
Abstract
Recent studies demonstrated that metformin exerts anti-neoplastic effect in a spectrum of malignancies. However, the mechanism whereby metformin affects various cancers, including gastric cancer, is poorly elucidated. Considering apoptosis plays critical role in tumorigenesis, we, in the present study, investigated the in vitro apoptotic effect of metformin on human gastric cancer cell and the underlying mechanism. Three differently-differentiated gastric cancer cell lines, MKN-28, SGC-7901 and BGC-823, along with one noncancerous gastric cell line GES-1 were used. We found that metformin treatment selectively induces apoptosis in the 3 cancer cell lines, but not the noncancerous one, as confirmed by flow cytometry, Caspase-Glo assay and western blotting against PARP and cleaved caspase 3. Moreover, the apoptotic effect of metformin seems to correlate negatively with the differentiation degree of gastric cancer. Metformin-induced apoptosis may be partially mediated through inhibition of anti-apoptotic survivin. Additionally, AMPK and mTOR, 2 important regulatory molecules responsible for metformin action, were investigated for their possible involvements in metformin-induced apoptosis of gastric cancer cell. AMPK knockdown by siRNA restores metformin-inhibited survivin expression and partially abolishes metformin-induced apoptosis. Similarly, forced overexpression of mTOR downstream effector p70S6K1 relieves metformin-induced inhibition of survivin and partly attenuates metformin-induced apoptosis. More importantly, survivin overexpression alleviates metformin-induced apoptosis. Xenograft nude mouse experiment also confirmed that AMPK/mTOR-mediated decrease of suvivin is in vivo implicated in metformin-induced apoptosis. Taken together, these evidences suggest that AMPK/mTOR-mediated inhibition of survivin may partly contribute to metformin-induced apoptosis of gastric cancer cell.
Abbreviation
| PARP | = | Poly Adenosine Diphosphate Ribose Polymerase |
| AMPK | = | Adenosine Monophosphate Activated Protein Kinase |
| mTOR | = | Mammalian Target of Rapamycin |
| siRNA | = | Small interfering Ribonucleic Acid |
Introduction
Gastric cancer is one of the most commonly-diagnosed malignancies of the human digestive tract, and the second-leading cause of cancer-related death worldwide.Citation1 Apart from surgery, no satisfactory chemo-preventive and therapeutic strategies are currently available for this lethal disease.Citation2 In addition, the prognosis of advanced gastric cancer is generally poor.Citation1 Thus there are still needs to seek out novel effective therapy for gastric cancer.
Metformin, a biguanide drug clinically used as first-line anti-diabetic therapy for the treatment of type 2 diabetes,Citation3 has attracted great attentions owing to its anti-neoplastic effect in a variety of malignancies, including breast,Citation4 colon,Citation5 prostate,Citation6 glial and endometric cancers.Citation7,8 A growing body of epidemiological surveys suggested beneficial effect of metformin in prevention and improved prognosis of solid tumor malignancies in diabetic patients.Citation9 Numbers of mechanisms underlying metformin's anti-tumorigenic action have been proposed. Studies showed that the anti-tumorigenic action of metformin may be attributed to increased insulin sensitivity, reduced liver gluconeogenesis and decreased hyperglycemia and insulin levels.Citation10 AMPK (Adenosine Monophosphate Activated Protein Kinase) /mTOR (Mammalian Target Of Rapamycin) cascade is the pivotal signaling pathway that underlies metformin action; metformin use activates AMPK, resulting in inhibition of mTOR that in turn represses protein synthesis, cell proliferation, cell cycle progression and angiogenesis.Citation11 Although mechanism responsible for metformin action has been investigated, its role in gastric cancer remains partly understood; the effect of metformin on gastric cancer has not been adequately elucidated.
Apoptosis, or programmed cell death, is a tightly-regulated biological event that maintains cell number in many normal physiological activities.Citation12 Dysregulated apoptosis is shown to be implicated in the onset and progression of various human cancers.Citation13 Down-regulated apoptosis is a hallmark of cancer.Citation14 Therapy aiming to restore apoptotic pathway in cancer may be of clinical potentials. Considering the critical involvement of apoptosis in tumorigenesis, we herein explored the apoptotic effect of metformin on human gastric cancer cell in vitro and in vivo as well as the underlying mechanism.
Results
Metformin selectively induces apoptosis in human gastric cancer cell lines, but not the noncancerous one
Flow cytometry, Caspase-Glo assay and protein gel blotting were used to explore the in vitro apoptotic effect of metformin on human gastric cancer cell lines. Metformin treatment significantly increases caspase 3/7 activities of the 3 gastric cancer cell lines, as compared to that of the noncancerous GES-1 (All P<0.05, ). In addition, metformin treatment induces cleavages of PARP and caspase 3 in the 3 gastric cancer cell lines, but not the noncancerous GES-1 (). Result of flow cytometry also showed that, after metformin treatment, apoptotic cells increase in the 3 gastric cancer cell lines in contrast with the counterpart of GES-1 (All P<0.05, ). Interestingly, as illustrated in , the apoptotic effect of metformin on gastric cancer cell seems to correlate negatively with the differentiation degree; BGC-823, the poorly-differentiated gastric cancer cell line, is most sensitive to the apoptotic effect of metformin.
Figure 1. Metformin selectively induces apoptosis in 3 human gastric cancer cell lines, but not the noncancerous one. (A): 48 h after addition of 5 mM metformin, caspase 3/7 activities were measured with Caspase-Glo assay as described in Materials and methods. Cells without metformin treatment were served as the control. Data are shown as mean (Rectangular box) ± SD (Error bar) and represent 3 independent experiments. (B): 48 h after 5 mM metformin treatment, cleavages of PARP and caspase 3 were examined by protein gel blotting, β-actin was probed as the loading control. Panel B is the representative image of 3 independent experiments.
