
Abstract
VEGF/VEGFR signal axis has been proven to be an important target for development of novel cancer therapies. One challenging aspect in small molecular VEGFR inhibitors is to achieve sustained target inhibition at tolerable doses previously seen only with the long-acting biologics. It would require high potency (low effective drug concentrations) and sufficient drug exposures at tolerated doses so that the drug concentration can be maintained above effective drug concentration for target inhibition within the dosing period. Fruquintinib (HMPL-013) is a small molecule inhibitor with strong potency and high selectivity against VEGFR family currently in Phase II clinical studies. Analysis of Phase I pharmacokinetic data revealed that at the maximum tolerated dose of once daily oral administration fruquintinib achieved complete VEGFR2 suppression (drug concentrations were maintained above that required to produce >85% inhibition of VEGFR2 phosphorylation in mouse) for 24 hours/day. In this article, the preclinical data for fruquintinib will be described, including kinase enzyme activity and selectivity, cellular VEGFR inhibition and VEGFR-driven functional activity, in vivo VEGFR phosphorylation inhibition in the lung tissue in mouse and tumor growth inhibition in a panel of tumor xenograft and patient derive xenograft models in mouse. Pharmacokinetic and target inhibition data are also presented to provide a correlation between target inhibition and tumor growth inhibition.
Abbreviations
| VEGF | = | vascular endothelial growth factor |
| VEGFR | = | vascular endothelial growth factor receptor |
| KDR | = | Kinase insert domain-containing receptor, also named as VEGFR2 |
| PI3K | = | phosphoinositide 3-kinase |
| AKT | = | protein kinase B |
| PKC | = | protein kinase C |
| ERK | = | extracelluar signal-regulated kinase |
| CAM | = | chorioallantoic membrane |
| PK/PD | = | pharmacokinetics/pharmacodynamics |
Introduction
Angiogenesis is a process in which new blood vessels grow from existing vasculature. It plays a pivotal role in embryonic development and tumorigenesis.Citation1-3 Tumor angiogenesis has been shown to be critical during tumor growth by supplying nutrients and oxygen, facilitating dysregulated growth, and providing a route for metastatic dissemination.Citation4 VEGF/VEGFR is one of the most important pro-angiogenesis pathways. VEGF/VEGFR mediates angiogenesis by stimulating endothelial cell proliferation, migration and enhancing new vessel tube formation.Citation5 Production of VEGF by tumor cells can be disproportionately up-regulated via oncogenic activation or loss of tumor suppressor functionCitation6, 7 and hypoxia or glucose level change.Citation8 The amount of VEGF-A expressed by tumor cells correlates with poor prognosis in many types of tumors including lung, gastric, colon, prostate, ovary and melanoma.Citation9-14
Of the 3 closely related members of the VEGFR family (VEGFR1, 2, 3), VEGFR2 (also named as KDR or Flk1) and VEGFR3 mainly expressed in vascular and lymphatic endothelial cells, plays major roles in VEGF-A and VEGF-C induced angiogenesis and lymphangiogenesis.Citation15,16 Binding VEGF ligands to the extracellular domain of VEGFRs on the cell surface of the endothelial cells leads to dimerization of the receptor, and consequently, phosphorylation of the intracellular kinase domain of VEGFRs and triggers activation of downstream cell signaling cascades, including PI3K/AKT, PKC, Raf/Ras and ERK pathways, resulting in vascular/lymphatic endothelial cell proliferation and formation of new vessel branches.Citation16,17 There are 2 major approaches targeting VEGF/VEGFR axis as anti-cancer therapies: neutralization of VEGF or VEGFR by monoclonal antibodies and blockage of VEGFR kinase activity with small molecule inhibitors. The most successful example in the antibody approach is the VEGF-A trap antibody bevacizumab (Bevacizumab®; Genentech Inc.). Bevacizumab in combination with 5-fluorouracil–based chemotherapy has been approved as first/second line therapy for advanced colorectal cancer (CRC) and in combination with carboplatin and/or paclitaxel has been approved as first line therapy for advanced non-small cell lung cancer (NSCLC).Citation18-21 The success of bevacizumab can be attributed to its sustained target inhibition resulted from its long half-life and good safety profile due to its high specificity allowing it to combine with a variety of chemotherapies. However, bevacizumab has its limitations in the clinic, such as its intravenous dosing, immunogenicity and potential to induce autoimmune diseases after long term treatment. Furthermore, the high cost of bevacizumab limited patients’ access. It would be highly desirable if an orally active small molecule VEGFR inhibitor could achieve sustained target inhibition and is capable of combining with chemotherapies and at the same time avoid limitation of antibodies.
The earlier generation small molecule VEGFR inhibitors, such as sunitinib,Citation22 sorafenib,Citation23 regorafenibCitation24 and pazopanibCitation25 suffer from poor kinome selectivity. In fact, many of them inhibit more than 10 kinases at similar potency. As a consequence, the drug exposures at the maximum tolerated dose (MTD) are limited due to inhibition of multiple targets, resulting in less optimal and/or short duration of inhibition of any targets, in particular VEGFR. A potent and highly selective small molecule inhibitor could hopefully minimize off-target toxicity and afford high drug exposures at MTD leading to sustained VEGFR target inhibition similar to biologics and flexibility for combination therapies due to narrow target-base toxicity profile.
Fruquintinib represents a new generation of small molecule VEGFR inhibitors with strong potency and high kinome selectivity targeting VEGFR1, 2, 3. In in vitro enzymatic and cellular assays, it inhibited VEGFR family kinases and suppressed VEGF/VEGFR cell signaling in human umbilical vein endothelial cell (HUVEC) and human lymphatic endothial cell (HLEC) with IC50 at low nanomolar level. Consequently it blocked HUVEC function and produced anti-angiogenesis effect in CAM model. Few kinases were inhibited other than VEGFRs in a panel of 253 kinases test. Fruquintinib demonstrated favorable pharmacokinetic profile in multiple animal species,Citation26 oral administration of fruquintinib strongly suppressed VEGF-induced VEGFR2 phosphorylation in the lung tissue in mice. The extent and duration of the inhibition of VEGFR2 phosphorylation correlated well with drug exposures. The strong anti-angiogenic effect resulted in robust anti-tumor efficacy in a number of human tumor xenograft models with good dose response. In addition, fruquintinib demonstrated enhanced anti-tumor activity when combined with chemotherapeutic agents with good tolerance in the patient derived xenograft (PDX) models. All these provide fruquintinib as a promising anti-angiogenesis inhibitor deserving further development. It is currently being evaluated in Phase II clinical trials for multiple cancers including lung, gastric and colon.
