
Abstract
Aneuploidy and chromosomal instability (CIN) are common features of gastric cancer (GC), but their contribution to carcinogenesis and antitumour therapy response is still poorly understood. Failures in the mitotic checkpoint induced by changes in expression levels of the spindle assembly checkpoint (SAC) proteins cause the missegregation of chromosomes in mitosis as well as aneuploidy. To evaluate the possible contribution of SAC to GC, we analyzed the expression levels of proteins of the mitotic checkpoint complex in a cohort of GC cell lines. We found that the central SAC proteins, Mad2 and BubR1, were the more prominently expressed members in disseminated GC cell lines. Silencing of Mad2 and BubR1 in MKN45 and ST2957 cells decreased their cell proliferation, migration and invasion abilities, indicating that Mad2 and BubR1 could contribute to cellular transformation and tumor progression in GC. We next evaluated whether silencing of SAC proteins could affect the response to microtubule poisons. We discovered that paclitaxel treatment increased cell survival in MKN45 cells interfered for Mad2 or BubR1 expression. However, apoptosis (assessed by caspase-3 activation, PARP proteolysis and levels of antiapoptotic Bcl 2-family members), the DNA damage response (assessed by H2Ax phosphorylation) and exit from mitosis (assessed by Cyclin B degradation and Cdk1 regulation) were activated equally between cells, independently of Mad2 or BubR1-protein levels. In contrast, we observed that the silencing of Mad2 or BubR1 in MKN45 cells showed the induction of a senescence-like phenotype accompanied by cell enlargement, increased senescence-associated β-galactosidase activity and increased IL-6 and IL-8 expression. In addition, the senescent phenotype is highly increased after treatment with PTX, indicating that senescence could prevent tumorigenesis in GC. In conclusion, the results presented here suggest that Mad2 and BubR1 could be used as prognostic markers of tumor progression and new pharmacological targets in the treatment for GC.
Abbreviations
| BMC | = | bleomycin |
| BubR1 | = | budding uninhibited by benzimidazoles 1 homolog B protein (gene BUB1B) |
| CDDP | = | cisplatin |
| CIN | = | chromosome instability |
| DDR | = | DNA damage response |
| Mad2 | = | mitotic arrest deficient-like-1 protein (gene Mad2L1) |
| PTX | = | paclitaxel |
| SAC | = | spindle assembly checkpoint |
| SASP | = | senescence associate secretory phenotype |
| γH2AX | = | phosphorylated H2AX |
| Monopolar Spindle kinase | = | MPS1 |
Introduction
Gastric cancer (GC) is a highly lethal malignancy.Citation1 Surgery is the main therapeutic option for patients with localized disease, followed by adjuvant chemotherapy, which enhances clinical responses.Citation2 However, many patients experience disease recurrence and eventually succumb due to chemotherapy resistance and/or metastasis. Paclitaxel (PTX) is widely used in the treatment of GC, usually in combination with other agents, depending on the state of the tumor at diagnosis.Citation3 This agent targets microtubules and provokes mitotic arrest, causing cell death. However, cells have developed various mechanisms to avoid apoptosis during mitosis through a process called “slippage” and by inducing senescence-like phenotypes due to abnormal mitosis in a variety of tumor cellsCitation4 or through mitotic catastrophe.Citation5,6 The spindle assembly checkpoint (SAC) is a regulatory mechanism present in all eukaryotes, which prevents chromosome missegregation during mitosis, thereby preventing aneuploidy.Citation7 SAC is the checkpoint through which PTX and other microtubule poisons exert their effects. The reduced expression of SAC proteins such as BubR1 (budding uninhibited by benzimidazoles 1 homolog B; BUB1B) or Mad2 (mitotic arrest deficient-like-1; Mad2L1) is associated with acquired paclitaxel resistance in ovarian carcinoma cell lines.Citation8,9
Chromosome instability (CIN) and aneuploidy are hallmarks of aggressive solid tumors such as GC.Citation10,11 Aneuploidy can result from inaccurate chromosome segregation; therefore, SAC has a crucial function in genetic integrity. Impaired SAC function has been suggested as one of the causes of aneuploidy in human cancers.Citation7,12 SAC is a complex of proteins that includes Mad1, Mad2, Bub1, BubR1, Bub3, and MPS1.Citation13 Among all SAC components, Mad2 and BubR1 have a pivotal function in checkpoint signaling due to their central role in the inhibition of the anaphase-promoting complex/cyclosome (APC/C). The discovery of mutations in BubR1 and Bub1 in a subset of colon cancer cell linesCitation14 and in mosaic variegated aneuploidy (a rare human disorder characterized by a predisposition to childhood cancers)Citation15 suggests that a weakened SAC contributes to the oncogenic process. In addition, mouse models that carry mutations in the SAC genes have been reported to develop spontaneous tumors. These heterozygous animals are viable and apparently healthy but develop spontaneous tumors.Citation16–22 There are examples of no tumor induction, as seen in mice with reduced Bub3Citation23 or BubR1 expression.Citation18 However, Mad2Citation19 or Mad1Citation21 haploinsufficiency caused a mild increase in the rate of spontaneous tumors. At the other end of the spectrum, Bub1−/H (null over hypomorphic)Citation24 and Mad2-overexpressing miceCitation20 all displayed high rates of spontaneous tumourigenesis. Coincidently, aberrant expression of the mitotic checkpoint proteins has been observed in human cancer cells, suggesting that SAC protein levels play an important role in cancer initiation and progression.
Senescence and apoptosis are two important mechanisms that protect cells against cellular transformation and cancer development. Cellular senescence is considered an alternative tumor suppressor mechanism,Citation25 and a correlation has been reported between senescence cells found in premalignant lesions (but not in malignant tumors) after oncogene activation.Citation26 However, senescent cells can interfere with their microenvironment by secreting proteases and mitogenic, antiapoptotic and antigenic factors, which can promote carcinogenesis in neighboring cells. This entity was recently described as the senescence-associated secretory phenotype (SASP)Citation27 and is gaining considerable interest in cancer research.Citation28
We have previously demonstrated that pretreatment with PTX, which induces a prolonged arrest in mitosis, increases cisplatin (CDDP) sensitivity in MKN45 GC cells due to impaired DNA repair.Citation29 In this study, we hypothesized that SAC dysfunction would be associated with GC and with PTX therapy response. We studied SAC protein levels in GC cell lines and found that Mad2 and BubR1 are frequently overexpressed in gastric adenocarcinoma. To clarify whether BubR1 or Mad2 levels play a role in GC, we generated GC gastric cell lines with depletion of these proteins. Our results show that these proteins are involved in proliferation, migration and tumor progression. However, after treatment with PTX the survival ratio of MKN45 cells lacking Mad2 or BubR1 is higher compared with MKN45 parental cells. We attempted to clarify the mechanism by which cells acquired resistance to PTX, and surprisingly, PTX monotherapy induced the apoptotic pathway independently of Mad2 or BubR1 protein levels, indicating that this pathway is unaffected. In the other hand, we observed that PTX treatment induced a senescence phenotype, which is exacerbated in cells that have been silencing for Mad2 or BubR1. The results are promising for the treatment of GC, as senescence could be acting as a suppressor of cancer progression. Although further in vivo studies are needed to clarify the role of senescence in this type of tumor, our results raise the possibility of a new treatment based on Mad2 or BubR1inhibitors in combination with PTX.