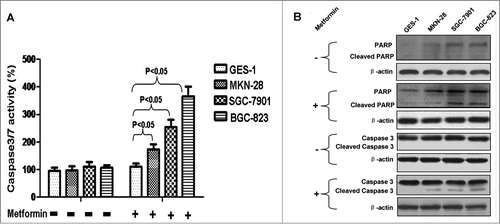
Figure 2. Metformin downregulates survivin in human gastric cancer cell lines. (A): Result of western blotting against survivin. Cells were incubated with or without 5 mM metformin for 48 h, and then immunoblotted with anti-survivin antibody. β-actin was served as the loading control. Representative image of 3 separate experiments is shown. (B): Densitometry and statistical analysis of panel A. Densitometry analysis was performed using the Image-J program as stated in Materials and methods. Data are shown as mean (Rectangular box) ± SD (Error bar), *, P < 0.05. (C): Metformin-induced reduction of relative survivin expressions in human gastric cancer cell lines. Relative survivin expression was similarly calculated as did in Panel B; reduction of relative survivin expression was computed by deducting the relative survivin expression of the metformin-treated from that of the corresponding metformin-untreated.
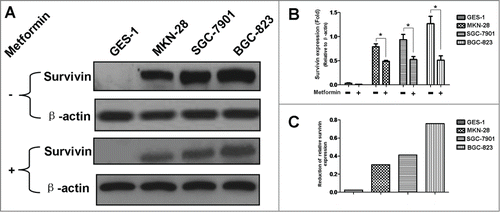
Figure 3. Metformin induces phosphorylation of AMPK in human gastric cancer cell lines. (A): Result of protein gel blotting. Cells were first treated with or without 5 mM metformin for 48 h, and then examined by western blotting against AMPK, P(Thr172)-AMPK and P(Ser79)-ACC (Acetyl-CoA Carboxylase). P(Ser79)-ACC, the classical downstream target of AMPK, was examined as an additional control to evaluate the functional activity of AMPK. β-actin was served as the loading control. Representative image of 3 separate experiments is shown. (B-D): Densitometry and statistical analysis of panel A. Densitometry analysis was performed using the Image-J program as stated in Materials and methods. Data are shown as mean (Rectangular box) ± SD (Error bar), *, P < 0.05.
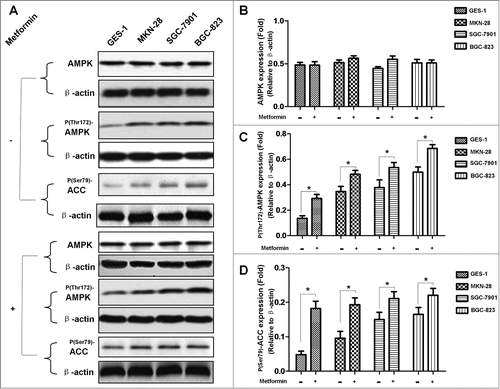
Figure 4. Metformin induces dephosphorylation of mTOR in human gastric cancer cell lines. (A): Result of protein gel blotting. Cells were first treated with or without 5 mM metformin for 48 h, and then examined by western blotting against mTOR and P(Ser2448)-mTOR. β-actin was served as the loading control. Representative image of 3 separate experiments is shown. (B-C): Densitometry and statistical analysis of panel A. Densitometry analysis was performed using the Image-J program as stated in Materials and methods. Data are shown as mean (Rectangular box) ± SD (Error bar), *, P < 0.05.
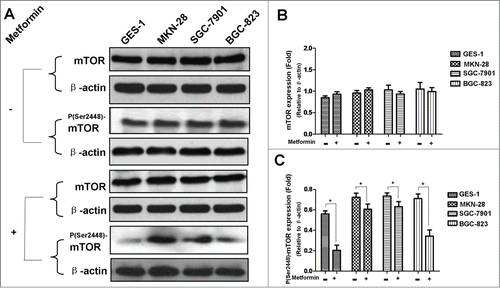
Figure 5. AMPK knockdown by siRNA alleviates metformin-induced inhibition of survivin in human gastric cancer cell lines. (A-B): Result of protein gel blotting. AMPK expression was abrogated by siRNA against α-1/α-2 AMPK isoforms or non-silencing control siRNA, and then subjected to 5 mM metformin treatment. AMPK, P(Thr172)-AMPK, P(Ser79)-ACC, P(Ser389)-p70S6K1 and survivin expressions were examined by western blotting. P(Ser79)-ACC and P(Ser389)-p70S6K1, 2 classical downstream targets of AMPK, were examined as additional controls to evaluate the functional activity of AMPK. β-actin was applied as the loading control. Representative image of 3 independent experiments is shown. (C-H): Densitometry and statistical analysis of panel A and B. Densitometry analysis was performed using the Image-J program as stated in Materials and methods. (C): Densitometry and statistical analysis of AMPK expression. (D): Efficacy of siRNA-induced AMPK inhibition. Relative AMPK expression was similarly calculated as did in Panel C; inhibition of relative AMPK expression (I) was computed by deducting the relative AMPK expression of the siRNA-affected from that of the corresponding siRNA-unaffected (II). The inhibition efficacy was then calculated according to the formula: Inhibition Efficacy=(I/II) × 100%. (E): Densitometry and statistical analysis of P(Thr172)-AMPK expression. (F): Densitometry and statistical analysis of P(Ser79)-ACC expression. (G): Densitometry and statistical analysis of P(Ser389)-p70S6K1 expression. (H): Densitometry and statistical analysis of survivin expression. Data are shown as mean (Rectangular box) ± SD (Error bar), *, P < 0.05.