Results
Fruquintinib is a highly potent and selective VEGFR 1, 2, 3 inhibitor
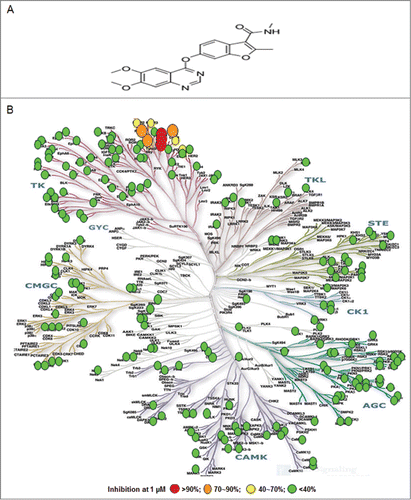
Fruquintinib was found to inhibit VEGFR2 (KDR) with an IC50 of 25 nmol/L in the Z-lyte assay. The kinase selectivity of fruquintinib was evaluated against a panel of 253 kinases using [Citation32p-ATP] incorporation assay by Upstate Biotechnology Inc. (UBI) (). The results showed that fruquintinib inhibited VEGFR family member (VEGFR1, 2, 3) with IC50s of 33 nmol/L, 35 nmol/L and 0.5 nmol/L, respectively with weak inhibition of RET, FGFR-1 and c-kit kinases. No significant inhibition was found against all other kinases at 1 μmol/L (). The selectivity of fruquintinib against FGFR-1 was further assessed in a cellular assay. Compared with the inhibition of KDR phosphorylation IC50 of 0.6 nmol/L, the cellular activity against FGFR phosphorylation was much weaker and over 1000 fold, confirming that fruquintinib is highly selective for VEGFR1, 2, 3 tyrosine kinase.
Table 1. VEGFR kinase activity and selectivity profile and bioactivities
Figure 1. Fruquintinib is a highly selective and potent VEGFR1, 2, 3 kinase inhibitor. (A) Chemical structure of fruquintinib. (B) Kinome selectivity of fruquintinib at 1 μmol/L against 253 kinases using Citation32p-ATP incorporation method generated at Millipore. The Kinome tree was downloaded from http://www.cellsignal.com. Percentage (%) denoted the inhibition of fruquintinib at 1 μmol/L to the recombinant kinases. Over 90% inhibition was observed for 3 VEGFR family members (1, 2, 3) and 70~90% inhibition on 4 other kinases, including Fms(Y969C), Ret, and FGFR1 and little effect on remaining kinases tested. IC50s were generated for the kinases of interest and shown in .
Fruquintinib suppresses VEGF/VEGFR signaling and cell proliferation in HUVECs and HLECs
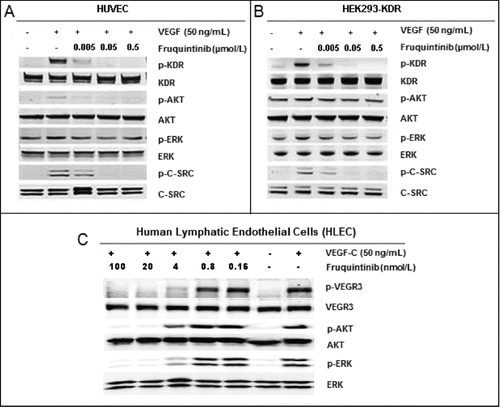
Consistent with the activity against KDR inhibition in the enzymatic assay, fruquintinib demonstrated potent inhibition on VEGF-A dependent KDR phosphorylation in HEK293-KDR cells and VEGF-A induced proliferation in primary HUVECs with IC50s of 0.6 ± 0.2 nmol/L and 1.7 nmol/L, respectively (). Similarly, potent VEGFR3 attenuation by fruquintinib was observed in primary HLECs, with IC50s of 1.5 nmol/L and 4.2 nmol/L for VEGF-C stimulated VEGFR3 phosphorylation and proliferation, respectively (). The inhibitory effect of fruquintinib on VEGF/VEGFR downstream signaling was evaluated in HUVEC, HEK293-KDR, and HLEC cells. As shown in , KDR was barely phosphorylated in the absence of VEGF in HEK293-KDR and HUVECs, but significant phosphorylation of KDR and its downstream signaling molecules, including Akt and Erk, were seen upon stimulation with VEGF. Fruquintinib inhibited VEGF stimulated phosphorylation of KDR and downstream signal molecules, e.g. Erk, in both HEK293-KDR cells and primary HUVEC cells in a concentration-dependent manner (). In HLEC, phosphorylation of VEGR3 and downstream molecules Akt and Erk was induced upon VEGF-C stimulation, and fruquintinib exhibited concentration dependent suppression (). Collectively, fruquintinib demonstrated equal potent inhibitory effect against KDR and VEGFR3.
Figure 2. Inhibition on VEGF stimulated activation of KDR and VEGFR3. (A) Fruquintinib inhibited VEGF-A-stimulated KDR phosphorylation and downstream signaling in HUVEC. (b) Fruquintinib abrogated VEGF-A-stimulated KDR phosphorylation and downstream signaling in HEK-293-KDR cell line. (C) Fruquintinib suppressed VEGF-C stimulated VEGFR3 phosphorylation in VEGF-C-stimulated HLEC.