Results
mRNA expression levels of SAC components in GC cell lines
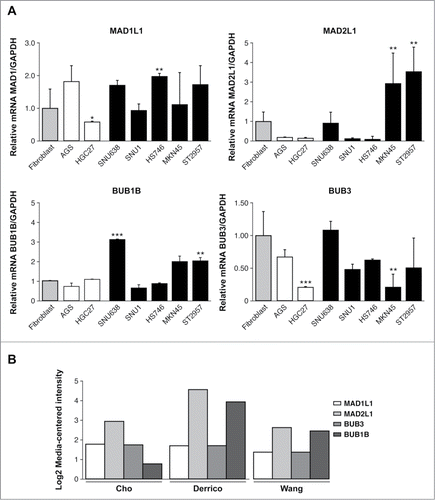
One of the characteristics of GC is the presence of a high number of aneuploidies.Citation30 To analyze the potential role of SAC defects in the generation of aneuploidies, we determined the BUB1B, BUB3, MAD1L1, MAD2L1 expression levels that participate in SAC regulation in 7 different GC cell lines. We also determined the levels of other genes linked to this checkpoint, such as CENPE and AURKB. The transcript levels of the selected genes were quantified by quantitative real time reverse-transcription polymerase chain reaction (qRT-PCR), and represented as fold changes between each GC cell line and the normal human colon fibroblast CCD18Co cell line. We compared the expression levels between primary (AGS and HGC27) and metastatic (SNU1, SNU638, Hs746t, MKN45, ST2957) GC cell lines, and we observed that MAD1L1 and BUB3 transcript levels were variable between all of them. Thus, MAD1L1 is significantly downregulated in HGC27 and BUB3 transcript levels were reduced in HGC27 and MNK45. However, MAD2L1 and BUB1B appeared to be upregulated in metastatic cells. Thus, MKN45 showed 3-fold greater expression of MAD2L1 and 1.5 fold times of BUB1B with respect to control fibroblasts. Moreover, ST2957 cells showed 4 times greater expression MAD2L1 and 2 times greater for BUB1B. Also SNU638 showed an increase of 3 times greater BUB1B expression. (). We analyzed MAD2L1, MAD1L1, BUB1B and BUB3 expression by using several published databases from the publicly available Oncomine database.Citation31–33 We required a P-value of below 0.05 and a fold-change of 2 for gene expression compared to the control. The results indicated that a significant increase in MAD2L1 and BUB1B mRNA level were observed in Gastric Intestinal Type Adenocarcinoma vs. Normal Gastric in 3 different clusters (). CENP-E transcription was reduced in HGC27 and Hs746t and increased in ST2957. Finally, when we studied AURK-B, we observed significantly increased transcript levels in the HGC27 and Hs746t cell lines (Fig. S1). These results suggest that MAD2L1 and/or BUB1B overexpression should be a common feature in gastric tumors and could be involved in modulating not only SAC activity but also the ability of adenocarcinoma gastric cells to proliferate and ultimately, their fate after specific drug treatments.
Figure 1. Relative levels of mRNA transcripts for the individual components of SAC. (A) Real-time q-PCR was performed to measure the relative levels of MAD1L1, MAD2L1, BUB1B, BUB3 mRNAs in the 7 GC cell lines established from primary tumor (white bars) and disseminated tumor (black bars). Each gene was normalized with GAPDH. Data are shown as relative to CCD18-Co, and the statistical significance was evaluated with ANOVA. ** P < 0.05 and *** P < 0.001. (B) Selected datasets from the Oncomine cancer microarray database were used to determine the alternations of MAD1L1, MAD2L1, BUB3 and BUB1B in mRNA expression levels. The graph represents the fold in gastric intestinal type adenocarcinoma versus normal gastric tissue based on studies reported by Cho et al., Derrico et al. and Wang et al. P < 0.001.
Interference of Mad2 and Bub1R1 expression modulates proliferation and cell migration in GC cells
To study the physiological functions of these proteins, we knocked down Mad2 and BubR1 expression with shRNA lentivirus (shMAD2L1 and shBUB1B) in 2 different cell lines: MKN45 and ST2957. After selecting with puromycin, we were able to obtain stable cultures of cells with reduced levels of Mad2L1 and Bub1B genes. We conducted a qRT-PCR study to select those clonal cultures with the highest knockdown of our genes of interest. We therefore selected the following for further study: MKN45-shBUB1B-2 and MKN45-shMAD2L1–2 from the MKN45 cells and ST2957-shBUB1B-3 and ST2957-shMAD2–1 from the ST2957 cell line. Western blot (WB) analysis corroborated the decrease in protein levels for Mad2 or BubR1, after infection with the specified lentivirus clones (Fig. S2).
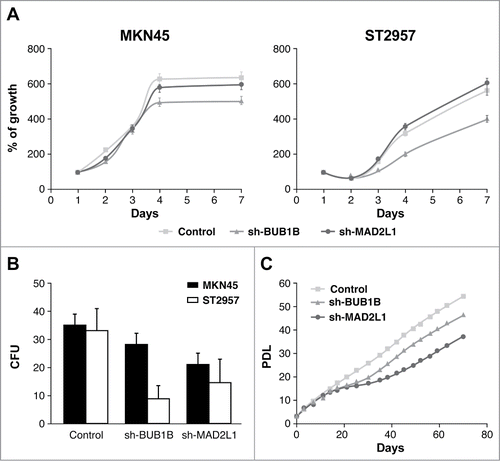
We evaluated the influence of MAD2L1 and BUB1B downregulation on cell proliferation in 7-day cultures and found that although there were no differences when knocking down BUB1B, the reduction of MAD2L1 appeared to regulate cell growth in both cell lines after 7 d However, there were no statistical differences (). To confirm the role of Mad2 in proliferation, we performed a colony-forming assay. As expected, our results showed that the absence of Mad2 reduced the number of surviving colonies at the end of the experiment (15 days) in both cells lines. We counted 25 colony-forming units (CFUs) in sh-MAD2L1 versus 35 CFUs in MKN45 and 15 CFUs in sh-MAD2L1versus 30 CFUs in control ST2957 (). Surprisingly, although we did not observe a change in proliferation at 7 days, the ability of the cells to form colonies in the absence of BubR1 was also reduced, with the reduction more significant in ST2957 cells (9 vs. 30 CFUs) than in the MKN45 cells (28 vs. 35 CFUs) (). To clarify this contradictory result, we compared the replicative life span of MKN45 cells with that of the cells with diminished Mad2 or BubR1 expression. Our experiment showed that after 15 days, both cell lines achieved lower population doubling levels (PDLs) than the parental MKN45 cells. After another 15 days, however, the cells overcame the crisis and were able to continue growing. Nevertheless, cells without BubR1 achieved lower PDLs than control cells (). These results suggest that Mad2 and BubR1 play a role in proliferation, which indicates that they act on the tumourigenesis process.
