Results of clonogenic cell survival assay confirmed the notion that XIAP depletion enhanced IR-induced cancer cell killing (). Clonogenic cell survival was minimal following IR with XIAP depletion in which Chk1 expression was most severely downregulated (). The results support the concept that downregulation of Chk1 by XIAP depletion enhances cancer cell killing when it is combined with IR treatment.
Figure 2. Enhancement of cellular radiation sensitivity by depletion of XIAP. (A) A549 cells were transfected with Chk1 construct and/or with SiRNAs against XIAP. Cells were treated with no IR or with 5 Gy of IR. Whole cell extracts were used to detect Chk1 and XIAP by protein gel blot analysis. β-actin was used as a loading control. The band intensity of Chk1 in each lane is normalized against that of the untreated control (lane 1). (B) Clonogenic cell survival was measured for the cells: untreated control (Con), Chk1-overexpressed cells by transient transfection (Chk1), XIAP-depleted cells by the corresponding SiRNA transfection (siXIAP), and Chk1-overexpressed cells with XIAP depletion (Chk1/siXIAP). Cell survival values were normalized to those of the unirradiated cells. (C) Colony formation of the cells, which were treated as in B, is shown.
Overexpressed XAF1 induces Chk1 downregulation
As modulation of Chk1 expression levels appears to be crucial to cellular radiation sensitivity, we reasoned that additional cellular factors might participate in the process in association with XIAP. XAF1 has been recognized as a regulator of both Chk1 and XIAP. For instance, XAF1 interacts with Chk1 and regulates G2/M cell cycle checkpoint.Citation25 Moreover, XAF1 has been known to downregulate Survivin through a complex containing XIAP.Citation22 Therefore, we sought to determine whether XAF1 is involved in the regulation of Chk1 in association with XIAP.
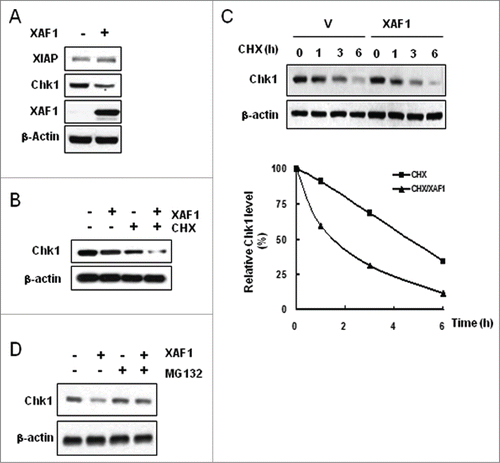
Overexpressed XAF1 by transient transfection downregulated endogenous Chk1, whereas XAF1 had little effect on XIAP (). Endogenous expression of XAF1 in vector-transfected A549 lung cancer cells was absent as reported.Citation31 XAF1-induced Chk1 downregulation seems to be caused by posttranslational mechanism, because cycloheximide (CHX) treatment in the XAF1-overexpressed cells promoted a reduction of Chk1 protein when compared to cells with CHX treatment alone (). Measurement of Chk1 half-life with or without XAF1 overexpression confirmed the notion by showing that Chk1 half-life was shortened about 3 times by XAF1 overexpression (). XAF1-induced Chk1 downregulation was restored in the presence of the proteasome inhibitor MG132, suggesting proteasomal degradation of Chk1 under XAF1 overexpression ().
Figure 3. XAF1-mediated degradation of Chk1. (A) A549 cells were transfected with control vector or with plasmid construct encoding XAF1, and incubated for 30 hr. Whole cell extracts were immunoblotted with the indicated antibodies. β-actin was used as a loading control. (B) XAF1- or empty vector-expressed cells were treated for 4 hr with DMSO or with 100μg/ml cycloheximide (CHX). Levels of Chk1 protein were determined by protein gel blot analysis. β-actin was used as a loading control. (C) XAF1- or empty vector-expressed cells were treated with 100μg/ml CHX for the indicated times. Levels of Chk1 protein were determined by protein gel blot analysis (upper panel). Band intensity of Chk1 protein was determined by densitometry, and normalized against the zero hour control, which was set as 100%. Graphical representation of the densitometric analysis of Chk1 allows measurement of Chk1 half-life (Lower panel). (D) XAF1- or empty vector-expressed cells were treated with 10 μM MG132 for 4 hr. Levels of Chk1 protein were determined by western blotting analysis. β-actin was used as a loading control.
XIAP-XAF1 complex mediates Chk1 downregulation
The RING domain of XIAP functions as an E3 ligase to catalyze the ubiquitination of target protein substrates.Citation20 Survivin, a target of XIAP, is downregulated by a mechanism that requires XAF1 and an intact XIAP RING domain.Citation22 Since this mechanism suggests that XAF1 affects Survivin stability via its interaction with XIAP, we explored the possibility whether XAF1 downregulates Chk1 via its interaction with XIAP.
Endogenous Chk1 was immunoprecipitated from vector- or XAF1 construct-transfected cells and precipitates were analyzed for the presence of XIAP and XAF1 by western blotting. As seen in , both XIAP and XAF1 were coprecipitated with Chk1 (lane 2) in XAF1-overexpressed cells, whereas only Chk1-XIAP interaction was observed in XAF1-absent control cells. We also examined whether XIAP or XAF1 expression alone or their coexpression affects formation of the XIAP-XAF1-Chk1 complex. More Chk1-XAF1 association was observed in XIAP/XAF1 coexpression condition than XAF1 expression alone () suggesting that XIAP determines degree of Chk1-XAF1 association. The Chk1-XAF1 association was inversely correlated with Chk1 protein levels. XAF1 expression alone was able to downregulate Chk1, whereas XIAP/XAF1 coexpression degraded more Chk1 protein (). In order to corroborate the idea that XIAP is a critical factor in XAF1-mediated Chk1 downregulation, we depleted XIAP in XAF1-overexpressed cells and examined Chk1-XAF1 association by immunoprecipitation. XAF1 was coprecipitated with endogenous Chk1 in XAF1-overexpressed cells as shown in (lane 2), whereas the XAF1-Chk1 association was barely detected when XIAP was depleted in XAF1-overexpressed cells ( lane 3). This abrogation of the XAF1-Chk1 association by depletion of XIAP underscores a critical role of XIAP in formation of the complex. Interestingly, depletion of XIAP partly prevented Chk1 downregulation caused by overexpression of XAF1 (). Together, these results suggest that XIAP plays a crucial role in formation of the Chk1-XAF1 complex and that the XIAP- XAF1 complex mediates Chk1 downregulation.