Fruquintinib inhibits tubule sprouting and prevents angiogenesis in vitro
Microvessel tube formation is one of the key functions in angiogenesis. The effect of fruquintinib on tube formation was investigated. Fruquintinib suppressed the tube branching, tube length and area in a concentration-dependent manner (). The tubule length of primary HUVECs decreased by 74% and 94% at 0.03 and 0.3 μmol/L of fruquintinib, respectively. In a parallel cell survival assay, fruquintinib did not significantly affect the viability of HUVEC cells at the tested concentrations (), suggesting that the observed effect on tube formation by fruquintinib is due to inhibition of VEGF/VEGFR axis, rather than a consequence of the cytotoxicity.
Figure 3. Fruquintinib inhibited HUVEC tubule growth and CAM angiogenesis. Tube formation was suppressed significantly after treatment with fruquintinib at 0.3 μmol/L for 18 hours (A and B) No cytotoxicity was seen at the same concentration of fruquinitib in HUVECs. The plates were incubated for 3 hours at 37°C and fluorescence value was read at Ex 530 nm and Em 590 nm on Tecan (C) Fruquintinib displayed strong inhibition on the development of new blood vessels in the chick embryo (D) Left and right panels, as arrows indicated, were treated with saline and compound, respectively. Pseudolarix acid B was used as a positive control.
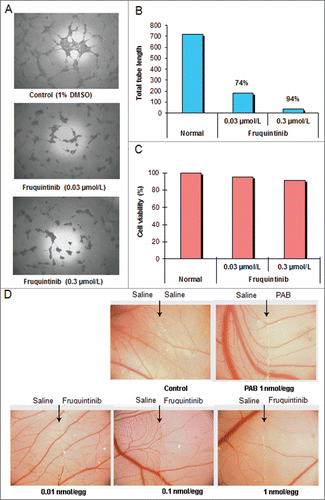
Ex vivo CAM assay was performed to further confirm the anti-angiogenic activity of fruquintinib. As shown in , the micro-vessel density was reduced by fruquintinib in a dose-dependent manner after incubation for 48 hours. Fruquintinib demonstrated significant inhibitory effect on micro-vessel sprouting at 0.1 nmol/egg concentration. Collectively, these data suggested that fruquintinib is a highly potent inhibitor of VEGF-induced angiogenesis.
Fruquintinib potently inhibits VEGF induced KDR phosphorylation in lung tissue of mouse
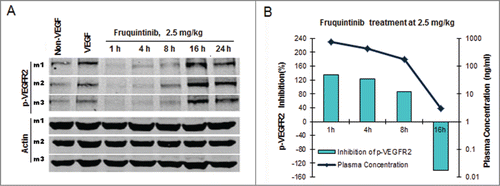
Fruquintinib inhibited KDR signaling in HUVEC and HEK293-KDR cells in vitro. To confirm such effect and establish PK/PD relationship in vivo, VEGF-A induced KDR phosphorylation in the lung tissue in mice was evaluated following oral administration of fruquintinib. As demonstrated in , after a single oral dose of fruquintinib at 2.5 mg/kg, VEGF stimulated KDR phosphorylation was completely suppressed and the effect sustained for at least 8 hours whereas the p-KDR level recovered at 16 hours after dose. Plasma samples were collected at 1, 4, 8, 16 and 24 hours post dosing to determine the concentrations of fruquintinib (). Fruquintinib reached Cmax at 1 hour and accordingly achieved maximum level of inhibition of p-KDR, taking the p-KDR level below the basal level. At 8 hours the p-KDR inhibition still maintained at 86%, and the corresponding fruquintinib concentration in plasma was 176 ng/mL. Consistent with lack of inhibition of KDR phosphorylation in the lung tissue at 16 hours, the plasma concentration of fruquintinib was found to be below 10 ng/mL. At 24 hours, the plasma concentration of fruquintinib was below the low limit of quantification. These results demonstrated that the p-KDR inhibition in lung directly correlated with drug exposures in plasma and 176 ng/mL of fruquintinib in plasma could achieve greater than 80% target inhibition in lung tissue.
Figure 4. Fruquintinib inhibited p-KDR in lung tissues of mice. (A) Fruquintinib inhibited VEGF-A induced p-KDR in lung tissues. Each group was composed of 3 mice (m1, m2, m3). Animals were treated as described in Method section. (B) Fruquintinib concentration in plasma (purple line) in relation to its inhibition on p-KDR phosphorylation (green bar).
Fruquintinib inhibits tumor growth in multiple human xenograft models
Anti-tumor activity of fruquintinib was evaluated in a variety of tumor xenografts along with measurements of plasma drug concentrations in an attempt to establish target inhibition-tumor growth inhibition relationship ( and ). The results from gastric cancer BGC-823 model seemed to indicate that the drug concentration needs to be at least maintained above EC85 (drug concentration required to inhibit KDR phosphorylation by 85%) for around 8 hours in order to achieve >80% tumor growth inhibition (clinically stable disease), and the longer target covering duration to achieve tumor regression (clinically partial response) (). BGC-823 was found to be most sensitive to fruquintinib. In this model, fruquintinib inhibited tumor growth by 62.3% and 95.4∼98.6%, at 0.5 and 2 mg/kg once daily dosing, respectively (, ). When the dose was elevated to 5 mg/kg and 20 mg/kg, the tumors regressed by 24.1% and 48.6%, respectively (, ) presumably due to longer duration of target inhibition (, plasma concentration of 5 and 20 mg/kg appeared to cover EC85 for about 12 and 18 hours, respectively). The level of anti-tumor growth activity of fruquintinib varied in different tumor xenograft models. In some cases, activation of tumor growth signaling pathways may play a role. For instance, in renal cell cancer Caki-1 xenograft model, fruquintinib at 2 mg/kg dose only produced a moderate TGI of 51.5%, comparing to almost complete tumor growth inhibition observed in BCG-823 model at this dose. (, ). Further analysis revealed that significant c-Met activation is present in Caki-1 cells. Rational combination in such tumors may provide optimal therapeutic effect. In fact, combination treatment of fruquintinib with a c-Met inhibitor in Caki-1 xenograft model produced significant synergy and led to complete tumor growth suppression (Data will be presented in a separate article).