Figure 2. Mad2 and BubR1 regulate cell proliferation. (A) The MKN45 and ST2957 cell lines were transduced with lentivirus expressing sh-MAD2L1 and sh-BUB1B. The graphs show the proliferation rate measured every 24 h up to 7 d after infection, measured by crystal violet method. Data were calculated relative to the staining obtained on the first day. The experiments were done in quadruplicate at least 3 times. (B) Clonogenic assay. The graph represents the colony counting average at day 14, in 3 different areas per well. The experiment was performed in duplicate. (C) Analysis of the accumulated number of duplications over time (PDLs) in the MKN45 cell line and its transduced sh-BUB1R and sh-Mad2 cell lines. Each condition was done in duplicate.
SAC downregulation decreases cell migration and invasion in MKN45 cells
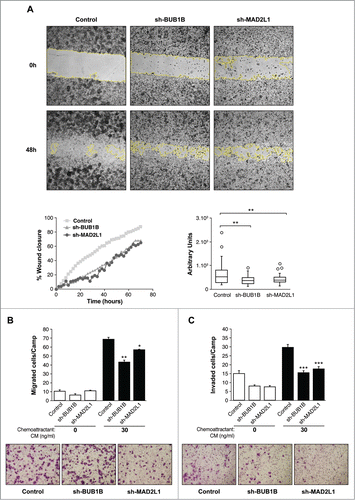
The capacity for cells lacking Mad2 or BubR1 to migrate was evaluated with an in vitro wound-healing assay. In control MKN45 cells, 88% of the wound area had been filled within 48 h postscratch. However, when BubR1 or Mad2 expression was diminished in these cells, only 66% of the wound area was filled during that time for both sh-MAD2L1 and sh-BUB1B cells (, Images and left graph). The right graph of shows the speed at which cells were able to repopulate the scratched area in arbitrary units: 659 ± 100; 434 ± 160 and 388 ± 95 for MKN45, sh-MAD2L1 and sh-BUB1B, respectively. The ST2957 cell line required more than 50 h to heal the scratch completely. ST2957-shBUB1B and ST2957-shMAD2L1 cells showed impaired migration in the same time period in contrast with the wild type ST2957 cells, with 36% of the area covered compared with 73% for the control cells (; Fig. S3). These observations were confirmed by migration assays (). The number of migrating cells was decreased in the absence of Mad2 or BubR1, compared with MKN45 cells. (). Both wound-healing and transwell migration assays revealed that Mad2 overexpression contributed to cell migration in the MKN45 cells. The invasive properties of the cells were assessed by a matrigel-coated transwell assay. There was a basal level of invasion in the control MKN45 cells (30 cells/field). However, the rate was significantly decreased to 18 cells/field in sh-BUB1B and 15 cells/field in sh-MAD2L1 (), demonstrating that the overexpression of Mad2 signaling confers invasive properties to adenocarcinoma parental cell lines. These findings imply that activation of SAC signaling promotes migration and invasion and could be associated with a poor prognosis for GC patients.
Figure 3. Silencing of Mad2 and Bub1B reduces migration and invasion in MKN45 cells. (A) Representative images of the first (0 h) and last picture (48 h) of control MKN45, sh-Mad2L1 and sh-BUB1B cells taken during the wound healing experiment. Images were taken at 10× magnification, every 2 h for 48 h. The yellow line represents the wound border. Left panel: The graph shows the percentage of wound closure over the study time using the ImageJ program. Right panel: Assessment of speed variations within the interfered cell lines and the control. The table shows the average speed for each line. The experiment was performed twice, and statistical differences were assessed by one-way ANOVA (* P < 0.05, ** P < 0.005, *** P < 0.001). (B) Transwell migration assay. The graph shows the quantification of stained migratory cells using the transwell assay without chemoattractant (basal migration control) and 30 ng/ml chemoattractant presence at 24 h. Representative photographs of stained cells attached to the bottom membrane of a transwell at the bottom. Statistical differences were assessed by a one-way ANOVA (* P < 0.05, ** P < 0.005, *** P < 0.001) (C) Transwell invasion assay. Graph shows the quantification of stained invading cells using the transwell assay with 6 μg of Matrigel, without chemoattractant (basal invading control) and 30 ng/ml chemoattractant presence at 24 h. Representative photographs of stained cells attached to the bottom membrane of a transwell at the bottom. Statistical differences were assessed by one-way ANOVA (* P < 0.05, ** P < 0.005, *** P < 0.001
Silencing of Mad2 and Bub1R1 increases survival of MNK45 cells treated with PTX
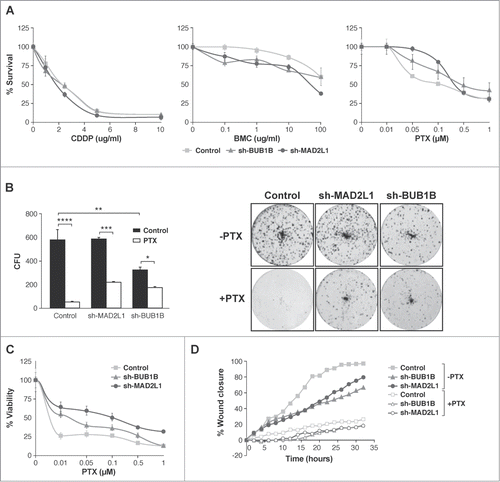
Next, we ascertained whether Mad2 upregulation could play a role in drug responses in GC. First, we studied cell survival after exposure to 3 different antitumoural agents: cisplatin (CDDP), paclitaxel (PTX) and the radiomimetic drug bleomycin (BMC). Our MTS viability assay showed that, in response to increasing amounts of CDDP (0–20 μg/ml), the behavior of all cell lines was almost the same; however, after BMC treatment (0–100 μg/ml), the reduction in Mad2 or BubR1 levels appeared to sensitize cells, given that a reduction in IC50 was observed. Surprisingly, cells with reduced Mad2 or BubR1 levels are more resistant to PTX (0–1 μM) than the control cells (). To support this observation, we studied the viability of cells after exposure to varying doses of PTX using the crystal violet method and we achieved the same result (). To confirm these observations, we performed a colony survival assay. MKN45, sh-MAD2L1 and sh-BUB1B cells were treated with solvent or PTX (100 nM) for 20 h. Ten days later, the number of surviving colonies was recorded. We found that the number of colonies in the absence of Mad2 was similar to MKN45, although the cells with reduced BubR1 had a lower number of surviving colonies compared with the other cultures (574 ± 89, 575 ± 22 and 319 ± 25.5 for MKN45, sh-MAD2L1 and sh-BUB1B, respectively). The number of colonies was higher (also in the absence of Mad2 or BubR1) after PTX treatment than in the control cells (43 ± 1.4, 216 ± 3.5 and 168 ± 8.5 for MKN45, sh-MAD2L1 and sh-BUB1B, respectively) (). However, our data show that migration is not modified by PTX treatment (). These results confirm the crucial role of SAC in cell death induction in response to PTX treatment.