Figure 4. Ubiquitin-dependent Chk1 proteolysis is mediated by the XIAP-XAF1 complex. (A) A549 cells were transfected with XAF1-encoding construct or with empty vector. Whole cell extracts were prepared, immunoprecipitated with Chk1 antibody, and immunoblotted with XIAP and with XAF1 antibodies. (B) A549 cells were transfected with the constructs encoding the proteins as specified (V, XIAP, XAF1, and XIAP/XAF1). XIAP/XAF1 indicates co-transfection with both constructs. V is an empty vector transfection. Cell extracts were prepared and then immunoblotted (IB) with the indicated antibodies (upper panel). Also Chk1 or XIAP was immunoprecipitated (IP) and co-IP of XAF1 was assessed by protein gel blot analysis (IB) (lower panel). (C) Cells with or without XAF1 overexpression were transfected with XIAP siRNA or scrambled siRNA as a control. Cell extracts were subjected to protein gel blot analysis (IB) (upper panel) and immunoprecipitation (IP) with the indicated antibodies (lower panel). Co-IP of XAF1 was assessed by protein gel blot analysis (IB). (D) A549 cells with or without co-overexpression of XAF1 and XIAP were transfected with a plasmid construct encoding ubiquitin. Ubiquitination of total proteins was determined by protein gel blot analysis of whole cell extracts (left panel). Ubiquitination of endogenous Chk1 was monitored by immunoprecipitation (IP) with antibody against ubiquitin. Co-IP of Chk1 was assessed by protein gel blot analysis (IB) (right panel). (E) A549 cells were transfected with XAF1 alone or co-transfected with XAF1 and XIAP (XAF1/XIAP). Vector (V)-transfection is a control. Cells were fixed, incubated with anti-Chk1 antibody, and stained with Alexa 488-conjugated secondary antibody. Nuclei were stained with DAPI (blue, x1000). Fluorescent images were obtained using a fluorescence microscope.
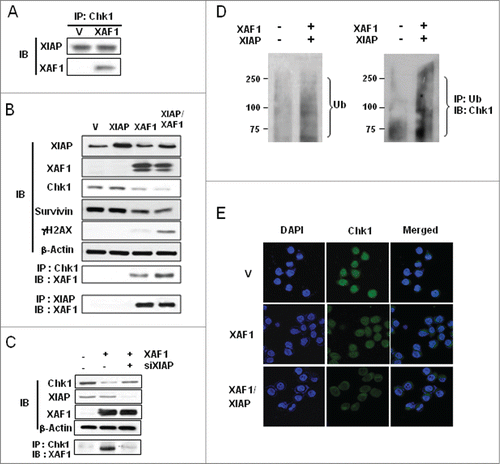
XIAP/XAF1 coexpression increased ubiquitination of cellular proteins when whole cell lysates were examined by protein gel blot analysis (, left panel). More importantly the same condition increased ubiquitination of Chk1, which is evidenced by the appearance of higher molecular weight species of Chk1 (, right panel). These results suggest involvement of the ubiquitin-proteasome pathway in the XIAP-XAF1-mediated Chk1 turnover.
Interestingly, Chk1 was mainly localized in the cytoplasm in XIAP/XAF1-transfected cells, whereas Chk1 was present in the nucleus in vector-transfected control cells in accord with previous results on Chk1 localization ().Citation32 This might have implication in enhancement of radiation sensitivity in which the XIAP-XAF1-induced degradation and abnormal localization of Chk1 might result in enhanced cancer cell killing following IR treatment.
Involvement of DDB1 and CUL4A in Chk1 degradation by the XIAP- XAF1 complex
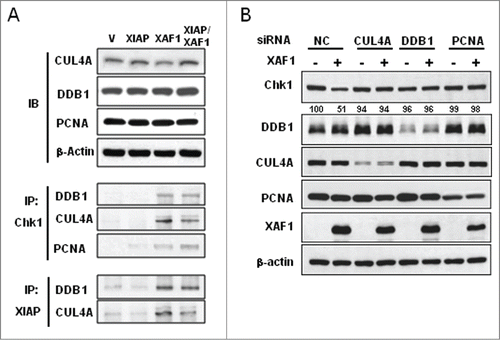
It was previously reported that ubiquitination and degradation of Chk1 is regulated by several E3 ligase complexes such as DDB1-CUL4A or Skp1-Cul1-Fbx6.Citation10-12,33 We were interested in whether those components of E3 ligases are involved in the XIAP-XAF1 complex-mediated turnover of Chk1. Immunoprecipitation experiments showed that association of DDB1, CUL4A, and PCNA with Chk1 was prominent when XAF1 alone or both XAF1 and XIAP were coexpressed (, lane 3 and 4). Similarly, association of XIAP with DDB1 and CUL4A proteins was enhanced under the same conditions (). XIAP overexpression alone had little effect on the complex formation of Chk1-DDB1-CUL4A-PCNA or of XIAP-DDB1-CUL4A (, lane 2) suggesting that XAF1 overexpression is required to form such complexes. Association of different classes of E3 ligases suggests possible collaboration in Chk1 turnover under XAF1 overexpression. The XIAP-XAF1 complex might collaborate with the DDB1-CUL4A E3 ligase complex to degrade Chk1. To test the idea, we depleted CUL4A, DDB1 or PCNA in the cells with or without XAF1 overexpression. Knockdown of DDB1, CUL4A or PCNA by using corresponding siRNA prevented Chk1 degradation even in XAF1-overexpressed cells (). This result suggests that DDB1, CUL4A and PCNA might collaborate with the XIAP-XAF1 complex in degrading Chk1.