Table 2. Anti-tumor activity of fruquintinib in multiple xenograft tumor models
Figure 5. Fruquintinib inhibited BGC-823 and Caki-1 tumor growth and anti-angiogenesis in tumor tissues. (A) Dose-dependent tumor growth inhibition in BGC-823 tumor following once daily oral treatment of fruquintinib at 0.5, 1 and 2 mg/kg. (B) Anti-tumor effect of fruquintinib at 2, 5 and 20 mg/kg in BGC-823. BGC-823 tumor was regressed by fruquintinib at 5 and 20 mg/kg. (C) Plasma exposures of fruquintinib at 2, 5 and 20 mg/kg in BGC-823 tumor bearing mice following consecutive oral daily dosing. The red line indicated the plasma EC85 for p-KDR inhibition in nude mouse. (D) Dose-dependent tumor growth inhibition in Caki-1 tumor model. (E) CD31 immunohistochemistry staining in Caki-1 xenografts. The pictures were taken at 200× magnification under microscope. Vascular vessels were stained as brown, representing the tumor angiogenesis. (E) Semi-quantification of IHC image. The percentage of CD31 positive area to tumor area was analyzed with NIKON BR-3.0 software. *P < 0.05, **P < 0.01 compared with vehicle group.
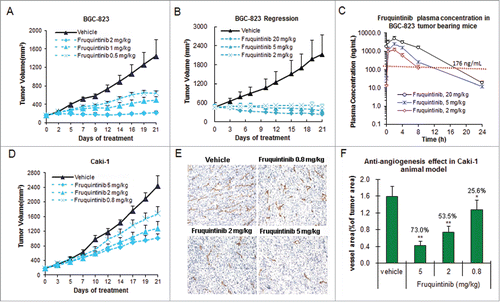
To further confirm anti-angiogenic effect of fruquintinib in vivo, an endothelial cell surface marker CD31 (PECAM-1) in the Caki-1 tumor tissue was measured by immunohistochemistry (IHC) method after treatment with fruquintinib for 3 weeks. The results are shown in . A clear dose-dependent anti-angiogenic effect was observed in the Caki-1 tumor tissue, and the inhibition rate was 73.0%, 53.5% and 25.6% at 5, 2, and 0.8 mg/kg, respectively. Comparing to the vehicle-treated group, fruquintinib significantly decreased the micro-vessel density even at the lowest dose of 0.8 mg/kg (P < 0.05). These results suggested that fruquintinib inhibited tumor growth through inhibition of tumor angiogenesis.
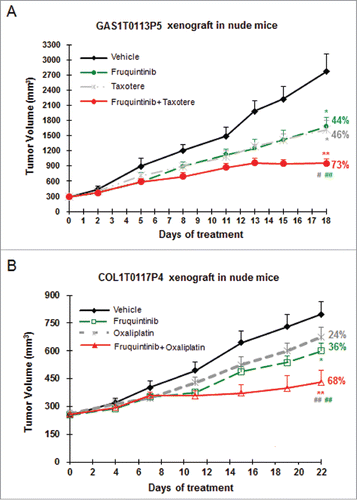
Anti-tumor efficacy of fruquintinib combining with chemotherapeutic agents was also investigated in PDX models. As shown in , combination of fruquintinib with Taxotere (docetaxel) showed significantly enhanced anti-tumor effect in gastric cancer GAS1T0113P5, with a TGI of 73%, comparing to 44% and 46% when treated with fruquintinib and docetaxel alone, respectively. Similarly, the enhanced inhibitory effect of fruquintinib with oxaliplatin was seen in the colon cancer PDX model COL1T0117P4 (). It was worth noting that fruquintinib did not increase animal body weight loss caused by docetaxel or oxalipatin in combinational use ().
Figure 6. Combination of fruquintinib with chemo drugs shows enhanced anti-tumor effect in PDX models. (A) Fruquintinib in combination with taxotere (docetaxel) in gastric cancer PDX model. (B) Fruquintinib in combination with oxaliplatin in colon cancer PDX model. Percentage (%) represented tumor growth inhibition (TGI) rate. Two-tailed t-test for fruquintinib treatment versus the control group: *, P < 0.05; **, P < 0.01. The combination vs. single agent: #, P < 0.05; ##, P < 0.01.
In summary, fruquintinib is a potent anti-tumor agent against a broad range of tumors either used alone or in combination settings.
Discussion
Inhibition of VEGF/VEGFR signaling is considered to be an important therapeutic strategy to suppress tumor growth through mechanism of anti-angiogenesis, including effects on neovascular survival, and vascular permeability in tumors.Citation27-29 Several products targeting VEGF/VEGFR pathway have already been approved by FDA that provide significant benefits for certain cancer patients.Citation22-24 However, these therapies are far from perfect. The antibody drugs have long plasma half-life that can provide sustained target inhibition. However, VEGF and/or VEGFR levels increase with the increase of tumor size, making it difficult to provide complete target coverage with a fixed dose potentially compromising the efficacy. The long half-life sometimes can make the toxicity management a challenge. In addition, inconvenient dosing and high cost further add to the problem with the antibody drugs. The small molecule VEGFR inhibitors are orally active and much more cost effective in theory. However, in poor kinase selectivity associated toxicities, relatively short plasma drug half-life limiting their ability to cover the target well and challenges in combining with chemotherapies due to poor tolerability lead to far less than optimal clinical efficacy. It is highly desirable to develop an anti-angiogenic product that combines the advantages of long half-life/sustained target inhibition of an antibody with the convenient oral dosing and lower cost of a small molecule kinase inhibitor that is well tolerated and highly effective.
Fruquintinib is a potent compound targeting VEGFR1, 2, 3 at the enzyme level. The high selectivity was demonstrated in a panel of 253 kinases in which fruquintinib was found to inhibit VEGFR1, 2, 3 potently and weak to no inhibitory effect on all other kinases. In cellular assays, fruquintinib was found to inhibit VEGFR2 and VEGFR3 with almost equal potency.