Figure 4. Interference of Mad2 and BubR1 expression increases survival to PTX in MKN45 cells. (A) Indicated cell lines were seeded on MW96 and treated with increasing doses of CDDP, PTX or BMC. Graphs show survival curves measured by MTS 48 h after treatment. The experiments were performed in quadruplicate and repeated twice. (B) Left panel: Clonogenic assay. The graph represents the average of all clones in each experimental condition, in 3 independent experiments performed in duplicate. Statistical significance was studied by a 2-way anova (* P < 0.05, ** P < 0.005, *** P < 0.001). Cells were plated at low density and treated with 0.05 μM of PTX. Right panel: Representative images of colonies from a 10-day assay in control MKN45, sh-Mad2L1 and sh-BUB1B cell lines. (C) Control MKN45, sh-MAD2L1 and sh-BUB1B cells were treated with increasing concentrations of PTX (0–100 nM). Viability was quantified using the crystal violet after 48 h of treatment. Results are presented as percentage of viable cells relative to untreated cells. Data show the results from 3 independent experiments, performed in quadruplicate. Statistical differences were tested using Student's t-test (* P < 0.05, ** P < 0.005, *** P < 0.001). (D) The effect of PTX (0.1 μM) on control MKN45, sh-Bub1B and sh-Mad2L1 cell migration in the wound healing assay was measured up to 48 h. The wound closure was quantified every 2 h postwounding by measuring the remaining unmigrated area using ImageJ.
Silencing of Mad2 and Bub1b induces senescence in MKN45 cells treated with PTX
After prolonged arrest in mitosis, cells can die by apoptosis and re-enter on cycle G1 in a tetraploid state, through a process known as slippage. Death in mitosis is principally mediated by a caspase-dependent apoptotic pathway requiring mitochondrial outer membrane permeabilisation. A flow cytometry analysis demonstrated that the percentage of apoptosis was equivalent in MKN45 cells in response to PTX, independent of SAC (data not shown). We then studied the various molecular markers of apoptosis after PTX treatment in MKN45 cells and those with Mad2 or BubR1 depletion. We found that levels of the antiapoptotic member of the Bcl-2 family Mcl-1 (myeloid cell leukemia-1), a key molecule in cell death during mitosis, decreased 24 h after PTX exposure in MKN45 and sh-BUB1B. However, in the absence of Mad2, the protein remains at basal levels at least 48 h after treatment. Nevertheless, we observed the same behavior in Bcl-xl, with a shift indicative of inhibitory phosphorylation 24 h after PTX treatment (). Mcl1 degradation is controlled by phosphorylation mediated by Cdk1 or the stress kinases p38 and JNK. Our WB analysis showed that Cdk1 is phosphorylated at Y15 (inactive), with the same kinetics in MKN45 cells and cells depleted of BubR1 or Mad2, which achieved maximum levels 24 h after treatment. We then examined the impact of PTX on the stress-related JNK and p38 pathways. Treatment of cells with PTX resulted in a transient increase in phospho-JNK at 24 h after PTX exposure, with a modest increase in Mad2-depleted cells. Moreover, p38 showed persistent activation in MKN45 cells, transient activation in sh-BUB1B and virtually no activation in sh-MAD2L1 cells (). Treatment of cells with PTX induced cleavage/activation of procaspase-3 and degradation of PARP to an 85 kDa species in all cell lines 48 h after PTX exposure (). Given that Cyclin B levels control the mitotic state, we found Cyclin B degradation 48 h after PTX treatment, indicating that apoptosis occurs after exit mitosis. A prolonged arrest in mitosis could also induce DNA damage. Our data showed that the marker of DNA damage H2Ax is activated in MKN45 cells to a lesser degree than in the case of SAC compromised cells. These results suggest that arrest in mitosis increases DNA damage and induces the intrinsic apoptotic pathway in MKN45 cells, regardless of the strength of the SAC. Mcl-1 degradation depends mainly on p38, and its degradation is delayed in depleted Mad2 cells but probably not enough to inhibit apoptosis.
Figure 5 (See previous page). Interference of Mad2 o BubR1 expression induces senescence phenotype in MKN45. (A and B) Western blotting analysis of MKN45, sh-MAD2L1 and sh-BUB1B cultures treated with 0.1 μM PTX during several time intervals. Twenty micrograms (20 μg) of WCE protein were resolved in 15% or 8% SDS-PAGE. Expression/activation of Bcl-xl and Mcl-1 proteins (A-left panel), Cdk1Y15, JNK1/2, phosphorylated P38 and total P38 (A, right panel), Caspase-3 proteolysis, H2AX (b-left panel), PARP, cleaved-PARP, cyclin-B1 (B, right panel) were detected by using specific antibodies against each one. α-tubulin was used as a loading control. (C) Left upper panel: β-Galactosidase activity in MKN45 control and cells with Mad2 and BubR1 knockdown in 3-day cultures. Down panel: PTX effect in senescence phenotype was studied measuring β-Galactosidase activity in MKN45 control and interfered cell lines. Cells were treated with PTX (0.1 μM) for 3 d. Bars represent average of 3 independent experiments. (* P < 0.05,** P < 0.005, ***P < 0.001. Representative images of β-Galactosidase stained cell culture with and without PTX in 3 d. Arrows point to senescent cells. (D) Q-PCR analysis was performed using Taqman to measure p53 and p21 mRNAs levels in shBUB1B and sh-MAD2L1 knockdown cells referring to MKN45 cells. β-actin was used as endogenous gene control. Western blotting analysis of p53 expression in cells treated with 0.1 μM of PTX for 72 h, using a specific antibody against phosphorylated Ser15 and an antibody against the native protein. α-tubulin was used as a loading control. (E) IL-6 e IL-8 expression levels were analyzed by Q-PCR using Taqman in all cell lines in the 3-day culture. β- actin was employed as an endogenous gene control and the graph shows the fold induction referred to the MKN45 control cell line.