Figure 5. Involvement of DDB1 and CUL4A in the XIAP-XAF1 complex-mediated Chk1 degradation. (A) A549 cells were transfected with constructs that are specified (Vector (V), XIAP, XAF1, XIAP/XAF1). XIAP/XAF1 indicates co-transfection with both constructs. Whole cell extracts were prepared and subjected to protein gel blot analysis (IB) to detect CUL4A, DDB1 and PCNA (upper panel). β-actin was used as a loading control. Immunoprecipitation (IP) was performed by using antibodies against Chk1 or XIAP. Co-IP of DDB1, CUL4A, or PCNA was detected by protein gel blot analysis (lower panel). (B) CUL4A, DDB1 or PCNA was depleted by transfection of the specified SiRNA in A549 cells with or without XAF1 overexpression. Levels of the specified proteins were determined by protein gel blot analysis. Band intensity of Chk1 protein was determined by densitometry, and normalized against that of the non-treated control (NC), which was set as 100%.
RING domain of XIAP is required for Chk1 degradation by the XIAP-XAF1 complex
Since the RING domain in XIAP functions as an E3 ligase to catalyze the ubiquitination of substrate proteins,Citation20 we examined whether the XIAP-XAF1-mediated downregulation of Chk1 might engage the RING domain of XIAP. For the purpose, we prepared XIAP construct with deletion of the RING domain (XIAPΔRING), and then transfected the construct with XAF1 in A549 cells. Overexpression of XIAPΔRING prevented XAF1-mediated Chk1 degradation (, lane 4) suggesting a crucial role of the domain in the process. Moreover, overexpression of XIAPΔRING prevented formation of the XIAP-XAF1-DDB1-CUL4A complex even in the presence of XAF1 overexpression, whereas expression of XAF1 alone or together with that of intact XIAP enhanced formation of the complex (). These results suggest that the RING domain of XIAP plays crucial roles in XAF1-mediated Chk1 degradation as well as in the formation of the XIAP-XAF1-DDB1-CUL4A complex by which Chk1 degradation might be processed.
Figure 6. Role of the RING domain of XIAP in the XIAP-XAF1-mediated Chk1 degradation. (A) Vector (V) or His-XIAPΔRING-expressed cells were transfected with a construct that encodes XAF1. XIAPΔRING is histidine-tagged and detected by western blot analysis with antibody against the tag (His). Chk1, XAF1 and β-actin were also detected by western blot analysis. (B) A549 cells were transfected with constructs that are specified in the figure (XAF1, His-XIAP and ΔR). ΔR indicates XIAPΔRING. Cell extracts were prepared and immunoprecipitated with antibody against XIAP. Co-immunoprecipitated proteins were detected with the specified antibodies by western blot analysis (IB). (C) A549 cells were transfected with constructs that are specified in the figure. Cells were treated with or without 5 Gy IR and incubated for 12 hr. Cells were harvested and whole cell extracts were prepared to detect Chk1 and PARP1 cleavage by western blot analysis (upper panel). An arrow indicates cleaved PARP1. Levels of Chk1 protein in each lane, as determined by densitometry, were normalized to those of β-actin and represented as a graph (lower panel). Values are means ± SD of 3 experiments. **P < 0.01 vs. XIAP/XAF1 coexpression. (D) Measurement of cell death in XAF1-, XIAP/XAF1-, and XIAPΔRING/XAF1-expressed cells following 5 Gy IR. Treated cells were incubated for 24 hr and stained with Annexin V-FITC and propidium iodide (PI). Fluorescence intensity of Annexin V-FITC and propidium iodide was analyzed by flow cytometry. Values are means ± SD of 3 independent experiments. **P < 0.01 vs. XIAP/XAF1 coexpression.
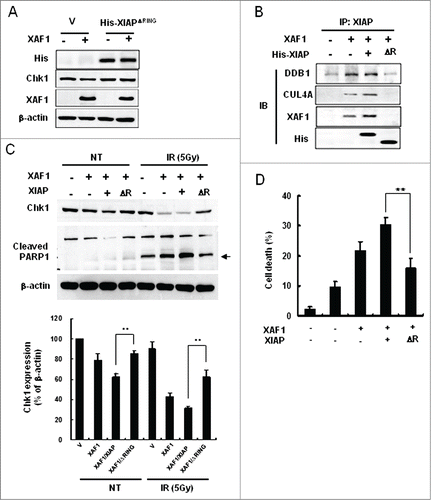
Since Chk1 is an essential factor for cell survival in cellular radiation responses, we examined whether Chk1 protein expression and apoptotic cell death are affected following irradiation in cells with overexpression of various constructs such as XAF1, XIAP, or XIAPΔRING. XAF1 expression with or without XIAP reduced Chk1 levels which were further decreased following IR treatment (). Overexpression of XIAPΔRING partly protected Chk1 from XAF1-mediated degradation either in non-irradiated control or in irradiated samples underscoring a crucial role of the RING domain in the regulation of Chk1 decrease.
PARP1 cleavage, which was assessed as an indicator of apoptosis, was inversely proportional to Chk1 levels following IR suggesting an essential role of Chk1 in cell survival (). Prevention of Chk1 degradation by overexpression of XIAPΔRING was accompanied by less PARP1 cleavage following IR, which may explain an essential role of Chk1 in cell survival. Indeed, IR treatment enhanced cell death in XAF1 or XAF1/XIAP-coexpressed cells, whereas IR-induced cell death was attenuated in cells with XAF1/XIAPΔRING overexpression ().
Taken together, the results show that Chk1 degradation by the XIAP-XAF1 complex increases radiation sensitivity by enhancing cell death and that the RING domain of XIAP plays key roles in the processes.
Interferon-gamma treatment enhances IR sensitivity via XAF1 induction
In search of physiological relevance to our findings, we examined effect of IFN-γ on XAF1, XIAP and Chk1 in lung cancer cells. As previously shown in HeLa cells,Citation26 IFN-γ treatment of various lung cancer cells, such as A549, H1299 and NCI-H460, induced XAF1 expression (). Interestingly, Chk1 protein levels were inversely proportional to those of XAF1 under the condition, which is reminiscent of XAF1-mediated Chk1 degradation ().