VEGFR3 is activated through binding to its specific ligand VEGF-C. VEGFR3 signaling is involved in regulating the development and growth of lymphatic system.Citation30,31 Clinical investigations have demonstrated that overexpression of VEGF-C/VEGFR3 in tumors is associated with lymphangiogenesis, lymph node metastasis and poor prognosis in several types of tumor.Citation32-36 VEGF-C promotes lymphangiogenesis and lymphatic vessel-mediated metastasis to regional lymph nodes, and it could be inhibited by blocking VEGFR3 receptor.Citation37 Given the importance of lymphangiogenesis in tumor metastasis and overall survival for certain cancer patients, it would seem to be an attractive approach to target VEGFR3, although unlike targeting VEGFR2, there have been no successful products developed yet specifically targeting VEGFR3. Fruquintinib, with its balanced activity against VEGFR2 and VEGFR3 and high selectivity over other kinases, could prove advantageous by simultaneously shutting down both angio- and lymphaniogenesis affecting tumor growth and metastasis over antibody drugs targeting VEGF-A/VEGFR2 alone.
The potent in vitro activity against VEGFR of fruquintinib was confirmed in vivo following oral administration. Fruquintinib was found to inhibit VEGF stimulated VEGFR2 phosphorylation (p-KDR) in lung tissue of mice in an exposure dependent manner. A single oral dose of fruquintinib at 2.5 mg/kg in mice resulted in near complete (>85%) inhibition of p-KDR and the inhibition was maintained for at least 8 hours in the lung tissue. At that time point the corresponding fruquintinib plasma concentration was determined to be 176 ng/mL (Effective Concentration for 85% target inhibition, or EC85). In anti-tumor efficacy studies, at the dose of around 2 mg/kg given orally once daily, statistically significant tumor growth inhibition was achieved in a variety of human tumor xenograft models in mice, indicating that maintaining complete suppression of VEGFR2 pathway approximately 8 hours or longer could produce significant anti-tumor efficacy, although it would be preferable to sustain the complete target inhibition for 24 hours/day similar to antibodies to achieve the maximum anti-tumor activity. The EC85 (176 ng/mL for fruquintinib) is useful for estimating the duration of target inhibition at the recommended doses in clinical trials. According to pharmacokinetic data in phase I clinical trial, the plasma exposures of fruquintinib at the maximum tolerated dose achieved the EC85 concentration for 24 hours per day with once daily dosing schedule,Citation38 indicating that fruquintinib potentially could introduce continuous target inhibition in cancer patients.
In summary, fruquintinib is a potent, highly selective and orally active inhibitor of VEGFR1, 2, 3 tyrosine kinases. Fruquintinib inhibited VEGF-induced VEGFR2 phosphorylation, endothelial cell proliferation, and tubule formation in vitro and VEGFR2 phosphorylation in lung tissue. Fruquintinib demonstrated potent tumor growth inhibition activity in a panel of human tumor xenografts in mice. Enhanced tumor growth inhibition was observed in several PDX models when fruquintinib was combined with chemotherapeutic agents. The equally potent inhibitory activity against VEGFR2 and VEGFR3 of fruquintinib could potentially offer strong anti-tumor growth and metastasis benefits. These attractive attributes coupled with favorable pharmacokinetic propertiesCitation26 and toxicity profile make fruquintinib a promising candidate for clinical investigation.
Materials and Methods
Chemical compound
Fruquintinib, 6-(6,7-dimethoxyquinazolin-4-yloxy)-N,2-dimethyl benzofuran-3-carboxamide () with a molecule weight 393.39, was synthesized by Hutchison MediPharma Limited (HMP). The structure of fruquintinib was illustrated in Figure 1A. Fruquintinib was prepared initially as a 10 mmol/L stock solution in DMSO and diluted in appropriate assay media for all in vitro assays. Fruquintinib was suspended in aqueous 0.5% CMC-Na (w/v) and stored at 4°C for all in vivo studies.
Cell lines and cell cultures
HEK-293 was purchased from American Type Culture Collection (ATCC, CRL-1573). The plasmid carrying full-length kdr cDNA was kindly provided by Dr. Bruce I. Terman (Department of Medicine, Albert Einstein College of Medicine, Bronx, New York, USA). HEK-293 cell line with stably-transfected KDR (HEK-293-KDR) was established in HMP. Primary HUVEC cells and HLEC were purchased from Allcell (cat# HUVEC-001F) and Sciencell (cat# 2500), respectively.
Biochemical assays
For human kinase assay in HMP, all recombinant kinases were purchased from Life Technologies. The kinase activity was determined by Z’-lyte or Transcreener fluorescence polarization according to manufacturers’ instruction. Fruquintinib selectivity was further assessed at 1 μmol/L against a panel of 253 kinases using [Citation32p-ATP] incorporation method. The UBI protocols are available at www.millipore.com/drugdiscovery/ KinaseProfiler.
KDR and VEGFR3 phosphorylation in cells
Inhibition of the receptor phosphorylation at a cellular level was determined by DELFIA or Western blotting. In DELFIA assay, HEK293-KDR cells were seeded in 96-well plates at a density of 2 × 104 cells/well in media and starved overnight. The cells were treated with the compound for 30 minutes, stimulated with 50 ng/mL recombinant human VEGF-A165 (R&D, 293-VE) for 8 minutes at 37°C and then lysed. p-KDR was captured with a monoclonal anti-KDR antibody (Sigma, V9134) and detected by DELFIA Eu-N1 labeled anti-phosphotyrosine antibody PT66 (PerkinElmer, AD0040).
HEK-293-KDR, primary HUVEC and HLEC cells were used in the study of VEGF-A or VEGF-C stimulated activation of KDR or VEGFR3 and downstream signaling in Western blot assays. Cells were seeded at 5 × 105 cells/well in 6-well plates and starved for 16 hours. The cells were then treated with the compound for 30 minutes before stimulation by 50 ng/mL of VEGF-A165 or VEGF-C for 8 minutes. VEGF-A-dependent VEGFR2 phosphorylation, VEGF-C-stimulated VEGFR3 phosphorylation, AKT and ERK1/2 phosphorylation were analyzed by Western blotting analyses. All antibodies were purchased from Cell Signaling, with the exception of anti-KDR rabbit antibody (Upstate, 07-716) and anti-actin mouse antibody (Sigma, A1978).
