
Abstract
The human tumor suppressor neurofibromin contains a cysteine and serine-rich domain/Ras-GTPase activating protein domain (CSRD/RasGAP) and a C-terminal domain (CTD). Domain studies of neurofibromin suggest it has other functions in addition to being a RasGAP, but the mechanisms underlying its tumor suppressor activity are not well understood. The budding yeast Saccharomyces cerevisiae is a good model system for studying neurofibromin function because it possesses Ira1 and Ira2, which are homologous to human neurofibromin in both sequence and function. We found that overexpression of CTD or a neurofibromin CTD-homologous domain (CHD) of Ira1/2 in budding yeast delayed degradation of the securin protein Pds1, whereas overexpression of CSRD/RasGAP did not affect Pds1 degradation. We also found that when CTD or CHD was overexpressed, the number of cells in metaphase was higher than in the control. These results demonstrate that CTD and CHD function in the metaphase to anaphase transition. In addition, Δira1Δira2 cells bypassed mitotic arrest in response to spindle damage, indicating that Ira1 and Ira2 may be involved in the spindle assembly checkpoint (SAC). However, Δira1Δira2Δmad2 cells are more sensitive to spindle damage than Δmad2 or Δira1Δira2 cells are, suggesting that Ira1/2 and Mad2 function in different pathways. Overexpression of CTD but not CSRD/RasGAP partially rescued the hypersensitivity of Δira1Δira2Δmad2 cells to microtubule-destabilizing drugs, indicating a role for CTD in the SAC pathway. Taken together, independently of RasGAP activity, the C-terminal domains of neurofibromin, Ira1, and Ira2 regulate the metaphase to anaphase transition in a Mad2-independent fashion.
Abbreviations
| NF1 | = | neurofibromatosis type 1 |
| APC | = | anaphase promoting complex |
| CSRD/RasGAP | = | cysteine and serine-rich domain/Ras-GTPase activating protein |
| CTD | = | C-terminal domain |
| CHD | = | CTD-homologous domain |
| SAC | = | spindle assembly checkpoint |
Introduction
Neurofibromatosis type 1 (NF1) is a human genetic disorder affecting 1 in 3000–4000 individuals. It is caused by mutations in NF1 and increases the risk of tumor development in the central and peripheral nervous systems.Citation1-3 NF1 encodes neurofibromin, a large protein (2,839 amino acids; 319 kDa) that contains several domains, including a cysteine and serine-rich domain/Ras-GTPase activating protein domain (CSRD/RasGAP) and a C-terminal domain (CTD).Citation4-6 CSRD/RasGAP is thought to inhibit the GTPase Ras by accelerating hydrolysis of active Ras-GTP to inactive Ras-GDP.Citation7-9 Li et al. suggested that loss-of-function mutations in CSRD/RasGAP might contribute to the development of tumors through aberrantly activating Ras signaling.Citation10 However, analyses of NF1 patients found mutations not only in CSRD/RasGAP but also in CTD.Citation11-13 Thus, CTD is strongly implicated in tumor suppression.
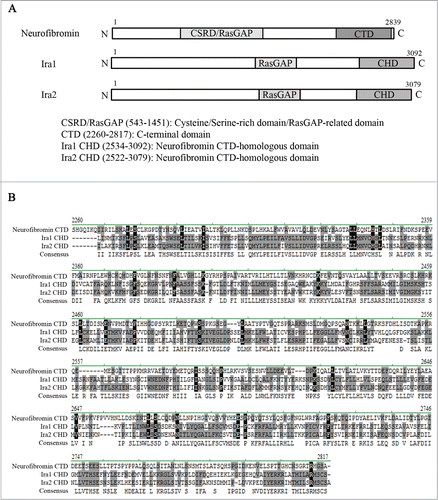
As NF1 is so large, it is difficult to clone and express. Thus, although NF1 was identified more than 25 years ago, the molecular function of neurofibromin in cell cycle progression has yet to be fully elucidated. However, the budding yeast Saccharomyces cerevisiae is a good model system for studying neurofibromin function, since it contains Ira1 and Ira2, which are homologous to human neurofibromin in both sequence and function ().Citation7,14,15
Figure 1. Neurofibromin and its yeast homologs, Ira1 and Ira2. (A) Schematic diagram of the full-length neurofibromin, Ira1, and Ira2. The cysteine and serine-rich domain/Ras-GTPase activating protein domain (CSRD/RasGAP) and C-terminal domain (CTD) of neurofibromin are conserved in its yeast homologs Ira1 and Ira2. CSRD/RasGAP is shown in light gray, and the neurofibromin CTD and Ira 1/2 neurofibromin CTD-homologous domain (CHD) in dark gray. (B) Alignment of the CTD and CHD amino acid sequences using Vector NTI software (Invitrogen). Identical amino acids are shown in black, conserved amino acids in gray.