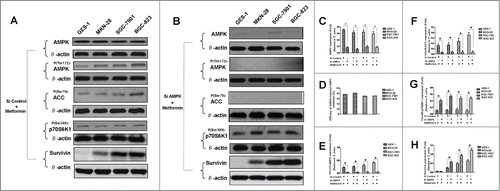
Figure 6. p70S6K1 overexpression ameliorates metformin-induced inhibition of survivin in human gastric cancer cell lines. (A-B): Result of protein gel blotting. Cells were induced to overexpress p70S6K1 before 5 mM metformin treatment. p70S6K1 and survivin expressions were examined by western blotting. β-actin was applied as the loading control. Representative image of 3 independent experiments is shown. (C-D): Densitometry and statistical analysis of panel A and B. Densitometry analysis was performed using the Image-J program as stated in Materials and methods. Data are shown as mean (Rectangular box) ± SD (Error bar), *, P < 0.05.
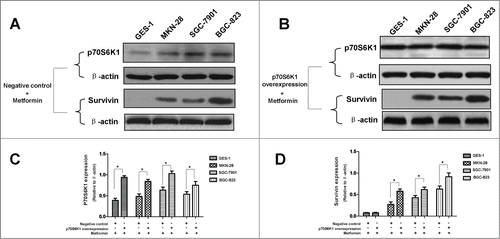
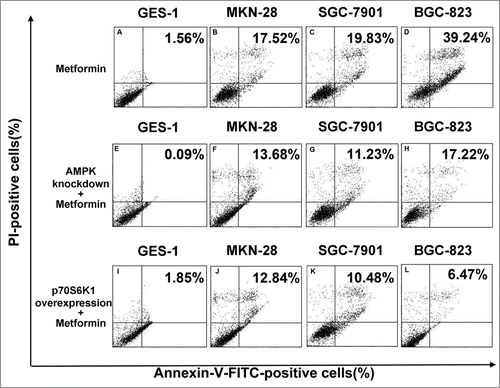
Figure 7. Blockade of AMPK/mTOR cascade partially antagonizes metformin-induced apoptosis in human gastric cancer cell lines. (A-D): Cells were treated with 5 mM metformin for 48 h and then stained with Annexin-V-FITC and PtdIns as described in Materials and methods. Apoptosis was analyzed by flow cytometry. (E-H): AMPK expression of cells was abrogated by siRNA against α-1/α-2 AMPK isoforms prior to 48 h incubation in 5 mM metformin, and then stained similarly with Annexin-V-FITC and PI. Apoptosis was analyzed by flow cytometry. (I-L): Cells were induced to overexpress p70S6K1 before the metformin treatment as detailed in Materials and methods, and then stained similarly with Annexin-V-FITC and PtdIns. Apoptosis was analyzed by flow cytometry. Percentage represents positive cells for Annexin-V-FITC and PI. Representative image of 3 independent experiments is shown.
Downregulation of survivin expression may contribute to metformin-induced apoptosis in human gastric cancer cell lines
As stated above, we found that metformin selectively induces apoptosis in gastric cancer cells, but not the noncancerous one. However, mechanism behind this interesting phenomenon still remains unknown. We next investigated the possible underlying mechanism.
Survivin belongs to the inhibitor of apoptosis protein family that prevents the execution of apoptosis.Citation15 Survivin inhibits apoptosis and protects cancerous cell against various cytotoxic factors, thus conferring immortalization and immune privilege to cancer cell.Citation16,17 Moreover, survivin is highly-expressed in a plethora of human malignancies but absent in normal cell.Citation15 These characteristics suggest that survivin may likely be a possible mediator of the above-mentioned underlying mechanism. Hence, we explored the role of survivin in metformin-induced apoptosis. As indicated in , in absence of metformin treatment, survivin was found to express only in the 3 human gastric cancer cell lines, with the highest survivin expression found in the poorly-differentiated BGC-823 cell line. However, in presence of metformin treatment, survivin expression of the 3 human gastric cancer cell lines declines, to various degrees, as compared to the counterpart in absence of metformin treatment. These results seem to suggest that metformin-induced apoptosis may be partially mediated through inhibition of survivin expression.
AMPK/mTOR-mediated survivin inhibition partly contributes to metformin-induced apoptosis in human gastric cancer cell lines
AMPK /mTOR cascade is the pivotal signaling pathway that underlies metformin action; metformin activates AMPK and subsequently leads to inhibition of mTOR which in turn represses protein synthesis, cell proliferation, cell cycle progression and angiogenesis.Citation11 In order to further elucidate the underlying mechanism responsible for metformin-induced apoptosis observed in this study, we went on investigating the involvement of AMPK/mTOR cascade in metformin-induced apoptosis. As shown in , metformin treatment results in increased phosphorylation of AMPK () but decreased phosphorylation of mTOR () in 4 cell lines when compared to the untreated counterpart.
Our study showed that metformin-induced inhibition of survivin expression may contribute, in part, to the apoptosis of gastric cancer cell. However, it is still unclear whether there are crosstalks between survivin expression and AMPK/mTOR cascade, the fundamental signaling pathway that governs metformin action. We therefore abrogated AMPK expression by siRNA prior to metformin treatment, and found that abrogation of AMPK expression restores metformin-inhibited survivin expression () and partially attenuates metformin-induced apoptosis ( and ). Additionally, we also deliberately induced overexpression of p70S6K1, an important downstream substrate of mTOR, prior to metformin treatment. Intriguingly, we discovered that p70S6K1 overexpression relieves metformin-induced inhibition of survivin () and counteracts, in part, the metformin-induced apoptosis in a similar manner ( and ).
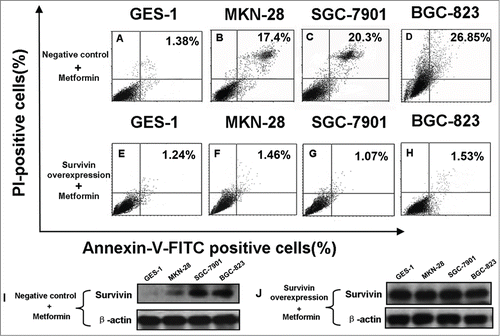
In order to further ascertain the role of survivin in metformin-induced apoptosis of gastric cancer cell, we explored the effect of survivin overexpression on metformin-induced apoptosis. As illustrated in , survivin overexpression alleviates metformin-induced apoptosis, indicative of important involvement of survivin in the pro-apoptotic effect of metformin on gastric cancer cell.