Figure 7. Enhancement of radiation sensitivity in interferon gamma-treated lung cancer cells. (A) Indicated lung cancer cells were treated with interferon-gamma (IFN-γ). Whole cell extracts were prepared and subjected to western blot analysis. Antibodies against XAF1, XIAP, and Chk1 were used for the analysis. β-actin was used as a loading control. (B) Graphical representation of levels of Chk1 expression in . Expression of Chk1 protein following IFN-γ treatment was measured by densitometric analysis of Chk1 bands in and normalized against non-treated control (NT) in each cell line. Values are means ± SD with *P < 0.01. (C) Clonogenic cell survival following a combined treatment with IFN-γ and IR in lung cancer cell lines. Cells were treated with 200 Units/ml IFN-γ for 1 hour before IR. IR doses were 0, 1, 2, and 4 Gy. Cell plates were incubated about 2 weeks for colony formation. Cell survival values were normalized to those of the unirradiated cells.
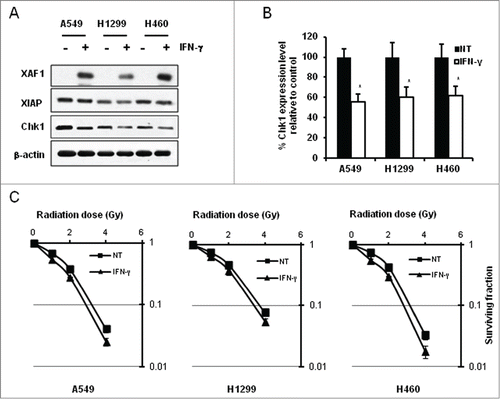
Since XAF1-mediated Chk1 downregulation enhanced radiation-induced cancer cell killing, we tested whether combined treatments of the cancer cells with IFN-γ and IR produce similar results. In all three cancer cell lines tested, IFN-γ treatment enhanced radiation sensitivity as shown by reduced clonogenic cell survival following IR ().
These findings suggest that IFN-γ treatment might enhance IR sensitivity in lung cancer cells, at least in part, via the XIAP-XAF1 complex-mediated Chk1 degradation.
Discussion
Since Chk1 is a promising target for anti-cancer therapy,Citation29 we have examined modulation of Chk1 stability by the XIAP-XAF1 complex in order to devise a strategy to enhance radiation sensitivity in lung cancer cells. The present study elucidates mechanism of Chk1 downregulation by the XIAP-XAF1 complex in lung cancer cell lines. Although XIAP expression alone stabilizes Chk1 under genotoxic treatments, coexpression with XAF1 induces Chk1 downregulation. XAF1 expression promotes ubiquitination and proteasomal degradation of Chk1 via formation of the XIAP-XAF1-Chkl complex, thereby reversing the stabilizing effects of XIAP for Chk1. The XIAP-XAF1 complex seems to mobilize additional factors, such as DDB1 and CUL4A, in a manner that requires the RING domain of XIAP to facilitate Chk1 degradation. As a result, downregulation of Chk1 by the XIAP-XAF1 complex sensitizes cancer cells to ionizing radiation. Moreover, we showed physiological relevance of the results by demonstrating that IFN-γ treatment enhanced IR sensitivity, which is accompanied by XAF1 induction as well as Chk1 degradation.
Although XIAP or Chk1 has been associated with tumor resistance to chemo- or radiotherapy, modulation of Chk1 stability by XIAP and its implication in regulation of radiation sensitivity have been rarely studied. We found that levels of Chk1 expression are correlated with those of XIAP in lung cancer cells and that overexpression of XIAP prevents downregulation of Chk1 under genotoxic treatments. More importantly, maintenance of Chk1 abundance by XIAP was critical for cell survival following IR treatment. By contrast, when XAF1 is overexpressed, the binding of XAF1 to XIAP reversed the Chk1 protective role of XIAP and the XIAP-XAF1 complex facilitated Chk1 degradation via proteasomal pathway, which suggests a dual function of XIAP in modulation of Chk1 stability in a manner dependent on XAF1 expression.
Previously, interactions between the proteins, e.g. XIAP, XAF1 or Chk1 have been reported.Citation21,22,24,25 For instance, Chk1 binds the BIR3 (baculoviral IAP repeat 3) domain of XIAP, whereas XAF1 binds the RING domain in XIAP.Citation21,24 In our study, we found that a complex, which is composed of XIAP, XAF1 and Chk1, is formed upon XAF1 overexpression and that the complex downregulates Chk1 indicating that XAF1 reverses the stabilizing effect of XIAP on Chk1 and that the XIAP-XAF1 complex mediates Chk1 turnover (). Thus in the absence of XAF1 expression as in most cancer cells, upregulated XIAP may interact with Chk1 () and prevent Chk1 destabilization under genotoxic conditions contributing to tumor resistance to chemo- or radiotherapy. Upon XAF1 expression, however, XAF1 binding to XIAP may trigger Chk1 degradation by activating the E3 ligase activity of the RING domain of XIAP. The XIAP-XAF1 complex-mediated downregulation of Chk1 in cancer cells is in accord with the notion that XAF1 is a candidate tumor suppressor. Overexpression of XAF1 in cancer cells induces mitotic catastrophe.Citation25 Similarly, depletion or instability of Chk1 leads to mitotic catastrophe.Citation34,35 Thus, it is of interest whether XAF1-induced mitotic catastrophe involves similar mechanism to our findings. Since Chk1 and XAF1 directly bind different domains in XIAP, the BIR3 and RING domain respectively,Citation21,24 and XIAP possesses E3 ligase activity, XIAP alone without linker proteins might be able to serve as an E3 ligase for Chk1, if not the only E3 ligase. In the case, Chk1 and XAF1 might be tethered to XIAP and activated E3 ligase activity of XIAP might ubiquitinate Chk1 for targeting in proteasomal degradation. In our experiment, however, XAF1-mediated Chk1 downregulation was only partly recovered by XIAP depletion () suggesting possible involvement of additional factors in the process. Indeed, DDB1, CUL4A and PCNA were associated with Chk1 or with XIAP in XAF1-overexpressed cells (). These proteins appear to participate in Chk1 degradation which is mediated by the XIAP-XAF1 complex because depletion of the proteins significantly prevented Chk1 degradation (). Moreover, these proteins seem to collaborate with XIAP in Chk1 degradation as overexpression of XIAPDRING (deletion of the RING domain in XIAP) prevented association of DDB1/CUL4A with XIAP as well as Chk1 degradation in XAF1-overexpressed cells (). DDB1, CUL4A and PCNA are components of E3 ligases.Citation36 Especially, the CUL4A/DDB1 ubiquitin E3 ligase is known to regulate Chk1 abundance.Citation10 Almost complete prevention of Chk1 degradation by depletion of CUL4A, DDB1 or PCNA in XAF-1 overexpressed cells raises the possibility that many additional E3 ligases which mobilize such factors are activated in XAF1-overexpressed cells and that these in turn might participate in Chk1 degradation either alone or in collaboration with the XIAP-XAF1 complex. CUL4A- and CUL1-based E3 ubiquitin ligases are involved in Chk1 proteolysis.Citation10-12 These cullin proteins serves as scaffolds in cullin-RING ligases complexes (CRLs).Citation37 CUL4A CRL is known to recruit substrates via DDB1 (damaged DNA binding protein 1) adaptor molecule. Or CUL4A-DDB1 complex requires additional substrate receptors such as CDT2.Citation33 We may speculate whether XAF1 plays a role as a substrate receptor for CUL4A CRL. If so, XAF1-mediated collaboration between different E3 ligases, such as XIAP and CUL4A CRL, might be further investigated.