Transition from metaphase to anaphase is only activated if duplicated chromosomes are properly attached to the mitotic spindle.Citation16 A surveillance mechanism called spindle assembly checkpoint (SAC) prevents segregation of duplicated chromosomes in the presence of spindle damage in all eukaryotes.Citation16 Mad1, Mad2, Mad3, Bub1, Bub3 and Mps1 are key components in the SAC pathway, and each mutation of these genes bypasses the mitotic arrest in response to spindle damage in budding yeast.Citation17-19 The SAC ensures the onset of anaphase via the securin Pds1. Pds1 must be degraded for the metaphase to anaphase transition, and this degradation is mediated through the ubiquitination of Pds1 by anaphase promoting complex (APC). APC-dependent ubiquitination of Pds1 is promoted by its direct interaction with Cdc20 in budding yeast.Citation20-24 Mad2 lies the most downstream in SAC to regulate the degradation of Pds1.Citation25 In response to spindle damage, the SAC stabilizes Pds1 by the Mad2-dependent sequestration of Cdc20 to delay the metaphase to anaphase transition in budding yeast.Citation16,26
In this study, we found that the neurofibromin CTD can regulate the metaphase to anaphase transition in budding yeast independently of CSRD/RasGAP and that this function is conserved in CHD. Furthermore, Ira1 and Ira2 are involved in the SAC pathway in a Mad2-independent manner. These results suggest that neurofibromin CTD and Ira1/2 CHD regulate the metaphase to anaphase transition during mitosis independently of Mad2 in the SAC pathway.
Results
CSRD/RasGAP shows RasGAP activity in budding yeast, but its CTD functions independently of RasGAP activity
Neurofibromin is a huge protein composed of 2839 amino acids and contains several domains including the CSRD/RasGAP and the CTD ().Citation5 Ira1 and Ira2 are yeast homologues of human neurofibromin and their residues of both the RasGAP and the CTD domains are conserved ().Citation7,14,15 Due to its huge size, the full-length NF1 gene is difficult to be cloned and studied as a whole. In this study, we examined the function of distinct neurofibromin domains in the budding yeast cell cycle.
We first examined whether CSRD/RasGAP would have RasGAP activity in budding yeast. Δira1 and Δira2 cells are sensitive to heat shock stress since the lack of RasGAP hyperactivates Ras-PKA signaling (), and this heat sensitivity is rescued by overexpressing the CSRD/RasGAP domain of neurofibromin ().Citation6,7 Therefore, we investigated whether overexpression of CSRD/RasGAP under the GAL1 promoter could rescue the heat-sensitive phenotypes of Δira1 and Δira2. As shown in , the induced expression of CSRD/RasGAP by galactose rescued the heat shock sensitivity of Δira1 and Δira2 cells. In contrast, Δira1 and Δira2 cells did not proliferate following heat shock when CTD was overexpressed (). These observations demonstrate that CSRD/RasGAP has RasGAP activity in budding yeast, but CTD does not.