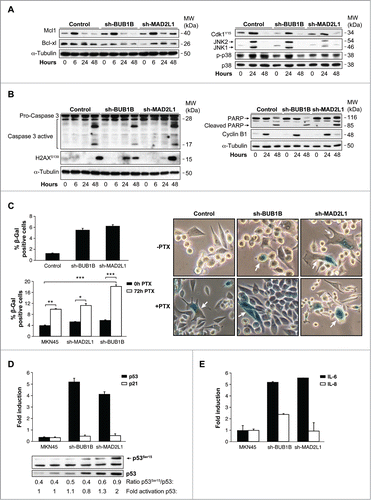
To determine whether senescence was an alternative process to explain the results obtained in survival, we performed an in situ senescence-associated (SA) β-Gal assay. We observed the induction of the senescence-like phenotype of MKN45 cells, manifested by changes in morphology and β-galactosidase (SA-β-gal) activation. With this experiment, we determined that the fraction of SA-β-gal-positive cells increases 3 d after Mad2 or BubR1 depletion (). Moreover, SA-β-galactosidase activity staining showed a clear increase in cellular senescence in these cells after PTX treatment compared with control MKN45 cells (). PTX causes a prometaphase arrest cell cycle that, if sustained, generates DNA damage and senescence. p53 is a marker for both events. When we analyzed its activation after 72 h of PTX treatment, we observed an increase in p53 mRNA levels in cells lacking Mad2 or BubR1 (). On the contrary, we observed no increase in p21waf1 WB analysis confirmed these results, as well as a sharp increase in p53Ser15, induced by PTX. To determine whether the SASP is also under SAC protein control, we monitored SASP IL-6 and IL-8 expression in control cells and after 50 d of gene interference (). Both IL-6 and IL-8 mRNA levels were increased during senescence in the absence of BUB1B, but only IL-6 (and not IL-8) expression was attenuated when Mad2 was decreased. The results of this study suggest that the weaker SAC in MKN45 cells increases the levels of a senescent phenotype involved in the PTX response in these cells.
Discussion
GC is a disease with a very poor prognosis at diagnosis. Finding new targets that increase our understanding of the disease presents a challenge for researchers, but can ultimately contribute to improved and more personalized treatments. However, the biology of this tumor is very complex at the molecular level, and there is no defined pattern of markers.Citation34 Our results show that GC cells undergo uncontrolled SAC protein expression, which is the case in other types of cancers such as lung, breast, head and neck, ovarian and cervical cancer.Citation35 This study has shown that two essential proteins in the mitotic checkpoint, Mad2 and BubR1, are overexpressed in the majority of disseminated gastric cancer cell lines, and these results are in agreement with the public database ONCOMINE. (http:oncomine.org). We have characterized the effects of a reduction in Mad2 or BubR1 expression and found that these proteins control cell proliferation, migration and tumor progression in MKN45 and ST2957 cell lines. Accordingly, overexpression of BubR1 in bladder cancerCitation36 or Mad2 in colorectal mucosaCitation37 has been associated with high proliferation. Moreover, ovarian carcinoma showed overexpression and correlation of Mad2 and BubR1, which are related to cellular proliferation and time to recurrenceCitation38. Taken together our data indicated that overexpressed Mad2 and BubR1 participate in the carcinogenesis of GC and high Mad2 and Bub1B could be used as a prognostic marker; in fact BubR1 have recently been suggested as a prognostic marker in ovarian cancer.Citation39
There is some controversy in the literature regarding the importance of correct SAC function and therapy response. Mad2 downregulation in MCF7 and A2780 cellsCitation9,40 resulted in resistance to PTX. Similarly, we also detected an increase in cell survival after treatment with PTX in MKN45 cells. Inhibition of apoptosis is a frequent cause of drug resistance, and it has been suggested that Mad2 interference could inhibit anticancer drug-induced apoptosis by upregulating Bcl-2 and interfering with the mitochondria apoptosis pathway.Citation41 Here, we demonstrated that depletion of Mad2 delay Mcl-1 degradation, suggesting that exit to mitosis is delayed; however, no differences have been found in apoptotic induction because activation/proteolysis of caspase-3/PARP occurs at the same time in cells, regardless of the presence of Mad2 or BubR1. We could say that the difference in survival among MKN45 cells is not due to apoptotic inhibition. More importantly, our data show that the molecular mechanism related to survival correlates with the induction of cellular senescence-like phenotype. The contribution of these proteins to PTX therapy has been previously studied. For example, BubR1 downregulation has been related to PTX sensitivity in esophageal squamous cell carcinoma,Citation42 but BubR1 inhibition promotes resistance to microtubule inhibitors in colorectal cancer (CRC).Citation43 Mad2 and BubR1 suppression in PTX-treated ovarian and breast cancer cells results in PTX resistance.Citation40 Our interpretation is that senescence in GC could act as a tumor suppressor, although in vivo studies are needed in order to confirm the contribution to cell death. Cellular senescence can be induced by oncogenes (oncogene-induced senescence [OIS]) and is a crucial anticancer mechanism that prevents the growth of cells that are at risk of neoplastic transformation. Signaling pathways known to regulate OIS include the p16/RB and p19/p53/p21 pathways. Our results show that Mad2 depletion increases activation of the p53 pathway; however, p53 activation does not result in induction of p21 expression. These results do not agree with those from studies showing that Mad2 partial depletion in IMR90 primary human fibroblasts induces the p53-p21 pathway. However, other cells (such as MCF10 A) show low activation of this pathway when Mad2 is downregulated. Another possibility is that the p53 activation and stability is a reflection of DNA damage in cells, and senescence is associated with the mTORC1 route, through a senescence-associated role for 4E-BP1 in crosstalk with the transcription factor p53.Citation44 Further studies are warranted to explore this possibility. Recent studies have demonstrated that normal human diploid fibroblasts (HDFs) exposed to various senescence-inducing stimuli undergo a mitosis skip before entry into permanent cell cycle arrest. This mitosis skip is mediated by both, p53 and pRb family protein-dependent transcriptional suppression of mitotic regulators,Citation45 which could be the case in our cells. On the other hand, we can argue that in the absence of Mad2 or BubR1, the PTX treatment leads to a prolonged arrest, probably increasing free radicals, which are crucial for SASP regulation.Citation46 The secretome of senescent cells is complex, consisting of a range of cytokines, chemokines and proteases, among other substances, and is associated with inflammation, proliferation and modulation of the extracellular matrix (ECM).Citation47 Our results show that senescent cells with Mad2 or BubR1 depletion undergo IL-6 and IL-8 expression induction. This led us to consider that an increase in IL-6 and IL-8 levels could favor paracrine signaling and increase senescence and ultimately cell death or, by contrast, that SASP contributes to cell transformation, which could ultimately be an advantage for tumor growth.Citation28 Further studies are needed to clarify the clinical outcome of patients in relation to Mad2 or BubR1 levels and PTX response. On the other hand, a small population of cells could instead become aneuploid, which could ultimately be an advantage for tumor growth.Citation28 In addition, we should consider studying the relationship between the recently described protein LZTS1 (leucine zipper putative tumor suppressor 1), which plays a critical role in resistance to paclitaxel, by controlling Cdk1 activity, in breast cancer.Citation48 Therefore, understanding exactly how common environmental and cellular stresses affect mitosis is critical to understanding how and why some cancer cells are sensitive and others are resistant to this important class of chemotherapy. The positive news is that sensitivity to cisplatin or BMC appears to be unaffected in the absence of these proteins, which opens the possibility of using them in combination with new drugs that target Mad2 or BubR1.
In conclusion, we have demonstrated that reduced Mad2 and BubR1 protein levels appear to contribute to cellular senescence induction, a mechanism that could prevent tumourigenesis. This is an exciting result because it opens a new line of research, aimed at designing new drugs that can target these proteins.