The RING domain in XIAP is required for formation of E3 complex and Chk1 degradation as well as IR-induced cell death in XAF1-overexpressed cells ().
XAF1 binding to the XIAP RING domain might have caused conformational changes in XIAP and consequent activation of E3 ligase activity. Although the molecular and structural mechanism is unknown, our data show that overexpression of XIAPΔRING prevents Chk1 degradation in XAF1-overexpressed cells. When one considers the fact that cells with endogenous levels of XIAP expression together with XAF1-overexpression was sufficient for Chk1 degradation (), intact level of Chk1 in cells with co-overexpression of XIAPΔRING and XAF1 raises interesting possibilities (). First XIAP without the RING domain might bind Chk1 via its intact BIR3 domain and be able to protect Chk1 even under the condition of XAF1 overexpression. Second, Overexpressed XIAPΔRING might interfere with endogenous XIAP in E3 complex formation that are composed of XIAP, XAF1, DDB1 and CUL4A (), thereby preventing Chk1 degradation. Since the RING domain of XIAP is required for many activities such as E3 ligase activity and interaction with other proteins,Citation20 lack of those activities in XIAPΔRING might provide some clues. XIAP is capable of binding various proteins such as c-IAP1, Smac/DIABLO or caspases-3, 7, and 9.Citation20 Some of these XIAP-binding factors share the same domain in XIAP with Chk1 for their binding. For instance, Suvivin, caspase-9 or Smac/DIABLO binds to the BIR3 domain of XIAP as Chk1 does.Citation20,38 Or c-IAP1 binds the RING domain of XIAP as XAF1. A multitude of XIAP binding proteins may indirectly affect the XIAP-XAF1 complex-mediated Chk1 degradation. For example, preoccupation of the BIR3 domain in XIAP by Survivin might limit Chk1 binding to the domain and attenuate Chk1 degradation by XIAP-XAF1 complex. Similarly, abundant binding of c-IAP to the RING domain of XIAP might hinder XAF1 binding to the domain and thus prevent Chk1 degradation. It is also possible that XIAP-binding proteins might affect Chk1 activity in cells without XAF1 expression as in certain cancer cells. As an example, preoccupation of the BIR3 domain in XIAP by Smac/DIABLO might hinder Chk1 binding to the domain, thereby protection of Chk1 by XIAP in non-XAF1-expressing cells might be compromised. Therefore it is likely that modulation of Chk1 protein by XIAP might be affected by expression of XAF1 as well as by abundance of XIAP-binding proteins. Degradation of Survivin by the XIAP-XAF1 complex had been reported.Citation22 Interestingly, downregulation of Chk1 by the XIAP-XAF1 complex exhibits similar features with that of Survivin. First, overexpressed XAF1 degrades Chk1 and Survivin via ubiquitin-dependent proteasomal pathway. It is likely that XAF1 interacts with XIAP and activates the E3 ligase activity of XIAP to target the protein substrates. In the processes, the RING domain of XIAP might serve as a XAF1-binding domain as well as an E3 ligase. Second, the binding of XAF1 to XIAP reverses protective role of XIAP and thereby facilitates degradation of both Chk1 and Survivin. Third, degradation of Chk1 and Survivin by the XIAP-XAF1 complex may affect activity of various caspases upon genotoxic treatments and thus cell death. A stabilized Survivin-XIAP complex is known to suppress caspase-9 activity and apoptosis,Citation38 whereas ectopic expression of XAF1 results in caspase-3 activation.Citation22 Similarly, cells with Chk1 depletion exhibited activated forms of caspase-3 and -9.Citation34 In addition, depletion or inhibition of Chk1 hyperactivates caspase-2 activity following IR.Citation9 Therefore, maintenance or degradation of Chk1 and Survivin by XIAP alone or by the XIAP-XAF1 complex respectively, might determine cell fates following genotoxic treatments. Indeed, many reports identify Chk1, Survivin and XIAP as determinants of radiation sensitivity.Citation39-41
Similar to our previous report,Citation26 we observed that IFN-γ treatment in lung cancer cells enhances radiation sensitivity via XAF1-mediated downregulation of Chk1 (). The rationale for enhancement of radiation sensitivity by the combined treatments is supported by the concept of synthetic lethality. Compromised checkpoint regulation by downregulated Chk1 would result in sensitization of cancer cells to ionizing radiation. Indeed, according to the rationale, several Chk1-targeting drugs, such as AZD7762, PF47736, and XL844, have been combined with gemcitabine for phase I studies.Citation42 Therefore, our findings of IFN-γ on Chk1 and XAF1 might be utilized to enhance radiation sensitivity of lung cancer cells when the cells are subjected to combined treatments with IFN-γ and IR.