Cell proliferation assay
Primary HUVECs or HLECs in exponential phase were suspended in 100 μL of RPMI-1640 media containing 0.5% FBS, and seeded at 5 × 103 cell/well in 96-well plates pre-coated with 0.2% gelatin or fibronectin, and incubated overnight in a 5% CO2, 37°C incubator. Fruquintinib and VEGF-A165 or VEGF-C (50 ng/mL) were added and incubated for 48 hours. Viability of the cells was determined using CCK-8 assay format (Dojindo, CK04-13).
HUVEC cytotoxicity assay
Primary HUVEC cells at 2 × 104 cells/well were seeded in flat bottomed 96-well plates with 100 μL media containing 0.5% FBS. The next day, cells were treated with fruquintinib for 18 hours at 37°C. The cell survival was determined by AlamarBlue assay (Life Technologies, DAL1025). The plates were incubated for 3 hours at 37°C and fluorescence value was read at Ex 530 nm and Em 590 nm on Tecan.
HUVEC Tube formation
Flat bottomed 96-well plates were pre-coated with 70 μL of basement membrane matrix (BD Biosciences, 354234) for 30 minutes at 37°C to form gelling. Primary HUVECs in exponential phase were seeded at 2 × 104 cells/well in 100 μL RPMI-1640 media containing 0.5% FBS. The cells, with or without fruquintinib treatment, were incubated in a 5% CO2, 37°C incubator for 18 hours. The result was recorded by photographing under a microscope with 40× magnification. The total length of tubes in the presence of the compound was compared to that in the absence of the compound by using Image-Pro Plus software, and the inhibition rate was calculated based on the total tube length (TTL) under the microscope using the formula below: Inhibition rate (%) = (1 − TTL of compound treatment/TTL of control) ×100%.
Chick embryo chorioallantoic membrane (CAM) assay
Fertilized chicken eggs with 6 day incubation were obtained from Breeder Farm, Shanghai, China. Forty eggs were divided into 4 groups and incubated at 37°C with 50% humidity for 24 hours. On the following day, a small window (1 × 1 cm2) was made in the shell under aseptic conditions. The slides loaded with 10 μL of physiological saline containing various concentrations of fruquintinib were placed on the top of the growing CAMs. Pseudolarix acid B (PAB) was applied as a positive control. The window was resealed with an adhesive tape and the eggs were returned to the incubator. Upon 48 hours of incubation, the CAMs were photographed using an Olympus Live View Digital camera.
In vivo target inhibition assay (p-KDR inhibition)
All animal experiments were performed in accordance with the protocols approved by the Hutchison MediPharma Limited Animal Care and Use Committee (HMPLACUC). Female Balb/c nude mice at the age of 10∼11 weeks (Shanghai Slac Laboratory Animal Co. Ltd) were treated with a single oral dose of fruquintinib at 2.5 mg/kg suspended in 0.5% aqueous CMC-Na. At 1, 4, 8, 16 and 24 hours post dose, the plasma and lung samples were harvested for fruquintinib exposure and p-KDR analyses, respectively. Each group of designated time points was composed of 3 animals. Recombinant mouse VEGF (R&D, 493-MV/CF) was intravenously injected to the animals at the dose of 0.5 μg/mouse in the study groups, while animals in the control group received the same volume of PBS instead. All animals were anaesthetized with CO2 and sacrificed 5 minutes after VEGF injection. The left lobes of the harvested lungs were lysed to determine p-KDR (Cell Signaling Technology, 2478s) and β-actin (Cell Signaling Technology, 4967) by Western blots. The p-KDR and β-actin bands were visualized with Odyssey Infrared Imaging system and quantified with QuantityOne4.31 software (Bio-Rad). The inhibitory effect of fruquintinib was evaluated by quantification of normalized p-KDR signal over β-actin of the fruquintinib-treated groups relative to that derived from VEGF stimulated vehicle-treated group.
In vivo anti-tumor efficacy assessment in human tumor xenograft models
Human gastric cancer BGC-823 cell line was obtained from Shanghai Institutes for Biological Sciences in China. Large cell lung cancer NCI-H460, colon cancer HT-29, and clear cell renal cancer Caki-1 cell lines were purchased from ATCC. The tumor cells, at a density of 1∼5 × 106 cells/mouse, were subcutaneously inoculated to the right flanks of nude mice. The patient derived xenograft models were established after the primary tumor adopted serial passages in vivo. Once tumors have grown to 100∼300 mm3, the animals were randomly assigned with 6∼8 animals per group. The mice were treated orally with the vehicle (control group) or fruquintinib at a dose range of 0.5∼20 mg/kg suspended in the vehicle (treated group) once daily for 3 weeks. In combination studies, docetaxel (Taxotere, 5 mg/kg) or oxaliplatin (10 mg/kg) was administered to nude mouse via intravenous injection, once a week. Tumor size and body weights were measured 3 times a week. Tumor volumes (TV) were calculated using the formula: TV = (length × [width]2)/2. The percentage of tumor growth inhibition (%TGI = 100 × [1− (TV final - TVinitial for drug treated group)/(TVfinal – TVinitial for the control group)]) was used for evaluation of anti-tumor efficacy.
In vivo anti-angiogenesis analysis
At the end of Caki-1 anti-tumor efficacy study, the subcutaneous tumors from control- and fruquintinib-treated groups were collected and fixed in Zinc-formalin, and paraffin embedded sections were prepared. Immunohistochemistry staining of CD31 (BD PharMingen, 550274) was analyzed. In each section, 5 non-necrosis areas were randomly chosen and CD31 positive area was determined by NIKON BR-3.0 software. The anti-angiogenic effect of fruquintinib was evaluated by CD31 inhibition calculated with the formula: Inhibition% = 100 × [1− (CD31 positive area/tumor area for compound treated group)/(CD31 positive area/tumor area for the control group)].
Analysis of drug concentration in plasma
Blood samples from animals were collected at various time points in all in vivo studies. All blood samples were analyzed for determination of fruquintinib concentration with liquid chromatography coupled with tandem mass spectrometric (LC/MS/MS) methods.Citation26
Disclosure of Potential Conflicts of Interest
All authors are employees of Hutchison MediPharma Limited.