Figure 2. The neurofibromin cysteine and serine-rich domain/Ras-GTPase activating protein domain (CSRD/RasGAP) but not the C-terminal domain (CTD) rescues heat sensitivity in Δira1 and Δira2 budding yeast cells. (A) The heat shock-sensitive Ras signaling pathway in budding yeast. (B) A complementation assay for heat sensitivity using the neurofibromin CSRD/RasGAP domain and CTD. pCEN-PGAL1-3HA-NF1-CTD, pCEN-PGAL1-3HA-NF1-CSRD/RasGAP, and pCEN-PGAL1-3HA were respectively transformed into Δira1 (strain YSK2620) and Δira2 (strain YSK2622) cells. pCEN-PGAL1-3HA was transformed into wild-type (W303a) cells and used as a control. Cells were serially diluted, spotted on plates containing either glucose or galactose, and then incubated at 30°C for 2 days. For heat shock (right panel), cells were incubated at 55°C for 30 min prior to the transfer to 30°C.

CTD regulates the metaphase to anaphase transition in budding yeast
We next examined whether overexpression of CTD and CSRD/RasGAP affects cell cycle progression in budding yeast. The cells were first synchronized at G1 with α-factor, and then CTD or CSRD/RasGAP expression was induced for 1 h. The cells were then released from G1 arrest into galactose medium and collected every 20 min for 3 h to count the number of cells in metaphase () or in anaphase () after staining with 4′,6-diamidino-2-phenylindole (DAPI). When either CTD or CSRD/RasGAP was overexpressed, metaphase cells began to accumulate approximately by 80 min after release, and by 120 min after release, the number of metaphase cells had greatly increased, particularly among cells expressing CTD. At 140 min after release, the number of metaphase cells expressing CSRD/RasGAP or the control vector sharply decreased, whereas the number of those expressing CTD remained relatively high. Similarly, the number of anaphase cells (large-budded cells with 2 divided nuclei) expressing CSRD/RasGAP or the control vector greatly increased until 140 min after release and sharply decreased thereafter, whereas the number of anaphase cells expressing CTD greatly increased until 160 min after release and then decreased thereafter. These observations suggest that CTD overexpression delays the metaphase to anaphase transition in budding yeast.
Figure 3. The neurofibromin C-terminal domain (CTD) and the Ira1/2 neurofibromin CTD-homologous domain (CHD) regulate the metaphase to anaphase transition in budding yeast. pCEN-PGAL1-3HA-NF1-CTD, pCEN-PGAL1-3HA-NF1-CSRD/RasGAP, pCEN-PGAL1-3HA-IRA1-CHD, pCEN-PGAL1-3HA-IRA2-CHD, or pCEN-PGAL1-3HA were transformed into cells expressing Myc-tagged Pds1 (strain YSK2202). Cells transformed with these plasmids were synchronized at G1 with α-factor (50 ng/mL), and then the expression of CTD, the neurofibromin cysteine and serine-rich domain/Ras-GTPase activating protein domain (CSRD/RasGAP), and the Ira 1/2 CHD under the GAL1 promoter were induced for 1 h prior to release into galactose medium at 25°C. (A, B, D, and E) The cell cycle progression and (C and F) Pds1 expression level in cells expressing CTD, CSRD/RasGAP, or CHD. (A, B, D, and E) Cells were collected every 20 min and stained with 4ʹ,6-diamidino-2-phenylindole, and then metaphase and anaphase cells were counted (n = 200). (C and F) Cells were collected every 20 min, and then Pds1 expression was detected by western blotting using an anti-Myc antibody (Pds1-9Myc). α-Tubulin is shown as a loading control (Tub1).