Materials and Methods
Materials
Antibodies against Chk1 (sc-8408), XAF1 (sc-19194), Survivin (sc-17779), DDB1 (sc-25367), PCNA (sc-56) and β-actin (sc-47778) were purchased from Santa Cruz Biotech Inc. Antibodies against XIAP (#2042), CUL4A (#2699), and Claspin (#2800) were obtained from Cell Signaling Technology Inc., and γ-H2AX antibody (#05-636) from Upstate Biotechnology. Anti-PARP1 antibody is from BD Biosciences (#552596) and anti-His antibody is from Sigma-Aldrich (H1029). The pCMV-Sport6 vectors encoding a full sequence of XIAP (MHS1010-7508457) and XAF (MHS1010-98051185) gene respectively were purchased from Open Biosystems Inc. XIAP construct with deletion of RING domain (XIAPΔRING) was generated by PCR with the following primers. Forward primer: atgacttttaacagttttgaaggat. Reverse primer: ctaaagcttctcctcttgcagg. PCR products were cloned into His-pcDNA3 TA-vector (Lifetechnologies, K4800) to make XIAPDRING construct. The protein product of XIAPDRING has deletion in amino acid residues from 451 to 497 of full length XIAP.
Cell culture and preparation of cell extract
A549, NCI-H1299, and NCI-H460 lung cancer cell lines were maintained in 100-mm culture plate at 37°C in 5% CO2 humidified incubator in RPMI1640 media supplemented with heat-inactivated 10% fetal bovine serum. WI38 normal lung fibroblast was cultured in DMEM media with 10% fetal bovine serum. Cells were washed with ice-cold PBS, lysed in 50 μl of ice-cold RIPA buffer (25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% NP-40, 0.5% sodium deoxycholate, 0.5% SDS, 1 mM Na3VO4, 5 mM NaF, and 1 mM phenylmethylsulfonyl fluoride), collected by scraping with a rubber policeman, incubated on ice for 30 min and centrifuged at 13,600 g for 15 min. Protein concentrations in the supernatants were quantified by the bicinchoninic acid (BCA) method (Pierce Biotechnology Inc., #23225) using bovine serum albumin as a standard. The volumes of the supernatants were adjusted for equal protein concentration.
Western blot analysis and immunoprecipitation
Proteins (35 μg) were separated on 10% SDS-polyacrylamide gels and then transferred to nitrocellulose membranes. The membranes were incubated with 5% skim milk solution in Tris-Tween Buffered Saline (TTBS) solution overnight at 4°C with the indicated antibodies. After washing 3 times in TTBS, horseradish peroxidase-conjugated secondary antibodies were applied. The proteins were visualized using enhanced chemiluminescence (GE Healthcare Life Sciences). Some membranes were stripped with an antibody stripping buffer (2% SDS, 100 mM β-mercaptoethanol, and 50 mM Tris–HCl, pH 7.0) at 55°C for 20 min. The membranes were then re-probed with a β-actin antibody as a control for protein loading.
For immunoprecipitation, cells were collected by centrifugation and resuspended in lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1% Triton X-100) containing protease inhibitors. Cell lysates were incubated on ice for 30 min and then centrifuged at 13,600 g for 15 min. Individual proteins were immunoprecipitated with anti-Chk1 or anti-XIAP antibodies respectively and specific proteins were detected as indicated. Preimmunized nonspecific mouse serum was used as a control.
Cell viability assays
Cell proliferation was measured with the MTT assay. Cells were seeded in 96-well plates at a density of 1 × 103 cells/well. After treatments, the cells were incubated with 1 mg/ml MTT [3-(4,5-dimethylthiazol-2yl)-2,5 diphenyltetrazolium bromide] solution for 2 h. The medium was aspirated and the resulting formazan product was solubilized with 100 μl of dimethyl sulfoxide. Viability was assessed by measuring absorbance at 570 nm with a BioRad microplate reader.
Annexin V-FITC (BD Biosciences, #556420) was used for analysis of apoptosis. Briefly, cells were seeded at 2 × 105 in 60 mm dishes. After transfection with various constructs such as vector, XAF1 or XIAP, the cells were irradiated, trypsinized, washed twice with cold PBS, and resuspended in 100 μl of 1x Binding buffer. The cells were added with 5 μl Annexin V-FITC and incubated for 15 min at room temperature in the dark. The FITC fluorescence intensity of each population of 10,000 cells was measured using a Becton-Dickinson FACS Caliber flow cytometer.
Clonogenic cell survival assay was performed by following standard protocols.Citation43
Cell cultures were irradiated using a 137 Cs gamma-ray source (Atomic Energy of Canada, Ltd.) at a dose rate of 3.81 Gy/min.
Immunofluorecence staining
Cells cultured on glass coverslips were washed with PBS, fixed with 3.7% (v/v) paraformaldehyde in PBS for 10 min, and permeabilized with 0.2% Triton X-100 in PBS for 5 min. For immunocytochemistry analysis of Chk1, cells were incubated with mouse anti-Chk1 antibody (Santa Cruz Biotechnologies), followed by Alexa488-conjugated anti-mouse IgG (A11001, Life Technologies). The Nuclei were fluorescently labeled with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich, Inc., D9542). The coverslips were then washed and mounted on glass slides. Fluorescent images were obtained using a fluorescence microscope (AE31, Motic).
Transfection of siRNAs
For knockdown of XIAP, transfection of siRNA against XIAP (100 nM, Dharmacon) was carried out using Lipofectamin 2000 transfection reagent (Invitrogen, #11668-019) according to the manufacturer's protocol. SiRNA against DDB1, CUL4A, PCNA, or siRNA negative control (Bioneer) were transfected similarly in cells for knockdown of the target genes, respectively. The cells were incubated for 48 hr at 37°C before cells were collected and subjected to western blotting and immunoprecipitation.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by nuclear research and development program of the national research foundation of Korea funded by the Korea government (grant number NRF-2012M2A2A7012377).