Figure 8. Survivin overexpression alleviates metformin-induced apoptosis in human gastric cancer cell lines. (A-E): Cells were transfected with empty plasmids prior to 48 h treatment of 5 mM metformin as stated in Materials and methods, and then stained with Annexin-V-FITC and PtdIns. Apoptosis was analyzed by flow cytometry. Percentage represents positive cells for Annexin-V-FITC and PI. (E-H): Cells were transfected with plasmids containing survivin cDNAs to induce survivin overexpression before 5 mM metformin treatment as detailed in Materials and methods, and then stained similarly with Annexin-V-FITC and PtdIns. Apoptosis was analyzed by flow cytometry. Percentage represents positive cells for Annexin-V-FITC and PI. (I-J): Cells were transfected with empty plasmids (I) or plasmids containing survivin cDNAs (J) before 5 mM metformin treatment as described in Materials and methods. Survivin expression was examined by protein gel blotting. β-actin was applied as the loading control. Representative image of 3 independent experiments is shown.
AMPK/mTOR cascade is in vivo implicated in metformin-induced apoptosis of gastric cancer cell
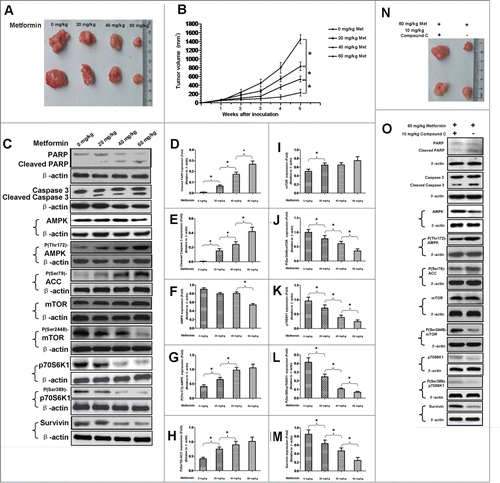
To explore whether or not metformin is able to induce apoptosis of gastric cancer cell in vivo, we established a xenograft tumor animal model by subcutaneously injecting BGC-823 gastric cancer cells into the rear flanks of nude mice. One week after the inoculation, mice were intraperitoneally administered once a day with metformin of different doses (0 mg/kg, 20 mg/kg, 40 mg/kg and 60 mg/kg, respectively) for 4 consecutive weeks. Throughout the experiment, metformin did not cause apparent side effects or changes of body weight in mice (Data not shown). Consistent with the aforementioned pro-apoptotic effect of metformin on gastric cancer cell in vitro, we found that metformin administration decreases, in a dose-dependent manner, the tumor volume and growth rate of BGC-823 cells in nude mice (). Moreover, in accordance with the in vitro finding that survivin expression is involved in metformin-induced apoptosis of gastric cancer cell, we discovered that metformin treatment reduces expression of survivin but up-regulates, concurrently, the expressions of cleaved PARP and caspase 3 in tumors obtained from xenograft mice (). In addition, similar to the increased expressions of cleaved PARP and caspase 3, metformin treatment also leads to increased phosphorylation of AMPK, inhibited phosphorylations of mTOR and p70S6K1 and reduced amount of p70S6K1 in tumors obtained from xenograft mice (), suggesting a possible association between AMPK/mTOR cascade and metformin-induced apoptosis in vivo. We next investigated whether or not AMPK/mTOR cascade is in vivo implicated in metformin-induced apoptosis of gastric cancer cell. Mice were treated with both metformin (60 mg/kg) and compound C, an AMPK inhibitor (10 mg/kg, as previously described).Citation18 As illustrated in , co-treatment of metformin and compound C results in decreased expressions of cleaved PARP and caspase 3, increased expression of survivin and reduced tumor volume, as compared to metformin treatment alone. These findings seem to indicate that AMPK/mTOR cascade is in vivo involved in metformin-induced apoptosis of gastric cancer cell.
Figure 9. AMPK/mTOR cascade is in vivo implicated in metformin-induced apoptosis of gastric cancer cell. (A): Nude mice were subcutaneously injected with BGC-823 gastric cancer cells at the rear flanks, and then intraperitoneally administered once a day with metformin of different doses as indicated in the photograph for the next 4 weeks. Tumors were excised after 4 weeks of metformin treatment. (B): Growth of tumors of BGC-823 cells in nude mice receiving different doses of metformin. Data are presented as mean (Solid dot) ± SD (Error bar), *, P < 0.05. (C): Expressions of cleaved PARP, cleaved caspase 3, AMPK, P(Thr172)-AMPK, P(Ser79)-ACC, mTOR, P(Ser2448)-mTOR, p70S6K1, P(Ser389)-p70S6K1 and survivin in tumors obtained from nude mice receiving different doses of metformin. Nude mice were similarly treated as described in panel A. The tumors were then subjected to western blotting. β-actin was applied as the loading control. Representative image of 3 independent experiments is shown. (D-M): Densitometry and statistical analysis of panel C. Densitometry analysis was performed using the Image-J program as stated in Materials and methods. Data are shown as mean (Rectangular box) ± SD (Error bar), *, P < 0.05. (N): Tumors of BGC-823 cells obtained from nude mice receiving co-treatment of metformin and compound C or metformin treatment alone. (O): Expressions of cleaved PARP, cleaved caspase 3, AMPK, P(Thr172)-AMPK, P(Ser79)-ACC, mTOR, P(Ser2448)-mTOR, p70S6K1, P(Ser389)-p70S6K1 and survivin in tumors obtained from nude mice receiving co-treatment of metformin and compound C or metformin treatment alone. β-actin was served as the loading control. Representative image of 3 separate experiments is shown.