Material and Methods
Cell culture and drugs
The MKN45 (poorly differentiated adenocarcinoma; DSMZ: Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH) cell line was maintained in RPMI 1640, supplemented with 20% FBS and 2-mM L-Glutamine. The ST2957 (lymph node metastases) and HEK293 cells were maintained in high glucose Dulbecco's modified Eagle Medium (DMEM; Gibco), supplemented with 2-mM L-Glutamine, 1-mM sodium pyruvate, 10% foetal bovine serum. All cell lines were grown at 37°C in a humid atmosphere containing 5% CO2.
Chemicals and antibodies
Antibodies against H2AX phosphorylated at Ser 139, p38, phosphorylated p38, caspase 3, cdk1 Y15, p53 and p53Ser15 were purchased from Cell Signaling Technologies (http://www.cellsignal.com). Antibodies against PARP, Cyclin B, Mcl-1 and Bcl-xl were purchased from Santa Cruz Technology (http://www.scbt.com). Antibody Anti-ACTIVE® JNK pAb, Rabbit, (pTPpY) was acquired from Promega Corporation-Spain (http://www.promega.es/). Paclitaxel and puromycin were acquired from Sigma-Aldrich (http://www.sigmaaldrich.com/sigma-aldrich/home.html). BMC was acquired from Calbiochem (http://www.merckmillipore.com/spain/life-science-research), and CDDP was donated from Ferrer FARMA.
Western blotting
Protein extracts (20 μg) were resolved on 4%– 20%– SDS-PAGE (BioRad) and transferred to nitrocellulose membranes. Western immunoblot analysis was performed as described previously.Citation29
Viral transduction of cells
Viral particles were generated according to the manufacturer's instructions using GIPZ Lentiviral shRNA for Mad2 or BubR1 (Thermo Scientific Open Biosystems). Briefly, 4.5 × 106 HEK 293 cells/plate in DMEM medium were transfected using lipofectamine 2000 (Invitrogen) with 15 μg of shMad2, 7 μg of envelope plasmid (VSV-G) and 7 μg of Helper plasmid (pCD/NL-BH). The supernatants were recovered 48 h and 72 h after transfection and frozen in small aliquots at -80°C until use. Transduction was conducted using 5 × 105 cells per well in a 6-well plate. The cells were examined microscopically 48 hours post-transduction for the presence of GFP reporter expression as an indicator of transduction efficiency. Cells were assayed 72 h later for reductions in gene expression by quantitative/real-time PCR (qRT-PCR) and compared with nonsilencing shRNA.
RT-PCR
Total cellular RNA was extracted using Tri-Reagent, as previously described. One microgram was primed with poly-T and cDNA synthesized with M-MLV reverse transcriptase following the manufacturers’ instructions (Promega). The quantitative expression of each gene was measured by SYBR Green polymerase chain reaction assay, using the following specific primer sets: 5′- TCGTGGCAATACAGCTTCAC-3′ and 5′- GGTCAATAGCTCGGCTTCC-3′ for BUB1B; 5′- GCTTGTAACTACTGATCTTG-3′ and 5′- GCAGATCAAATGAACAAGAA-3′ for Mad2L1; 5′-GCTCAGCAACAAACCATGG-3′ and 5′-GGAATATTCAACATAACATG-3′ for BUB; 5′- GGAGCTGGAGAACGAGAG -3′ and 5′- ACTGTGGTCCCGACTCTT -3′ for MAD1L1;5′ GTTGATCTTGCAGGCAGTGA -3′ and 5′- TGAAACCACCAACTTGTCCA -3′ for CENP-E; 5′ GGTGGTTCTGATGGCTTTGT -3′ and 5′- GCAAGCGTAGTCCCATCATT -3′ for BUB3; 5′ GGGAGAGCTGAAGATTGCTG -3′ and 5′- GCACCACAGATCCACCTTCT -3′ for AURK-B 5′-GAG; AGA CCC TCA CTG CTG-3′ and 5′-GAT GGT ACA TGA CAA GGT GC-3′ for GAPDH. Relative quantification (RQ) was performed using the delta-Ct method, where each 1-Ct difference equals a 2 fold change in transcript abundance. qRT-PCR analysis was employed to quantify the expression levels of IL-6, IL-8 and p21 by using Taq-man probes (Life technologies, https://www.lifetechnologies.com).
Cell viability, proliferation, clonogenic assay and PDLs
Viability and growth rate were determined using a crystal violet based staining method, as previously described.29 Survival was measured using an MTS assay (Promega). For the clonogenic assay, cells were cultured in a 6-well plate at a very low concentration (2000 cells per well). After 14 days, the cells were fixed with 1% glutaraldehyde and stained with crystal violet. The number of colonies was quantified directly using the 10× objective in 3 different areas per well. To measure the population doubling level (PDL), 300,000 cells were plated in p60, and the number of cells was quantified every 4 days. The calculation of the PDL was performed using the following formula: PDL = 3/ln(final no. of cells /300000)+1).
Cell migration and invasion assays
For the wound-healing assay, a confluent monolayer of MKN45 cells and ST2957 controls and knockdown for Mad2L1 or BUB1B cells was scratched into a 24-well plate with a sterile tip. The cell migration's ability to fill the wound was studied up to 50 h. The relative distance traveled by the leading cell edge was assessed by time-lapse microscopy using the Cell Observer Z1 (Zeiss) at 37°C and 5% CO2/95% air, using the imaging software Axiovision 4.8 and the Cascade 1 k camera. Images were taken every 2 h and further processed using the Digital Image Processing Software AXIOVISION (Zeiss). The wound closure was quantified by measuring the remaining unmigrated area using ImageJ. Migration and invasion assays were performed in modified Boyden chambers with polycarbonate filters (6.5 mm diameter, 8.0 μm pore size) (transwell migration and invasion assays) (Corning Inc., Corning, NY, USA) coated with 0.5% gelatin (migration assay) or 6 μg growth factor reduced Matrigel (BD Biosciences, Bedford, MA, USA) (invasion assay). Prior to the migration and invasion assays, cells were maintained for 16 h in serum-free medium and 1.5 × 106 cells resuspended in serum-free medium were seeded in the upper chamber. Serum-free conditioned medium from NIH3T3 cells at 30 ng/ml was used as chemoattractant and placed in the bottom chamber. Cells were allowed to migrate or invade for 24 h at 37°C with 5% CO2. Non-migrated and non-invaded cells were removed using a cotton swab, and the filters were stained with Diff Quik (Dade Behring, Newark, DE, USA). Migrated or invaded cells were counted in 10 fields of maximum migration or invasion under a light microscope at 40× magnification.
Senescence Analysis
A total of 150 × 103 cells were seeded on 6 MW plates, and the β-galactosidase (SA-β-Gal) activity was quantify using the Senescence detection kit (Biovision, http://www.biovision.com/)
Oncomine analysis
We searched the public cancer microarray database, Oncomine, to identify studies with MAD1L1, MAD2L1,BUB3 and BUB1B gene expression datasets and compared expression of gastric intestinal type adenocarcinoma vs. normal gastric tissue. In order to be included in our study, a data set was required to have significant gene expression with a P-value < 0.001.