References
- Sancar A, Lindsey-Boltz LA, Unsal-Kaçmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 2004; 73:39-85; PMID:15189136; http://dx.doi.org/10.1146/annurev.biochem.73.011303.073723
- Zhao H, Watkins JL, Piwnica-Worms H. Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc Natl Acad Sci USA 2002; 99(23):14795-800; PMID:12399544; http://dx.doi.org/10.1073/pnas.182557299
- Sørensen CS, Syljuåsen RG, Falck J, Schroeder T, Rönnstrand L, Khanna KK, Zhou BB, Bartek J, Lukas J. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 2003; 3(3):247-58; PMID:12676583; http://dx.doi.org/10.1016/S1535-6108(03)00048-5
- Falck J, Mailand N, Syljuåsen RG, Bartek J, Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 2001; 410(6830):842-7; PMID:11298456; http://dx.doi.org/10.1038/35071124
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006;444(7120):756-60; PMID:17051156; http://dx.doi.org/10.1038/nature05236
- Raleigh DR, Haas-Kogan DA. Molecular targets and mechanisms of radiosensitization using DNA damage response pathways. Future Oncol 2013; 9(2):219-33; PMID:23414472; http://dx.doi.org/10.2217/fon.12.185
- Patil M, Pabla N, Dong Z. Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cell Mol Life Sci 2013; 70(21):4009-21; PMID:23508805; http://dx.doi.org/10.1007/s00018-013-1307-3
- Dent P, Tang Y, Yacoub A, Dai Y, Fisher PB, Grant S. CHK1 inhibitors in combination chemotherapy: thinking beyond the cell cycle. Mol Interv 2011; 11(2):133-40; PMID:21540473; http://dx.doi.org/10.1124/mi.11.2.11
- Sidi S, Sanda T, Kennedy RD, Hagen AT, Jette CA, Hoffmans R, Pascual J, Imamura S, Kishi S, Amatruda JF, et al. Chk1 suppresses a caspase-2 apoptotic response to DNA damage that bypasses p53, Bcl-2, and caspase-3. Cell 2008; 133(5):864-77; PMID:18510930; http://dx.doi.org/10.1016/j.cell.2008.03.037
- Leung-Pineda V, Huh J, Piwnica-Worms H. DDB1 targets Chk1 to the Cul4 E3 ligase complex in normal cycling cells and in cells experiencing replication stress. Cancer Res 2009; 69(6):2630-7; PMID:19276361; http://dx.doi.org/10.1158/0008-5472.CAN-08-3382
- Zhang YW, Brognard J, Coughlin C, You Z, Dolled-Filhart M, Aslanian A, Manning G, Abraham RT, Hunter T. The F box protein Fbx6 regulates Chk1 stability and cellular sensitivity to replication stress. Mol Cell 2009; 35(4):442-53; PMID:19716789; http://dx.doi.org/10.1016/j.molcel.2009.06.030
- Zhang YW, Otterness DM, Chiang GG, Xie W, Liu YC, Mercurio F, Abraham RT. Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol Cell 2005; 19(5):607-18; PMID:16137618; http://dx.doi.org/10.1016/j.molcel.2005.07.019
- Salvesen GS, Duckett CS. IAP proteins: blocking the road to death's door. Nat Rev Mol Cell Biol 2002; 3(6):401-10; PMID:12042762; http://dx.doi.org/10.1038/nrm830
- Wilkinson JC, Cepero E, Boise LH, Duckett CS. Upstream regulatory role for XIAP in receptor-mediated apoptosis. Mol Cell Biol 2004; 24(16):7003-14; PMID:15282301; http://dx.doi.org/10.1128/MCB.24.16.7003-7014.2004
- Berezovskaya O, Schimmer AD, Glinskii AB, Pinilla C, Hoffman RM, Reed JC, Glinsky GV. Increased expression of apoptosis inhibitor protein XIAP contributes to anoikis resistance of circulating human prostate cancer metastasis precursor cells. Cancer Res 2005; 65(6):2378-86; PMID:15781653; http://dx.doi.org/10.1158/0008-5472.CAN-04-2649
- Holcik M, Yeh C, Korneluk RG, Chow T. Translational upregulation of X-linked inhibitor of apoptosis (XIAP) increases resistance to radiation induced cell death. Oncogene 2000; 19(36):4174-7; PMID:10962579; http://dx.doi.org/10.1038/sj.onc.1203765
- Tong QS, Zheng LD, Wang L, Zeng FQ, Chen FM, Dong JH, Lu GC. Downregulation of XIAP expression induces apoptosis and enhances chemotherapeutic sensitivity in human gastric cancer cells. Cancer Gene Ther 2005; 12(5):509-14; PMID:15706355
- Sasaki H, Sheng Y, Kotsuji F, Tsang BK. Down-regulation of X-linked inhibitor of apoptosis protein induces apoptosis in chemoresistant human ovariancancer cells. Cancer Res 2000; 60(20):5659-66; PMID:11059757
- Cao C, Mu Y, Hallahan DE, Lu B. XIAP and surviving as therapeutic targets for radiation sensitization in preclinical models of lung cancer. Oncogene 2004; 23(42):7047-52; PMID:15258565; http://dx.doi.org/10.1038/sj.onc.1207929
- Galbán S, Duckett CS. XIAP as a ubiquitin ligase in cellular signaling. Cell Death Differ 2010; 17(1):54-60; http://dx.doi.org/10.1038/cdd.2009.81
- Tse MK, Cho CK, Wong WF, Zou B, Hui SK, Wong BC, Sze KH. Domain organization of XAF1 and the identification and characterization of XIAP(RING) -binding domain of XAF1. Protein Sci 2012; 21(10):1418-28; PMID:22811387; http://dx.doi.org/10.1002/pro.2126