Discussion
To date, most publications exploring the anti-tumorigenic action of metformin mainly centered on suppression of cell proliferation.Citation19-21 However, apoptotic effect of metformin on cancerous cell has not yet been fully investigated and is still a controversy. Recently, Kato et al demonstrated that metformin represses proliferation of human gastric cancer cell,Citation21 but it is still unknown whether apoptosis is implicated in metformin-induced suppression of cell proliferation. We, in the present study, revealed that metformin treatment selectively induces apoptosis in human gastric cancer cell, but not the noncancerous one. This finding indicates that metformin-induced apoptosis may be associated with the suppressed proliferation of gastric cancer cell described in the study by Kato et al.
The interesting selectivity of metformin action observed here is consistent with several previous reports. An et al found that metformin decreases high-fat-induced cardiac cell death.Citation22 Likewise, Detaille et al showed that metformin is able to inhibit high-glucose-induced endothelial cell death.Citation23 In addition, recent studies by Deng et al and Feng et al discovered that metformin induces apoptosis in breast cancer cell and ESCC (esophageal squamous cell carcinoma) cell.Citation19,20 More importantly, the fact that metformin selectively suppresses growth of ESCC cell but not the immortalized noncancerous esophageal epithelial cell in Feng's study also supports our finding.Citation20
Based on the aforementioned finding, we went on exploring the underlying mechanism. Considering survivin is an inhibitor against apoptosis and only highly-expressed in a variety of cancers,Citation15 we thus evaluated the involvement of survivin in metformin-induced apoptosis of gastric cancer cell, and found that metformin-induced apoptosis may be mediated, at least in part, by inhibition of survivin expression. Interestingly, as revealed in , metformin-induced decrement of survivin expression seems to associate negatively with the differentiation degree of gastric cancer, which also coincides well with the negative correlation between the apoptotic effect of metformin and the differentiation degree (). This finding, together with the study by Vazquez-Martin et al in which metformin was reported to prevent up-regulation of survivin in MCF-7/HER2 lapatinib-refractory cell,Citation24 suggest that survivin may be a potential molecular target of metformin's anti-tumorigenic action. Furthermore, this finding also suggests that metformin may be a therapeutic strategy for patient with gastric cancer by inhibiting the expression of anti-apoptotic survivin and resensitizing cancer cell to chemo-radiation.
In order to further elucidate the underlying mechanism responsible for metformin-induced apoptosis observed in this study; we explored the possible crosstalks between survivin expression and AMPK/mTOR cascade, the pivotal signaling pathway that governs metformin action.Citation11 Our result showed that metformin treatment increases phosphorylation of AMPK () but decreases phosphorylation of mTOR (), which is in good agreement with numerous previous studies.Citation11,20,Citation25 Moreover, abrogation of AMPK expression by siRNA restores metformin-inhibited survivin expression () and partially attenuates metformin-induced apoptosis ( and ), indicatives of regulatory role of AMPK in metformin-induced apoptosis. However, forced overexpression of p70S6K1, an important downstream substrate of mTOR, relieves metformin-induced inhibition of survivin () and partly counteracts metformin-induced apoptosis ( and ) in a similar manner, suggesting an important involvement of mTOR in modulation of metforim-induced apoptosis. More importantly, we also found that survivin overexpression alleviates metformin-induced apoptosis, indicating an important involvement of survivin in the pro-apoptotic effect of metformin on gastric cancer cell. In addition, our xenograft nude mouse experiment also confirmed that AMPK/mTOR-mediated decrease of suvivin is in vivo implicated in metformin-induced apoptosis of gastric cancer cell (). Taken together, these evidences seem to indicate that AMPK/mTOR-mediated inhibition of survivin expression contributes, at least in part, to metformin-induced apoptosis of human gastric cancer cell. Our finding echoes with the results of several studies. Jin et al reported that deguelin activates AMPK, leading to suppression of mTOR-mediated survivin and enhanced apoptosis in premalignant human bronchial epithelial cell.Citation26 More importantly, Vazquez-Martin et al also suggested that metformin phosphorylates AMPK and inhibits phosphorylation of p70S6K1, thus resulting in downregualtion of survivin and increased apoptosis in MCF-7/HER2 lapatinib-refractory cell.Citation24 Besides, Vazquez-Martin et al believe that AMPK–p70S6K1–survivin axis is a major target for metformin-induced restoration of lapatinib efficacy in MCF-7/HER2 lapatinib-refractory cell.Citation24 Our finding acts in concert with these 2 studies to underscore the important involvement of AMPK/mTOR-mediated inhibition of survivin in modulating metformin-induced apoptosis.
Nevertheless, abrogation of AMPK or overexpression of p70S6K1 does not completely attenuate metformin-induced apoptosis of human gastric cancer cell, indicating an AMPK/mTOR-independent way whereby metformin induces apoptosis. It is likely that metformin, a biguanide drug, may have other unknown functions independent of its original action. In fact, Hawley et al found that biguanides do not directly activate AMPK in cell free assay,Citation27 Guigas et al proved that metformin is still functional in mice lacking α1 andα2 AMPK catalytic units.Citation28 Moreover, with non-transformed and transformed cells lacking AMPK expression, Vincent et al also argued in their recent publication that metformin effect is AMPK-independent and may be mediated via additional mechanism(s).Citation29 Taken together, these evidences seem to highlight the notion that action of metformin is mediated dependently and independently through AMPK pathway. Additional studies are therefore warranted to further elucidate the AMPK-independent way whereby metformin exerts its pro-apoptotic action on gastric cancer cell.