Statistical Analysis
All results were expressed as means ± SD of 3 independent experiments. Statistical analyses were performed using Graph path prims 5.0 ANOVA or Student's 2-tailed t-test. Values of *P < 0.05 were considered significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Javier Perez (photography facility), Diego Navarro and Lucia Sanchez (IIBM microscopy facility) for their technical assistance. Beatriz Morte for qPCR analysis.
962952_Supplementary_Materials.zip
Download Zip (17.1 MB)Funding
This work was supported by the following grants: PS09/1988, 595, PI11 -00949 supported by FEDER funds and UAM-Santander CEAL-AL/2013-29. JBI is a fellow of the Programa de Doctorado Doble en Ciencias Biomédicas UNAM, Mexico City, Mexico/Biociencias Moleculares UAM, Madrid, Spain. JBI was supported by a fellowship from Cátedra Isaac Costero, funded by Banco Santander.
Supplemental Material
Supplemental data for this article can be found on the publisher's website.
References
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin 2013; 63:11-30; PMID:23335087
- Garrido M, Fonseca PJ, Vieitez JM, Frunza M, Lacave AJ. Challenges in first line chemotherapy and targeted therapy in advanced gastric cancer. Expert Rev Anticancer Ther 2014: 14:887-900.
- Sakamoto J, Matsui T, Kodera Y. Paclitaxel chemotherapy for the treatment of gastric cancer. Gastric Cancern 2009; 12:69-78; PMID:19562460; http://dx.doi.org/10.1007/s10120-009-0505-z
- Goncalves A, Braguer D, Kamath K, Martello L, Briand C, Horwitz S, Wilson L, Jordan MA. Resistance to Taxol in lung cancer cells associated with increased microtubule dynamics. Proc Natl Acad Sci U S A 2001; 98:11737-42; PMID:11562465; http://dx.doi.org/10.1073/pnas.191388598
- Nitta M, Kobayashi O, Honda S, Hirota T, Kuninaka S, Marumoto T, Ushio Y, Saya H. Spindle checkpoint function is required for mitotic catastrophe induced by DNA-damaging agents. Oncogene 2004; 23:6548-58; PMID:15221012; http://dx.doi.org/10.1038/sj.onc.1207873
- Vakifahmetoglu H, Olsson M, Zhivotovsky B. Death through a tragedy: mitotic catastrophe. Cell Death Differ 2008; 15:1153-62; PMID:18404154; http://dx.doi.org/10.1038/cdd.2008.47
- Kops GJ, Weaver BA, Cleveland DW. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer 2005; 5:773-85; PMID:16195750; http://dx.doi.org/10.1038/nrc1714
- Fu Y, Ye D, Chen H, Lu W, Ye F, Xie X. Weakened spindle checkpoint with reduced BubR1 expression in paclitaxel-resistant ovarian carcinoma cell line SKOV3-TR30. Gynecol Oncol 2007; 105:66-73; PMID:17234259; http://dx.doi.org/10.1016/j.ygyno.2006.10.061
- Hao X, Zhou Z, Ye S, Zhou T, Lu Y, Ma D, Wang S. Effect of Mad2 on paclitaxel-induced cell death in ovarian cancer cells. J Huazhong Univ Sci Technolog Med Sci 2010; 30:620-5; PMID:21063845; http://dx.doi.org/10.1007/s11596-010-0553-y
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100:57-70; PMID:10647931; http://dx.doi.org/10.1016/S0092-8674(00)81683-9
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-4; PMID:21376230; http://dx.doi.org/10.1016/j.cell.2011.02.013
- Fang X, Zhang P. Aneuploidy and tumorigenesis. Semin Cell Dev Biol 2011; 22:595-601; PMID:21392584; http://dx.doi.org/10.1016/j.semcdb.2011.03.002
- Lara-Gonzalez P, Westhorpe FG, Taylor SS. The spindle assembly checkpoint. Curr Biol 2012; 22:R966-80; PMID:23174302; http://dx.doi.org/10.1016/j.cub.2012.10.006
- Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B. Mutations of mitotic checkpoint genes in human cancers. Nature 1998; 392:300-3; PMID:9521327; http://dx.doi.org/10.1038/32688
- Hanks S, Coleman K, Reid S, Plaja A, Firth H, Fitzpatrick D, Kidd A, Mehes K, Nash R, Robin N, et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat Genet 2004; 36:1159-61; PMID:15475955; http://dx.doi.org/10.1038/ng1449
- Li M, Fang X, Wei Z, York JP, Zhang P. Loss of spindle assembly checkpoint-mediated inhibition of Cdc20 promotes tumorigenesis in mice. J Cell Biol 2009; 185:983-94; PMID:19528295; http://dx.doi.org/10.1083/jcb.200904020
- Babu JR, Jeganathan KB, Baker DJ, Wu X, Kang-Decker N, van Deursen JM. Rae1 is an essential mitotic checkpoint regulator that cooperates with Bub3 to prevent chromosome missegregation. J Cell Biol 2003; 160:341-53; PMID:12551952; http://dx.doi.org/10.1083/jcb.200211048
- Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, Kumar R, Jenkins RB, de Groen PC, Roche P, et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet 2004; 36:744-9; PMID:15208629; http://dx.doi.org/10.1038/ng1382
- Michel LS, Liberal V, Chatterjee A, Kirchwegger R, Pasche B, Gerald W, Dobles M, Sorger PK, Murty VV, Benezra R. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature 2001; 409:355-9; PMID:11201745; http://dx.doi.org/10.1038/35053094
- Sotillo R, Hernando E, Diaz-Rodriguez E, Teruya-Feldstein J, Cordon-Cardo C, Lowe SW, Benezra R. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell 2007; 11:9-23; PMID:17189715; http://dx.doi.org/10.1016/j.ccr.2006.10.019
- Iwanaga Y, Chi YH, Miyazato A, Sheleg S, Haller K, Peloponese JM Jr, Li Y, Ward JM, Benezra R, Jeang KT. Heterozygous deletion of mitotic arrest-deficient protein 1 (MAD1) increases the incidence of tumors in mice. Cancer Res 2007; 67:160-6; PMID:17210695; http://dx.doi.org/10.1158/0008-5472.CAN-06-3326
- Dai W, Wang Q, Liu T, Swamy M, Fang Y, Xie S, Mahmood R, Yang YM, Xu M, Rao CV. Slippage of mitotic arrest and enhanced tumor development in mice with BubR1 haploinsufficiency. Cancer Res 2004; 64:440-5; PMID:14744753; http://dx.doi.org/10.1158/0008-5472.CAN-03-3119