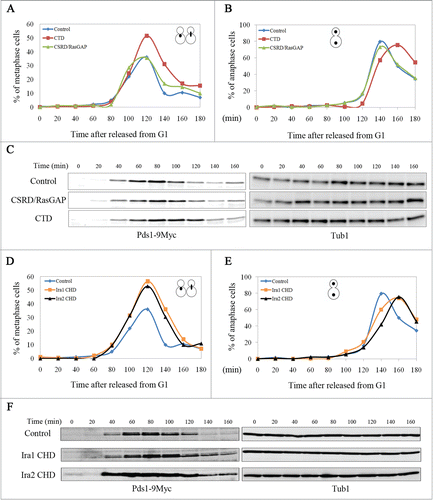
To verify the effects of CTD overexpression on the metaphase to anaphase transition in budding yeast, we examined the expression of yeast securin Pds1 by western blotting, as its degradation is required for the metaphase to anaphase transition.Citation20,21 When cells synchronized at G1 were released with the induction of CTD or CSRD/RasGAP, as shown in , Pds1 began to accumulate after 40 min, indicating that these strains have the same growth rate after release from G1 arrest. As expected from the results shown in and B, at 120 min, Pds1 was maintained in CTD-expressing cells but was mainly degraded in cells expressing CSRD/RasGAP or the control vector (). Taken together, these results demonstrate that CTD functions in the metaphase to anaphase transition in budding yeast by regulating Pds1 levels.
CHD also regulates the metaphase to anaphase transition
To test whether the CHD of Ira1/2 functions in the metaphase to anaphase transition, mitotic cell cycle progression during CHD overexpression was monitored. As with CTD overexpression, when CHD was overexpressed, the number of metaphase cells at 140 min was maintained at a relatively high level, and the number of anaphase cells was highest at 160 min. In contrast, the number of control cells in metaphase peaked at 120 min and declined sharply thereafter, and the number of control cells in anaphase peaked at 140 min (). The expression of Pds1 was consistent with the nuclear morphology observed in the cells: Pds1 began to accumulate at 40 min and was completely degraded by 140–160 min in the control, whereas in CHD-expressing cells, Pds1 began to accumulate at 40 min but was maintained at 140–160 min (). These results demonstrate that the function of CTD at the metaphase to anaphase transition is conserved in CHD.
Ira1 and Ira2 function in the SAC pathway in budding yeast
As Pds1 is a key target of the SAC in budding yeast, we next tested whether Ira1 and Ira2 were also involved in the SAC pathway. It had previously been reported that treatment of SAC mutants with benomyl, a microtubule-destabilizing compound, severely decreases cell viability.Citation17 Consistent with this report, we found that the viability of Δmad2 cells was sharply reduced in response to benomyl treatment, because Mad2 is a key component of the SAC pathway (). In contrast, the viability of Δira1 and Δira2 cells was partially decreased in response to benomyl, but not to the same extent as in (Δmad2 cells. As this result might have been due to the functional redundancy of Ira1 and Ira2, we next examined the sensitivity of the Ira1 and Ira2 double-knockout strain YMW208 in response to benomyl. Like Δmad2 mutant cells, Δira1Δira2 mutant cells had significantly decreased cell viability in response to benomyl, suggesting that Ira1 and Ira2 function redundantly in the SAC pathway.
Figure 4. Ira1 and Ira2 are involved in the spindle assembly checkpoint (SAC) pathway. (A) Δira1Δira2 cells are sensitive to spindle damage. Wild-type, Δira1 (strain YSK2620), Δira2 (strain YSK2622), Δira1Δira2 (strain YSK2866), and Δmad2 (strain YSK2668) cells were grown to mid-log phase at 25°C, serially diluted 10-fold, spotted onto either YPAD plates or YPAD plates containing 10 μg/mL benomyl, and incubated at 25°C. (B) Δgpb1Δgpb2 cells are proficient for SAC. Wild-type, Δgpb1Δgpb2 (strain YSK2929), Δira1Δira2 (strain YSK2866), and Δmad2 (strain YSK2668) cells were grown to mid-log phase at 25°C, serially diluted 10-fold, spotted onto either YPAD plates or YPAD plates containing 10 μg/mL benomyl and incubated at 25°C. (C) Δira1Δira2 cells bypassed mitotic arrest in response to spindle damage. Wild-type and Δira1Δira2 cells expressing Myc-tagged Pds1 (strains YSK2992 and YSK3001, respectively) were grown to early log phase at 25°C and then treated with 15 μg/mL nocodazole. Cells were collected every 1 h for 8 h, and Pds1 and Clb2 expression (Pds1-9Myc and Clb2, respectively) was detected by western blotting using anti-Myc and anti-Clb2 antibodies, respectively. α-Tubulin (Tub1) served as a loading control.