The study by Sambol et al showed that the C max of metformin in human plasma ranges from 0.9 ± 0.04 μg ml−1 to 1.75 ± 0.11 μg ml−1, and the AUC ranges from 7.65 ± 0.32 μg ml−1 h to 11.47 ± 0.61 μg ml-1 h. Citation30 The concentration of metformin used in our in vitro cell experiments (5 mM) is higher as compared to the C max reported by Sambol et al. The use of high concentration of metformin has been the subject of criticism of similar studies on various cancer cell types, including prostate, breast and colon cancer cell lines.Citation4,5,31 However, it is important to consider the fact that cells in culture are actually maintained under hyperglycemic conditions in the present study. In our study, cell culture medium alone contains high level of glucose, and 10% FBS is additionally supplemented, thus resulting in potent growth stimulation to the cells. This may explain why, in order to observe the anti-tumorigenic effect of metformin in cell culture system, it is necessary to use higher concentration of metformin than are clinically used in patients taking this drug.
In conclusion, our results revealed that metformin selectively induces apoptosis in gastric cancer cell rather than the noncancerous gastric cell. The metformin-induced apoptosis observed may be partly mediated by inhibition of survivin expression resulted from metformin treatment. AMPK/mTOR cascade, the pivotal signaling pathway that underlies metformin action, is involved in modulation of survivin expression. AMPK/mTOR-mediated inhibition of survivin expression contributes, at least in part, to metformin-induced apoptosis of gastric cancer cell. Metformin may be a promising therapy for treatment of gastric cancer.
Materials and methods
Chemicals and antibodies
Metformin was obtained from Sigma (USA) and dissolved in dimethyl sulfoxide (Sigma, USA). Metformin at the concentration of 5 mM was used in the present study as indicated by our preliminary experiments that showed 5 mM metformin maximally maintains the viability of the noncancerous gastric cell but decreases that of the gastric cancer cell (Data not shown). Anti-survivin and anti-β-actin antibodies were purchased from R&D Systems (Minneapolis, USA). Anti-cleaved caspase-3 and anti-PARP antibodies were purchased from Cell Signaling Technology (Danvers, USA). Antibodies against AMPK, P(Thr172)-AMPK, P(Ser2448)-mTOR, P(Ser79)-ACC (Acetyl-CoA Carboxylase), P(Ser389)-p70S6K1 and p70S6K1 were purchased from Santa Cruz Biotechnology (Santa Cruz, USA). The horseradish peroxidase (HRP)-conjugated secondary anti-mouse and anti-rabbit antibodies were obtained from Abcam (Cambridge, USA). Compound C was obtained from Merck (Darmstadt, Germany).
Cell culture
Three differently-differentiated human gastric cancer cell lines, MKN-28 (Well-differentiated), SGC-7901 (Moderately-differentiated) and BGC-823 (Poorly-differentiated), along with one immortalized noncancerous human gastric epithelial cell line GES-1, were purchased from American Type Culture Collection (ATCC, USA). Cells were then cultivated in DMEM medium (Invitrogen, USA) supplemented with 10% FBS (Invitrogen, USA), 100 U/ml penicillin (Sigma, USA) and 100 μg/ml streptomycin (Sigma, USA) at 37℃ in a 5% CO2 incubator.
Assessment of caspase 3 and 7 activities
The Caspase-Glo 3/7 assay kit (Promega, USA) was used to measure caspase 3 and 7 activities in accordance with manufacturer's instruction. Briefly, 100 μL assay agent was added to each well of the culture plate, and then incubated for 1 h at room temperature. The luminescent signal was captured using Veritas Microplate Luminometer (Turner Biosystems, USA).
Western blotting
Cells were harvested in RIPA buffer (Sigma, USA) supplemented with protease inhibitor cocktail (Roche, USA). Cell lysates were sonicated for 30 sec and centrifuged for 15 min at 4℃. The pellets were discarded and the supernatants were retained. Protein concentration was determined with BCA protein assay kit (Pierce, USA). 60 μg of total protein was mixed with equal volume of loading buffer, boiled for 10 min, resolved on 10% SDS-PAGE and electroblotted to nitrocellulose membrane (Millipore, USA). Membranes were blocked in TBS containing 0.05% Tween-20 and 5% (w/v) non-fat milk for 1 h, and then probed with primary antibodies overnight at 4°C. Subsequently, membranes were incubated with HRP-conjugated secondary antibodies for 2 h at room temperature. Signals were detected using enhanced chemiluminescence (Pierce, USA), and bands were visualized and imaged by ChemiDoc XRS Plus System (Bio-Rad, USA). Band intensity was quantified using the Image-J 1.46r program (NIH, USA), and protein expression was normalized to β-actin (The internal loading control) as previously described by Wu et al.Citation32
Apoptosis analysis by Annexin V-FITC
Apoptosis was detected with the Annex V-FITC apoptosis detection kit (Roche, Germany) in accordance with manufacturer's instruction. Cells were first washed with cold PBS, and then collected by trypsinization and centrifugation. Cell pellets were gently resuspended in 100 μL staining buffer containing 2 μL Annexin-V-FITC and 2 μL PI (prodium iodide), and incubated at room temperature for 30 min prior to assay. Flow cytometry was then performed on the Cytomics FC 500 flow cytometer (Beckman Coulter, USA). Data were analyzed by FlowJo software (TreeStar, USA).
AMPK knockdown by siRNA
siRNA against α-1 and α-2 AMPK isoforms, and non-silencing control siRNA were synthesized by and purchased from Shanghai GenePharma (Shanghai, China). siRNA transfection was performed using Lipofectamine 2000 kit (Invitrogen, USA) according to manufacturer's instruction. Twenty-four h after the transfection, 5 mM metformin was added to the medium. 48 h after the metformin addition, apoptosis was analyzed by flow cytometry and western blotting as stated above.
Overexpression of p70S6K1 and survivin
Cells were transfected with wild-type p70S6K1 or survivin cDNAs (OriGene, USA) using Lipofectamine 2000 kit (Invitrogen, USA) as described by the manufacturer. Cells similarly transfected with empty plasmids were served as the negative control. Twenty-four h after the transfection, 5 mM metformin was added to the medium. 48 h after the metformin addition, apoptosis was subsequently examined using flow cytometry and protein gel blotting as aforementioned.