Acknowledgments
We thank Linjiang Tong, Hua Xie, Jian Ding (Shanghai Institute of Materia Medica, Chinese Academy of Science) for conduction of chick embryo chorioallantoic membrane (CAM) assay.
References
- Karkkainen MJ, Petrova TV. Vascular endothelial growth factor receptors in the regulation of angiogenesis and lymphangiogenesis. Oncogene 2000; 19:5598-605; PMID:11114740; http://dx.doi.org/10.1038/sj.onc.1203855
- Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer 2003; 3:401-10; PMID:12778130; http://dx.doi.org/10.1038/nrc1093
- Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, Maira G, Parati EA, Stassi G, Larocca LM, et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010; 468:824-8; PMID:21102434; http://dx.doi.org/10.1038/nature09557
- Kerbel RS. Tumor angiogenesis. N Engl J Med 2008; 358:2039-49; PMID:18463380; http://dx.doi.org/10.1056/NEJMra0706596
- Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol 2002; 20:4368-80; PMID:12409337; http://dx.doi.org/10.1200/JCO.2002.10.088
- Rak J, Mitsuhashi Y, Bayko L, Filmus J, Shirasawa S, Sasazuki T, Kerbel RS. Mutant ras oncogenes upregulate VEGF/VPF expression: Implications for induction and inhibition of tumor angiogenesis. Cancer Res 1995; 55:4575-80; PMID:7553632
- Farhang Ghahremani M, Goossens S, Nittner D, Bisteau X, Bartunkova S, Zwolinska A, Hulpiau P, Haigh K, Haenebalcke L, Drogat B, et al. p53 promotes VEGF expression and angiogenesis in the absence of an intact p21-Rb pathway. Cell Death Differ 2013; 20:888-97; PMID:23449391; http://dx.doi.org/10.1038/cdd.2013.12
- Raja R, Kale S, Thorat D, Soundararajan G, Lohite K, Mane A, Karnik S, Kundu GC. Hypoxia-driven osteopontin contributes to breast tumor growth through modulation of HIF1α-mediated VEGF-dependent angiogenesis. Oncogene 2014; 33:2053-64; PMID:23728336; http://dx.doi.org/10.1038/onc.2013.171
- Chatterjee S, Heukamp LC, Siobal M, Schöttle J, Wieczorek C, Peifer M, Frasca D, Koker M, König K, Meder L, et al. Tumor VEGF:VEGFR2 autocrine feed-forward loop triggers angiogenesis in lung cancer. J Clin Invest 2013; 123:1732-40; PMID:23454747; http://dx.doi.org/10.1172/JCI65385
- Wang X, Chen X, Fang J, Yang C. Overexpression of both VEGF-A and VEGF-C in gastric cancer correlates with prognosis, and silencing of both is effective to inhibit cancer growth. Int J Clin Exp Pathol 2013; 6:586-97; PMID:23573305
- Martins SF, Garcia EA, Luz MA, Pardal F, Rodrigues M, Filho AL. Clinicopathological correlation and prognostic significance of VEGF-A, VEGF-C, KDR and VEGFR-3 expression in colorectal cancer. Cancer Genomics Proteomics 2013; 10:55-67; PMID:23603341
- Pan L, Baek S, Edmonds PR, Roach M, Wolkov H, Shah S, Pollack A, Hammond ME, Dicker AP. Vascular endothelial growth factor (VEGF) expression in locally advanced prostate cancer: secondary analysis of radiation therapy oncology group (RTOG) 8610. Radiat Oncol 2013; 8:100; PMID:23618468; http://dx.doi.org/10.1186/1748-717X-8-100
- Engels K, du Bois A, Harter P, Fisseler-Eckhoff A, Kommoss F, Stauber R, Kaufmann M, Nekljudova V, Loibl S. VEGF-A and i-NOS expression are prognostic factors in serous epithelial ovarian carcinomas after complete surgical resection. J Clin Pathol 2009; 62:448-54; PMID:19126566; http://dx.doi.org/10.1136/jcp.2008.063859
- Simonetti O, Lucarini G, Rubini C, Goteri G, Zizzi A, Staibano S, Campanati A, Ganzetti G, Di Primio R, Offidani A. Microvessel density and VEGF, HIF-1α expression in primary oral melanoma: correlation with prognosis. Oral Dis 2013; 19:620-7; PMID:23279259; http://dx.doi.org/10.1111/odi.12048
- Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-islandformation and vasculogenesis in Flk-1-deficient mice. Nature 1995; 376:62-6; PMID:7596435; http://dx.doi.org/10.1038/376062a0
- Shibuya M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: a crucial target for anti- and pro-angiogenic therapies. Genes Cancer 2011; 2:1097-105; PMID:22866201; http://dx.doi.org/10.1177/1947601911423031
- Ziche M, Donnini S, Morbidelli L. Development of new drugs in angiogenesis. Curr Drug Targets 2004; 5:485-93; PMID:15216914; http://dx.doi.org/10.2174/1389450043345371
- Zhou CC, Bai CX, Guan ZZ, Jiang GL, Shi YK, Wang MZ, Wu YL, Zhang YP, Zhu YZ. Safety and efficacy of first-line bevacizumab combination therapy in Chinese population with advanced non-squamous NSCLC: data of subgroup analyses from MO19390 (SAiL) study. Clin Transl Oncol 2014; 16:463-8; PMID:24002945; http://dx.doi.org/10.1007/s12094-013-1102-5
- Ciuleanu T, Tsai CM, Tsao CJ, Milanowski J, Amoroso D, Heo DS, Groen HJ, Szczesna A, Chung CY, Chao TY, et al. A phase II study of erlotinib in combination with bevacizumab versus chemotherapy plus bevacizumab in the first-line treatment of advanced non-squamous non-small cell lung cancer. Lung Cancer 2013; 82:276-81; PMID:23992877; http://dx.doi.org/10.1016/j.lungcan.2013.08.002