- Arora V, Cheung HH, Plenchette S, Micali OC, Liston P, Korneluk RG. Degradation of surviving by the X-linked inhibitor of apoptosis (XIAP)-XAF1 complex. J Biol Chem 2007; 282(36):26202-9; PMID:17613533; http://dx.doi.org/10.1074/jbc.M700776200
- Liston P, Fong WG, Kelly NL, Toji S, Miyazaki T, Conte D, Tamai K, Craig CG, McBurney MW, Korneluk RG. Identification of XAF1 as an antagonist of XIAP anti-Caspase activity. Nat Cell Biol 2001; 3(2):128-33; PMID:11175744; http://dx.doi.org/10.1038/35055027
- Galvan V, Kurakin AV, Bredesen DE. Interaction of checkpoint kinase 1 and the X-linked inhibitor of apoptosis during mitosis. FEBS Lett 2004; 558(1-3):57-62; PMID:14759516; http://dx.doi.org/10.1016/S0014-5793(03)01488-1
- Wang J, Gu Q, Li M, Zhang W, Yang M, Zou B, Chan S, Qiao L, Jiang B, Tu S. et al. Identification of XAF1 as a novel cell cycle regulator through modulating G(2)/M checkpoint and interaction with checkpoint kinase 1 in gastrointestinal cancer. Carcinogenesis 2009; 30(9):1507-16; PMID:19628579; http://dx.doi.org/10.1093/carcin/bgp155
- Kim KS, Choi KJ, Bae S. Interferon-gamma enhances radiation-induced cell death via downregulation of Chk1. Cancer Biol Ther 2012; 13(11):1018-25; PMID:22825336; http://dx.doi.org/10.4161/cbt.20990
- Holcik M, Gibson H, Korneluk RGXIAP: apoptotic brake and promising therapeutic target. Apoptosis 2001; 6(4):253-61; PMID:11445667; http://dx.doi.org/10.1023/A:1011379307472
- Dean EJ, Ranson M, Blackhall F, Dive C. X-linked inhibitor of apoptosis protein as a therapeutic target. Expert Opin Ther Targets 2007; 11(11):1459-71; PMID:18028010; http://dx.doi.org/10.1517/14728222.11.11.1459
- Toledo LI, Murga M, Fernandez-Capetillo O. Targeting ATR and Chk1 kinases for cancer treatment: a new model for new (and old) drugs. Mol Oncol 2011; 5(4):368-73; PMID:21820372; http://dx.doi.org/10.1016/j.molonc.2011.07.002
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, Donehower LA, Elledge SJ. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 2000; 14(12):1448-59; PMID:10859164
- Fong WG, Liston P, Rajcan-Separovic E, St Jean M, Craig C, Korneluk RG. Expression and genetic analysis of XIAP-associated factor 1 (XAF1) in cancer cell lines. Genomics 2000; 70(1):113-22; PMID:11087668; http://dx.doi.org/10.1006/geno.2000.6364
- Reinhardt HC, Hasskamp P, Schmedding I, Morandell S, van Vugt MA, Wang X, Linding R, Ong SE, Weaver D, Carr SA, Yaffe MB. DNA damage activates a spatially distinct late cytoplasmic cell-cycle checkpoint network controlled byMK2-mediated RNA stabilization. Mol Cell 2010; 40(1):34-49; PMID:20932473; http://dx.doi.org/10.1016/j.molcel.2010.09.018
- Huh J, Piwnica-Worms H. CRL4(CDT2) targets CHK1 for PCNA-independent destruction. Mol Cell Biol 2013; 33(2):213-26; PMID:23109433; http://dx.doi.org/10.1128/MCB.00847-12
- Niida H, Tsuge S, Katsuno Y, Konishi A, Takeda N, Nakanishi M. Depletion of Chk1 leads to premature activation of Cdc2-cyclin B and mitotic catastrophe. J Biol Chem 2005; 280(47):39246-52; PMID:16159883; http://dx.doi.org/10.1074/jbc.M505009200
- Jurvansuu J, Fragkos M, Ingemarsdotter C, Beard P. Chk1 instability is coupled to mitotic cell death ofp53-deficient cells in response to virus-induced DNA damage signaling. J Mol Biol 2007; 372(2):397-406; PMID:17663993; http://dx.doi.org/10.1016/j.jmb.2007.06.077
- Higa LA, Zhang H. Stealing the spotlight: CUL4-DDB1 ubiquitin ligase docks WD40-repeat proteins to destroy. Cell Div 2007; 2:5; PMID:17280619
- Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol 2005; 6(1):9-20; PMID:15688063; http://dx.doi.org/10.1038/nrm1547
- Dohi T, Okada K, Xia F, Wilford CE, Samuel T, Welsh K, Marusawa H, Zou H, Armstrong R, Matsuzawa S, et al. An IAP-IAP complex inhibits apoptosis. J Biol Chem 2004; 279(33):34087-90; PMID:15218035; http://dx.doi.org/10.1074/jbc.C400236200
- Pennati M, Folini M, Zaffaroni N. Targeting survivin in cancer therapy: fulfilled promises and open questions. Carcinogenesis 2007; 28(6):1133-9; PMID:17341657; http://dx.doi.org/10.1093/carcin/bgm047
- Meuth M. Chk1 suppressed cell death. Cell Div 2010; 5:21; PMID:20813042; http://dx.doi.org/10.1186/1747-1028-5-21
- Ohnishi K, Scuric Z, Schiestl RH, Okamoto N, Takahashi A, Ohnishi T. SiRNA targeting NBS1 or XIAP increases radiation sensitivity of human cancer cells independent of TP53 status. Radiat Res 2006; 166(3):454-62; PMID:16972754; http://dx.doi.org/10.1667/RR3606.1
- Liang Y, Lin SY, Brunicardi FC, Goss J, Li K. DNA damage response pathways in tumor suppression and cancer treatment. World J Surg 2009; 33(4):661-6; PMID:19034564; http://dx.doi.org/10.1007/s00268-008-9840-1
- Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc 2006; 1:2315-9; PMID:17406473; http://dx.doi.org/10.1038/nprot.2006.339