Animal studies
Animal experiments were performed in compliance with The Institutional Guidelines of Committee on Experimental Animals of Shanghai University of Traditional Chinese Medicine. Male nude mice (CD-1 nu/nu, 5∼6 weeks old) were obtained from Vital River Laboratory (Beijing, China) and then maintained under specific pathogen-free conditions, with free accesses to sterilized food and water. A total of 5 × 10 Citation6 BGC-823 gastric cancer cells suspended in 100 μL autoclaved PBS were injected subcutaneously into the rear flanks. The mice were randomly divided into 4 groups (n = 5), these groups were intraperitoneally administered once a day with 20 mg/kg metformin, 40 mg/kg metformin, 60 mg/kg metformin and equal volume of vehicle (0.9% w/v NaCl, injection grade), respectively. The daily administration of metformin or vehicle was initiated 7 d after the inoculation, and continued for the next 4 weeks until the mice were sacrificed. For the metformin and compound C co-treatment experiment, the experimental group (n = 5) was treated daily with intraperitoneal injections of metformin (60 mg/kg) and compound C (10 mg/kg, as previously described Citation18) for the next 4 weeks until the mice were sacrificed, whereas the control group (n = 5) received equal amount of metformin only. The mice were checked daily for any signs of discomfort and weighed every 3 d to evaluate healthy conditions. Bidimensional tumor size was measured weekly using caliper, and tumor volume was calculated according to the previously described formula: Tumor volume=(L × W2 × π/6) where L is the mid-axis length and W is the mid-axis width.Citation33 All mice were alive during the observation. When mice were sacrificed, tumors were excised, rinsed in cold PBS to remove blood and photographed. The tumors were then homogenized in RIPA buffer and subjected to western blotting as stated above.
Statistical analysis
Statistical analyses were conducted using Statistical Program for Social Science (SPSS) software 17.0 (SPSS, USA). Percentages and ratios were arcsine transformed prior to data analysis. A 2-sided P value of 0.05 was considered statistically significant. Comparisons between 2 groups were analyzed by Student's t-test. Data are presented as mean ± SD unless otherwise stated.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Smith JK, McPhee JT, Hill JS, Whalen GF, Sullivan ME, Litwin DE, Anderson FA, Tseng JF. National outcomes after gastric resection for neoplasm. Archives of Surgery 2007; 142:387-93; PMID:17441293; http://dx.doi.org/10.1001/archsurg.142.4.387
- Lordick F, Siewert JR. Recent advances in multimodal treatment for gastric cancer: a review. Gastric Cancer 2005; 8:78-85; PMID:15864714; http://dx.doi.org/10.1007/s10120-005-0321-z
- American Diabetes Association. Standards of medical care in diabetes-2010. Diabetes care 2010; 33:S11-S61; PMID:20042772; http://dx.doi.org/10.2337/dc10-S011
- Brown KA, Hunger NI, Docanto M, Simpson ER. Metformin inhibits aromatase expression in human breast adipose stromal cells via stimulation of AMP-activated protein kinase. Breast Cancer Res Treat 2010; 123:591-6; PMID:20300828; http://dx.doi.org/10.1007/s10549-010-0834-y
- Zhou XZ, Xue YM, Zhu B, Sha JP. Effects of metformin on proliferation of human colon carcinoma cell line SW-480. J Southern Med University 2010; 30:1935-8, 42; PMID:20813708
- Sahra IB, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le Marchand-Brustel Y, Bost F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 2008; 27:3576-86; PMID:18212742; http://dx.doi.org/10.1038/sj.onc.1211024
- Isakovic A, Harhaji L, Stevanovic D, Markovic Z, Sumarac-Dumanovic M, Starcevic V, Micic D, Trajkovic V. Dual antiglioma action of metformin: cell cycle arrest and mitochondria-dependent apoptosis. Cell Mol Life Sci 2007; 64:1290-302; PMID:17447005; http://dx.doi.org/10.1007/s00018-007-7080-4
- Ko EM, Walter P, Jackson A, Clark L, Franasiak J, Bolac C, Havrilesky LJ, Secord AA, Moore DT, Gehrig PA, et al. Metformin is associated with improved survival in endometrial cancer. Gynecol Oncol 2014; 132:438-42; PMID: 24269517; http://dx.doi.org/10.1016/j.ygyno.2013.11.021.