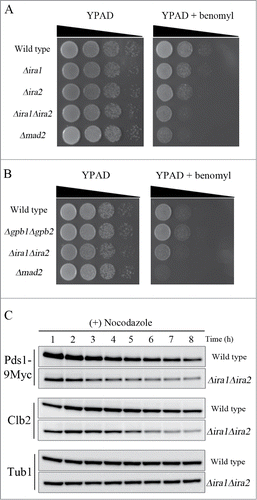
To determine whether the RasGAP activity of Ira1 and Ira2 is required for their function in the SAC pathway, we deleted both GPB1 and GPB2, which encode proteins that interact with Ira1 and Ira2 to maintain their RasGAP activity,Citation15 and then examined the viability of Δgpb1Δgpb2 cells in the presence of benomyl (). The Δgpb1Δgpb2 mutant cells were not as at all sensitive to benomyl as the Δira1Δira2 and Δmad2 mutants, indicating that the SAC function of Ira1 and Ira2 is independent of RasGAP activity.
To confirm that Ira1 and Ira2 function in the SAC pathway, we compared the expression of Pds1 and the mitotic cyclin Clb2 in Δira1Δira2 and wild-type cells in the presence of nocodazole, a microtubule-destabilizing compound. Since the metaphase to anaphase transition is delayed by microtubule defects, wild-type cells maintain expression levels of Pds1 and Clb2 when treated with microtubule-destabilizing drugs. However, Pds1 and Clb2 became degraded upon impairment of the SAC in budding yeast.Citation26,27 When wild-type and Δira1Δira2 cells were grown to mid-log phase and treated with nocodazole, the wild-type cells maintained high levels of Pds1 and Clb2 expression for 8 h, whereas the Δira1Δira2 cells began to degrade Pds1 and Clb2 at 3–4 h and 5–6 h, respectively (). This result confirms that Ira1 and Ira2 function in the SAC pathway to induce mitotic arrest in response to spindle damage.
Ira1 and Ira2 function independently of Mad2 in the budding yeast SAC pathway
Mad2 is a key downstream regulator of the SAC pathway that regulates the metaphase to anaphase transition in budding yeast; its deletion causes mitotic arrest in response to spindle damage to be bypassed.Citation16,17,25,26 Thus, we asked whether Ira1 and Ira2 function in the Mad2 pathway in response to spindle damage. To address this question, we constructed the triple Ira1, Ira2, and Mad2 knockout mutant Δira1Δira2Δmad2 and compared its sensitivity to benomyl with that of the Δira1Δira2 and Δmad2 mutants. Interestingly, Δira1Δira2Δmad2 cells were more sensitive to benomyl at either 25°C or 30°C than Δira1Δira2 and Δmad2 cells (). This result suggests that Ira1 and Ira2 might function independently of Mad2 in the SAC.
Figure 5. Ira1 and Ira2 function independently of Mad2 in the spindle assembly checkpoint (SAC) pathway. (A) Δira1Δira2Δmad2 was highly sensitive to spindle damage. Wild-type, Δira1Δira2 (strain YSK2866), Δmad2 (strain YSK2668), and Δira1Δira2Δmad2 (strain YSK2925) cells were grown to mid-log phase, serially diluted 10-fold, spotted onto either YPAD plates or YPAD plates containing 10 μg/mL benomyl and then incubated at either 25°C or 30°C. (B) Δira1Δira2Δmad2 cells are more sensitive to nocodazole than Δira1Δira2 or Δmad2 cells. Wild-type, Δira1Δira2, Δmad2, and Δira1Δira2Δmad2 cells expressing Myc-tagged Pds1 (strains YSK2992, YSK3001, YSK2984, and YSK3003, respectively) were grown to early log phase at 25°C and then treated with 15 μg/mL nocodazole for 130 min. Cells were collected every 10 min from 60 min, and then Pds1 expression (Pds1-9Myc) was detected by western blotting using an anti-Myc antibody. α-Tubulin (Tub1) served as a loading control.