- Baker DJ, Jeganathan KB, Malureanu L, Perez-Terzic C, Terzic A, van Deursen JM. Early aging-associated phenotypes in Bub3/Rae1 haploinsufficient mice. J Cell Biol 2006; 172:529-40; PMID:16476774; http://dx.doi.org/10.1083/jcb.200507081
- Jeganathan K, Malureanu L, Baker DJ, Abraham SC, van Deursen JM. Bub1 mediates cell death in response to chromosome missegregation and acts to suppress spontaneous tumorigenesis. J Cell Biol 2007; 179:255-67; PMID:17938250; http://dx.doi.org/10.1083/jcb.200706015
- Courtois-Cox S, Jones SL, Cichowski K. Many roads lead to oncogene-induced senescence. Oncogene 2008; 27:2801-9; PMID:18193093; http://dx.doi.org/10.1038/sj.onc.1210950
- Collado M, Serrano M. The senescent side of tumor suppression. Cell Cycle 2005; 4:1722-4; PMID:16294043; http://dx.doi.org/10.4161/cc.4.12.2260
- Young AR, Narita M. SASP reflects senescence. EMBO Rep 2009; 10:228-30; PMID:19218920; http://dx.doi.org/10.1038/embor.2009.22
- Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 2014; 15:482-96; PMID:24954210; http://dx.doi.org/10.1038/nrm3823
- Gutierrez-Gonzalez A, Belda-Iniesta C, Bargiela-Iparraguirre J, Dominguez G, Garcia Alfonso P, Perona R, Sanchez-Perez I. Targeting Chk2 improves gastric cancer chemotherapy by impairing DNA damage repair. Apoptosis 2013; 18:347-60; PMID:23271172; http://dx.doi.org/10.1007/s10495-012-0794-2
- Sanchez-Perez I, Garcia Alonso P, Belda Iniesta C. Clinical impact of aneuploidy on gastric cancer patients. Clin Transl Oncol 2009; 11:493-8; PMID:19661021; http://dx.doi.org/10.1007/s12094-009-0393-z
- Cho JY, Lim JY, Cheong JH, Park YY, Yoon SL, Kim SM, Kim SB, Kim H, Hong SW, Park YN, et al. Gene expression signature-based prognostic risk score in gastric cancer. Clin Cancer Res 2011; 17:1850-7; PMID:21447720; http://dx.doi.org/10.1158/1078-0432.CCR-10-2180
- D'Errico M, de Rinaldis E, Blasi MF, Viti V, Falchetti M, Calcagnile A, Sera F, Saieva C, Ottini L, Palli D, et al. Genome-wide expression profile of sporadic gastric cancers with microsatellite instability. Eur J Cancer 2009; 45:461-9; PMID:19081245; http://dx.doi.org/10.1016/j.ejca.2008.10.032
- Wang Q, Wen YG, Li DP, Xia J, Zhou CZ, Yan DW, Tang HM, Peng ZH. Upregulated INHBA expression is associated with poor survival in gastric cancer. Med Oncol 2012; 29:77-83; PMID:21132402; http://dx.doi.org/10.1007/s12032-010-9766-y
- Wu WK, Cho CH, Lee CW, Fan D, Wu K, Yu J, Sung JJ. Dysregulation of cellular signaling in gastric cancer. Cancer Lett 2010; 295:144-53; PMID:20488613; http://dx.doi.org/10.1016/j.canlet.2010.04.025
- Bharadwaj R, Yu H. The spindle checkpoint, aneuploidy, and cancer. Oncogene 2004; 23:2016-27; PMID:15021889; http://dx.doi.org/10.1038/sj.onc.1207374
- Yamamoto Y, Matsuyama H, Chochi Y, Okuda M, Kawauchi S, Inoue R, Furuya T, Oga A, Naito K, Sasaki K. Overexpression of BUBR1 is associated with chromosomal instability in bladder cancer. Cancer Genet Cytogenet 2007; 174:42-7; PMID:17350465; http://dx.doi.org/10.1016/j.cancergencyto.2006.11.012
- Burum-Auensen E, Deangelis PM, Schjolberg AR, Roislien J, Andersen SN, Clausen OP. Spindle proteins Aurora A and BUB1B, but not Mad2, are aberrantly expressed in dysplastic mucosa of patients with longstanding ulcerative colitis. J Clin Pathol 2007; 60:1403-8; PMID:17322345; http://dx.doi.org/10.1136/jcp.2006.044305
- McGrogan B, Phelan S, Fitzpatrick P, Maguire A, Prencipe M, Brennan D, Doyle E, O'Grady A, Kay E, Furlong F, et al. Spindle assembly checkpoint protein expression correlates with cellular proliferation and shorter time to recurrence in ovarian cancer. Hum Pathol 2014; 45:1509-19; PMID:24792619; http://dx.doi.org/10.1016/j.humpath.2014.03.004
- Lee YK, Choi E, Kim MA, Park PG, Park NH, Lee H. BubR1 as a prognostic marker for recurrence-free survival rates in epithelial ovarian cancers. Br J Cancer 2009; 101:504-10; PMID:19603021; http://dx.doi.org/10.1038/sj.bjc.6605161
- Sudo T, Nitta M, Saya H, Ueno NT. Dependence of paclitaxel sensitivity on a functional spindle assembly checkpoint. Cancer Res 2004; 64:2502-8; PMID:15059905; http://dx.doi.org/10.1158/0008-5472.CAN-03-2013
- Du Y, Yin F, Liu C, Hu S, Wang J, Xie H, Hong L, Fan D. Depression of MAD2 inhibits apoptosis of gastric cancer cells by upregulating Bcl-2 and interfering mitochondrion pathway. Biochem Biophys Res Commun 2006; 345:1092-8
- Hu M, Liu Q, Song P, Zhan X, Luo M, Liu C, Yang D, Cai Y, Zhang F, Jiang F, et al. Abnormal expression of the mitotic checkpoint protein BubR1 contributes to the anti-microtubule drug resistance of esophageal squamous cell carcinoma cells. Oncol Rep 2013; 29:185-92; PMID:23128493
- Swanton C, Tomlinson I, Downward J. Chromosomal instability, colorectal cancer and taxane resistance. Cell Cycle 2006; 5:818-23; PMID:16628000; http://dx.doi.org/10.4161/cc.5.8.2682
- Chao SK, Horwitz SB, McDaid HM. Insights into 4E-BP1 and p53 mediated regulation of accelerated cell senescence. Oncotarget 2011; 2:89-98; PMID:21399233
- Johmura Y, Shimada M, Misaki T, Naiki-Ito A, Miyoshi H, Motoyama N, Ohtani N, Hara E, Nakamura M, Morita A, et al. Necessary and Sufficient Role for a Mitosis Skip in Senescence Induction. Mol Cell 2014; PMID:24910096
- Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 2008; 6:2853-68; PMID:19053174; http://dx.doi.org/10.1371/journal.pbio.0060301
- Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev 2014; 28:99-114; PMID:24449267; http://dx.doi.org/10.1101/gad.235184.113
- Lovat F, Ishii H, Schiappacassi M, Fassan M, Barbareschi M, Galligioni E, Gasparini P, Baldassarre G, Croce CM, Vecchione A. LZTS1 downregulation confers paclitaxel resistance and is associated with worse prognosis in breast cancer. Oncotarget 2014; 5:970-7; PMID:24448468