- DeCensi A, Puntoni M, Goodwin P, Cazzaniga M, Gennari A, Bonanni B, Gandini S. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prevention Res 2010; 3:1451-61; PMID:20947488; http://dx.doi.org/10.1158/1940-6207.CAPR-10-0157
- Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, Pollak M, Regensteiner JG, Yee D. Diabetes and cancer: a consensus report. CA 2010; 60:207-21; PMID:20554718
- Jalving M, Gietema JA, Lefrandt JD, Jong Sd, Reyners AKL, Gans ROB, de Vries EG. Metformin: taking away the candy for cancer? Eur J Cancer 2010; 46:2369-80; PMID:20656475; http://dx.doi.org/10.1016/j.ejca.2010.06.012
- Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Disco 2008; 7:1013-30; PMID:19043451; http://dx.doi.org/10.1038/nrd2755
- Fulda S. Targeting apoptosis for anticancer therapy. Semin Cancer Biol 2014; pii: S1044-579X(14):00071-6; PMID:24859747; http://dx.doi.org/10.1016/j.semcancer.2014.05.002. [Epub ahead of print]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/10.1016/j.cell.2011.02.013
- Athanasoula K, Gogas H, Polonifi K, Vaiopoulos AG, Polyzos A, Mantzourani M. Survivin beyond physiology: orchestration of multistep carcinogenesis and therapeutic potentials. Cancer Lett 2014; 347:175-82; PMID:24560928; http://dx.doi.org/10.1016/j.canlet.2014.02.014
- Asanuma K, Tsuji N, Endoh T, Yagihashi A, Watanabe N. Survivin enhances Fas ligand expression via up-regulation of specificity protein 1-mediated gene transcription in colon cancer cells. J Immunol 2004; 172:3922-9; PMID:15004200; http://dx.doi.org/10.4049/jimmunol.172.6.3922
- Endoh T, Tsuji N, Asanuma K, Yagihashi A, Watanabe N. Survivin enhances telomerase activity via up-regulation of specificity protein 1- and c-Myc-mediated human telomerase reverse transcriptase gene transcription. Exp Cell Res 2005; 305:300-11; PMID:15817155; http://dx.doi.org/10.1016/j.yexcr.2004.12.014
- Kim EK, Miller I, Aja S, Landree LE, Pinn M, McFadden J, Kuhajda FP, Moran TH, Ronnett GV. C75, a fatty acid synthase inhibitor, reduces food intake via hypothalamic AMP-activated protein kinase. J Biol Chem 2004; 279:19970-6; PMID:15028725; http://dx.doi.org/10.1074/jbc.M402165200
- Deng XS, Wang S, Deng A, Liu B, Edgerton SM, Lind SE, Wahdan-Alaswad R, Thor AD. Metformin targets Stat3 to inhibit cell growth and induce apoptosis in triple-negative breast cancers. Cell Cycle 2012; 11:367-76; PMID:22189713; http://dx.doi.org/10.4161/cc.11.2.18813
- Feng Y, Ke C, Tang Q, Dong H, Zheng X, Lin W, Ke J, Huang J, Yeung SC, Zhang H. Metformin promotes autophagy and apoptosis in esophageal squamous cell carcinoma by downregulating Stat3 signaling. Cell Death Dis 2014; 5:e1088; http://dx.doi.org/10.1038/cddis.2014.59
- Kato K, Gong J, Iwama H, Kitanaka A, Tani J, Miyoshi H, Nomura K, Mimura S, Kobayashi M, Aritomo Y, et al. The antidiabetic drug metformin inhibits gastric cancer cell proliferation in vitro and in vivo. Mol Cancer Ther 2012; 11:549-60; PMID:22222629; http://dx.doi.org/10.1158/1535-7163.MCT-11-0594
- An D, Kewalramani G, Chan JK, Qi D, Ghosh S, Pulinilkunnil T, Abrahani A, Innis SM, Rodrigues B. Metformin influences cardiomyocyte cell death by pathways that are dependent and independent of caspase-3. Diabetologia 2006; 49:2174-84; PMID:16868748; http://dx.doi.org/10.1007/s00125-006-0338-9
- Detaille D, Guigas B, Chauvin C, Batandier C, Fontaine E, Wiernsperger N, Leverve X. Metformin prevents high-glucose-induced endothelial cell death through a mitochondrial permeability transition-dependent process. Diabetes 2005; 54:2179-87; PMID:15983220; http://dx.doi.org/10.2337/diabetes.54.7.2179
- Vazquez-Martin A, Oliveras-Ferraros C, del Barco S, Martin-Castillo B, Menendez JA. The antidiabetic drug metformin: a pharmaceutical AMPK activator to overcome breast cancer resistance to HER2 inhibitors while decreasing risk of cardiomyopathy. Ann Oncol 2009; 20:592-5; PMID:19153119; http://dx.doi.org/10.1093/annonc/mdn758
- Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond) 2012; 122:253-70; PMID:22117616
- Jin Q, Feng L, Behrens C, Bekele BN, Wistuba, II, Hong WK, Lee HY. Implication of AMP-activated protein kinase and Akt-regulated survivin in lung cancer chemopreventive activities of deguelin. Cancer Res 2007; 67:11630-9; PMID:18089792; http://dx.doi.org/10.1158/0008-5472.CAN-07-2401
- Hawley SA, Gadalla AE, Olsen GS, Hardie DG. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes 2002; 51:2420-5; PMID:12145153; http://dx.doi.org/10.2337/diabetes.51.8.2420
- Guigas B, Bertrand L, Taleux N, Foretz M, Wiernsperger N, Vertommen D, Andreelli F, Viollet B, Hue L. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside and metformin inhibit hepatic glucose phosphorylation by an AMP-activated protein kinase-independent effect on glucokinase translocation. Diabetes 2006; 55:865-74; PMID:16567505; http://dx.doi.org/10.2337/diabetes.55.04.06.db05-1178.
- Vincent EE, Coelho PP, Blagih J, Griss T, Viollet B, Jones RG. Differential effects of AMPK agonists on cell growth and metabolism. Oncogene 2014; PMID: 25241895; http://dx.doi.org/10.1038/onc.2014.301. [Epub ahead of print]
- Sambol NC, Brookes LG, Chiang J, Goodman AM, Lin ET, Liu CY, Benet LZ. Food intake and dosage level, but not tablet vs solution dosage form, affect the absorption of metformin HCl in man. Br J Clin Pharmacol 1996; 42:510-2; PMID:8904626; http://dx.doi.org/10.1111/j.1365-2125.1996.tb00017.x
- Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le Marchand-Brustel Y, Bost F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 2008; 27:3576-86; PMID:18212742; http://dx.doi.org/10.1038/sj.onc.1211024
- Wu S, Tan M, Hu Y, Wang JL, Scheuner D, Kaufman RJ. Ultraviolet light activates NFkappaB through translational inhibition of IkappaBalpha synthesis. J Biol Chem 2004; 279:34898-902; PMID:15184376; http://dx.doi.org/10.1074/jbc.M405616200
- Carrasco RA, Stamm NB, Marcusson E, Sandusky G, Iversen P, Patel BK. Antisense inhibition of survivin expression as a cancer therapeutic. Mol Cancer Ther 2011; 10:221-32; PMID:21216939; http://dx.doi.org/10.1158/1535-7163.MCT-10-0756