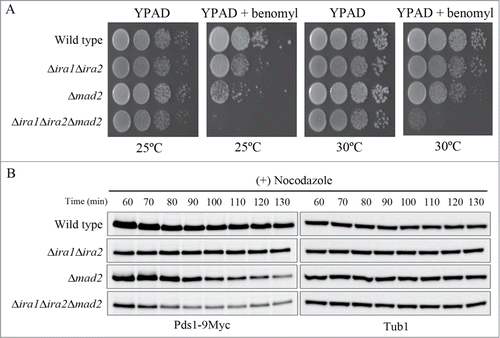
In order to verify that Ira1/2 and Mad2 delay the metaphase to anaphase transition in response to spindle damage via different pathways, we examined the kinetics of Pds1 degradation in wild-type, Δira1Δira2, Δmad2, and Δira1Δira2Δmad2 cells in response to nocodazole. As shown in , Δmad2 cells showed a drop in Pds1 levels at approximately 100–110 min after nocodazole treatment. Importantly, Pds1 degradation was exacerbated in Δira1Δira2Δmad2 triple mutant cells: it began at 80–90 min after nocodazole treatment (), suggesting that Ira1/2 and Mad2 respond to spindle damage via independent pathways. Consistent with the previous observation that Pds1 in Δira1Δira2 cells only begins to degrade approximately 3–4 h after nocodazole treatment (), the level of Pds1 expression in Δira1Δira2 cells was maintained for 130 min after nocodazole treatment (). These results strongly suggest that Ira1 and Ira2 act in the budding yeast SAC pathway independently of Mad2.
CTD functions in the budding yeast SAC pathway while RasGAP does not
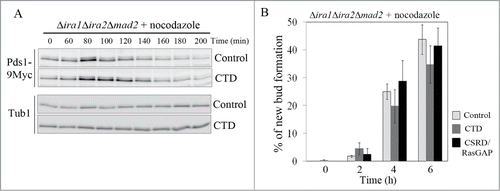
To determine whether CTD overexpression would rescue the SAC-defective phenotype of Δira1Δira2Δmad2 cells, the cells were synchronized at G1 with α-factor, and the expression of CTD or CSRD/RasGAP was induced for 1 h. Then, the cells were released into galactose medium containing nocodazole, and cell cycle progression was monitored. As expected, overexpression of CTD in Δira1Δira2Δmad2 cells partially restored their loss of mitotic arrest in response to spindle damage, whereas the expression of CSRD/RasGAP or vector control did not ( Fig. S3). As shown in , Pds1 accumulation peaked at 80 min and sharply decreased by 100 min in cells expressing the control vector. In cells overexpressing CTD, Pds1 accumulation also peaked at 80 min, but at 100–120 min, a relatively high level of Pds1 expression was maintained (). In contrast, the kinetics of Pds1 degradation in response to nocodazole in Δira1Δira2Δmad2 cells overexpressing CSRD/RasGAP was similar to that in cells expressing the control vector (Fig. S3). In addition, we counted the number of budded cells, a phenotype associated with bypass of mitotic arrest, in cells overexpressing CTD and CSRD/RasGAP. In this experiment, after the cells had been released from G1 arrest, they were collected every 2 h for 6 h and then examined for bud formation. Consistent with the results shown in and Fig. S3, the loss of mitotic arrest was partially relieved in a time-dependent manner in cells expressing CTD: 6 h after release, new bud formation was observed in 34.8 ± 6.7% of the CTD-expressing cells, as compared to 43.8 ± 5.3% of cells expressing the control vector and 41.5 ± 6.4% of CSRD/RasGAP-expressing cells (). These results show that CTD functions independently of RasGAP in the metaphase to anaphase transition.