- El-Rayes BF, Zalupski M, Bekai-Saab T, Heilbrun LK, Hammad N, Patel B, Urba S, Shields AF, Vaishampayan U, Dawson S, et al. A phase II study of bevacizumab, oxaliplatin, and docetaxel in locally advanced and metastatic gastric and gastroesophageal junction cancers. Ann Oncol 2010; 21:1999-2004; PMID:20332133; http://dx.doi.org/10.1093/annonc/mdq065
- Moehler M, Sprinzl MF, Abdelfattah M, Schimanski CC, Adami B, Godderz W, Majer K, Flieger D, Teufel A, Siebler J, et al. Capecitabine and Irinotecan with and without bevacizumab for advanced colorectal cancer patients. World J Gastroenterol 2009; 15:449-56; PMID:19152449; http://dx.doi.org/10.3748/wjg.15.449
- Grandinetti CA, Goldspiel BR. Sorafenib and sunitinib: novel targeted therapies for renal cell cancer. Pharmacotherapy 2007; 27:1125-44; PMID:17655513; http://dx.doi.org/10.1592/phco.27.8.1125
- Keating GM, Santoro A. Sorafenib: a review of its use in advanced hepatocellular carcinoma. Drugs 2009; 69:223-40; PMID:19228077; http://dx.doi.org/10.2165/00003495-200969020-00006
- Sartore-Bianchi A, Zeppellini A, Amatu A, Ricotta R, Bencardino K, Siena S. Regorafenib in metastatic colorectal cancer. Expert Rev Anticancer Ther 2014; 14:255-65; PMID:24559322; http://dx.doi.org/10.1586/14737140.2014.894887
- van Geel RM, Beijnen JH, Schellens JH. Concise drug review: pazopanib and axitinib. Oncologist 2012; 17:1081-9; PMID:22733795; http://dx.doi.org/10.1634/theoncologist.2012-0055
- Gu Y, Wang J, Li K, Zhang L, Ren HC, Guo LX, Sai Y, Zhang WH, Su WG. Preclinical pharmacokinetics and disposition of a novel selective VEGFR inhibitor fruquintinib (HMPL-013) and the prediction of its human pharmacokinetics. Cancer Chemoth Pharmacol 2014; 74:95-115; PMID:24817647; http://dx.doi.org/10.1007/s00280-014-2471-3
- Huang L, Huang Z, Bai Z, Xie R, Sun L, Lin K. Development and strategies of KDR/KDR inhibitors. Future Med Chem 2012; 4:1839-52; PMID:23043480; http://dx.doi.org/10.4155/fmc.12.121
- Takahashi S. Vascular endothelial growth factor (VEGF), VEGF receptors and their inhibitors for antiangiogenic tumor therapy. Biol Pharm Bull 2011; 34:1785-8; PMID:22130231; http://dx.doi.org/10.1248/bpb.34.1785
- Koch S, Tugues S, Li X, Gualandi L, laesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Biochem J 2011; 437:169-83; PMID:21711246; http://dx.doi.org/10.1042/BJ20110301
- Witzenbichler B, Asahara T, Murohara T, Silver M, Spyridopoulos I, Magner M, Principe N, Kearney M, Hu JS, Isner JM. Vascular endothelial growth factor-C (VEGF-C/VEGF-2) promotes angiogenesis in the setting of tissue ischemia. Am J Pathol 1998; 153:381-94; PMID:9708799; http://dx.doi.org/10.1016/S0002-9440(10)65582-4
- Karkkainen MJ, Petrova TV. Vascular endothelial growth factor receptors in the regulation of angiogenesis and lymphangiogenesis. Oncogene 2000; 19:5598-605; PMID:11114740; http://dx.doi.org/10.1038/sj.onc.1203855
- Huang YW, Xu LQ, Luo RZ, Huang X, Hou T, Zhang YN. VEGF-c expression in an in vivo model of orthotopic endometrial cancer and retroperitoneal lymph node metastasis. Reprod Biol Endocrinol 2013; 11:49; PMID:23693075; http://dx.doi.org/10.1186/1477-7827-11-49
- Kigure W, Fujii T, Sutoh T, Morita H, Katoh T, Yajima RN, Yamaguchi S, Tsutsumi S, Asao T, Kuwano H. The association of VEGF-C expression with tumor lymphatic vessel density and lymph node metastasis in patients with gastric cancer and gastrointestinal stromal tumor. Hepatogastroenterology 2013; 60:277-80; PMID:23574654
- Yao G, He P, Chen L, Hu X, Gu F, Ye C. MT1-MMP in breast cancer: induction of VEGF-C correlates with metastasis and poor prognosis. Cancer Cell Int 2013; 13:98; PMID:24119788; http://dx.doi.org/10.1186/1475-2867-13-98
- Wong SY, Haack H, Crowley D, Barry M, Bronson RT, Hynes RO. Tumor-secreted vascular endothelial growth factor-C is necessary for prostate cancer lymphangiogenesis, but lymphangiogenesis is unnecessary for lymph node metastasis. Cancer Res 2005; 65:9789-98; PMID:16267000; http://dx.doi.org/10.1158/0008-5472.CAN-05-0901
- Martins SF, Garcia EA, Luz MA, Pardal F, Rodrigues M, Filho AL. Clinicopathological correlation and prognostic significance of VEGF-A, VEGF-C, VEGFR-2 and VEGFR-3 expression in colorectal cancer. Cancer Genomics Proteomics 2013; 10:55-67; PMID:23603341
- Al-Rawi MA, Jiang WG. Lymphangiogenesis and cancer metastasis. Front Biosci 2011; 16:23-39; http://dx.doi.org/10.2741/3715
- Jin Li, Junning Cao, Jian Zhang, Zhiyu Chen, Wei Peng, Songhua Fan, Charlie Qi, Yi Gu, Yang Sai, Hua Mu. Phase I study of safety and pharmacokinetics of fruquintinib, a selective inhibitor of VEGF receptor-1, -2, and -3 tyrosine kinases in patients with advanced solid tumors. [Abstract]. In: Proceedings of the 104th annual meeting of the American association for cancer research. 2013 Apr 6–10; Washington, DC. Philadelphia (PA): AACR; Cancer Res 2013;73 (8 Suppl): Abstract nr 2413. doi:10.1158/1538-7445.AM2013-2413