Figure 6. The neurofibromin C-terminal domain (CTD) functions in the spindle assembly checkpoint (SAC) pathway. (A, B) CTD expression partially rescued the SAC-defective phenotype. pCEN-PGAL1-3HA-NF1-CTD, pCEN-PGAL1-3HA-NF1-CSRD/RasGAP, and pCEN-PGAL1-3HA were transformed into Δira1Δira2Δmad2 cells expressing Myc-tagged Pds1 to produce strain YSK3003. (A) Cells were synchronized at G1 with α-factor (5 μg/mL), the expression of CTD under the GAL1 promoter was induced for 1 h, and then the cells were released into galactose medium containing 15 μg/mL nocodazole at 25°C. After 60 min, cells were collected every 20 min for 200 min. Then, Pds1 expression (Pds1-9Myc) was detected by western blotting using an anti-Myc antibody. α-Tubulin (Tub1) served as a loading control. (B) Cells expressing CTD and the neurofibromin cysteine and serine-rich domain/Ras-GTPase activating protein domain (CSRD/RasGAP) were released from G1 arrest into galactose medium containing 15 μg/mL nocodazole and collected every 2 h for 6 h. New bud formation was scored at each time point (n = 400). Three independent experiments were performed, and the average was plotted with standard deviations.
Discussion
NF1 is a tumor suppressor, and mutations found in NF1 patients suggest a critical role for CTD as well as CSRD/RasGAP in cell cycle regulation.Citation11-13 However, no definite mechanism of the CTD in the control of cell proliferation has been elucidated. In this study, we showed that CTD regulates the metaphase to anaphase transition independently of RasGAP activity during mitosis in budding yeast and this function is conserved in the CHD of the neurofibromin yeast homologs Ira1 and Ira2. Furthermore, our results suggested that both CTD and CHD are involved in the SAC pathway.
The highly conserved SAC is a surveillance mechanism that monitors improper spindle attachment to sister chromatids to maintain genomic stability prior to chromosome segregation in mitosis.Citation28 SAC acts via delaying the degradation of the securin Pds1, which is an inhibitor of the metaphase to anaphase transition, through the Mad2-dependent sequestration of Cdc20 in response to spindle damage.Citation16 Interestingly, we observed that Ira1, Ira2, and CTD function independently of Mad2 to delay Pds1 degradation in response to spindle damage. This raises a question of what could be a molecular mechanism of Ira1, Ira2 and the CTD of neurofibromin in regulating Pds1 in a Mad2-independent manner.
Previous studies have shown that in response to DNA damage in budding yeast, protein kinase A (PKA) phosphorylates Cdc20 on S52 and S88 to inhibit the interaction of Cdc20 and Pds1, which then delays the APCCdc20-mediated degradation of Pds1.Citation29,30 In addition, it has been suggested that the Kelch proteins Gpb1 and Gpb2, which interact with Ira1/2 via CHD, inhibit PKA activity by promoting the interaction of the PKA regulatory and catalytic subunits.Citation15,31 Thus, we hypothesized that CHD and CTD might activate PKA by binding Gpb1 and Gpb2 and thus block the interaction between Gpb1 and Gpb2 with PKA. If this were the case, the expression of a phosphorylation-defective Cdc20 S52AS88A mutant should have facilitated Pds1 degradation in Δmad2 cells in response to spindle damage. However, when we overexpressed the phosphorylation-defective Cdc20 S52AS88A mutant in Δmad2 cells, Pds1 degradation in response to nocodazole was not accelerated (Fig. S1). As the phosphorylation of Pds1 inhibits its degradation by blocking ubiquitination from APCCdcCitation20 in response to DNA damage,Citation29,32 we also examined the possibility that Ira1 and Ira2 may promote the phosphorylation of Pds1 by an unknown pathway to induce the delay of Pds1 degradation in a Mad2-independent manner. However, we could not detect any Pds1 phosphorylation in response to spindle damage (data not shown), which is consistent with the previous report that Pds1 is phosphorylated by DNA damage but not by spindle damage.Citation32 Therefore, we speculate that the phosphorylation of Pds1 is not directly correlated with the SAC function of Ira1 and Ira2.
In addition, we also hypothesized that Ira1 and Ira2 may bind directly to Pds1 to inhibit the interaction between Pds1 and Cdc20, which could then block the APCCdc20-dependent ubiquitination of Pds1 in a Mad2-independent way. To test this idea, we first examined the physical interaction between Pds1 and Ira1 by co-precipitation in the presence or absence of nocodazole. However, as shown in Fig. S2A and B, Ira1 did not co-precipitate with Pds1. We also found that overexpressed CTD did not associate with Pds1 in budding yeast (Fig. S2C). Thus, the molecular mechanisms underlying the Mad2-independent regulation of Pds1 remain unclear and will be the subject of future studies. Nonetheless, this study reveals new insight into why mutations in the neurofibromin CTD can increase the risk for tumorigenesis in mammals.
Materials and Methods
Yeast strains, culture, and plasmids
The yeast strains and plasmids used in this study are described in and . All strains were constructed by PCR-based homologous recombination methods and verified by PCR and/or western blot analysis.Citation33,34 Yeast cells were grown in YPAD medium (1% yeast extract, 2% bacto-peptone, 100 μg/mL adenine and 2% dextrose) or in synthetic complete drop-out medium prepared with yeast nitrogen base and necessary supplements.Citation35 PCR-amplified CTD and CSRD/RasGAP, CHD, Cdc20, and Cdc20 S52AS88A were subcloned into pCEN-PGAL1-3HA. The expression of CSRD/RasGAP, CTD, CHD, Cdc20, and Cdc20 S52AS88A was induced using the GAL1 promoter as described by Kim et al.Citation35 Spindle damage was induced by adding either benomyl (10 μg/mL) or nocodazole (15 μg/mL) to the culture at 25°C.
Table 1. Yeast strains used in this study
Table 2. Plasmids used in this study
Yeast spotting assay
Cells were grown to mid-log phase and serially diluted 10-fold. The heat shock sensitivity assay was performed by incubating cells for 30 min at 55°C after being spotted on selective plates containing glucose or galactose.Citation36 The spindle damage assay was performed by spotting cells grown to mid-log phase on YPAD plates containing 10 μg/mL benomyl.Citation35
Microscopy
Harvested cells were fixed in 70% ethanol, washed once with distilled water, briefly sonicated, and stained with 1 μg/mL DAPI. Cells were observed by fluorescence microscopy using a 100× objective on an Axioplan2 (Zeiss), and the images were captured with an Axiocam CCD (Zeiss) camera using AxioVision software (Zeiss).Citation35
Co-immunoprecipitation
For co-immunoprecipitation, cells were extracted in lysis buffer [50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% nonidet P-40, 2 mM EDTA, 5 mM MgSO4, 50 mM NaF, 100 mM β-glycerophosphate, 1 mM Na3VO4, 1 mM dithiothreitol, 1 mM phenylmethylsulphonyl fluoride, and protease inhibitor cocktail (Roche)] by beadbeating (Biospec). To precipitate hemagglutinin (HA)-tagged Ira1, CTD, and CSRD/RasGAP, crude cell lysates (10 mg in lysis buffer) were incubated with an anti-HA antibody for 2 h at 4°C, followed by incubation with protein A agarose (Invitrogen) for 2 h.
Western blot analysis
Cellular lysates were extracted in sodium dodecyl sulfate (SDS) sample buffer [60 mM Tris-HCl (pH 6.8), 25% glycerol, 10% SDS, 5% 2-mercaptoethanol, and 0.1% bromophenol blue] by boiling for 5 min at 90–100°C. Detection of Myc, Clb2, HA, and α-tubulin was performed using anti-Myc mouse monoclonal antibody (Cell Signaling, 1:5000 dilution), anti-Clb2 rat polyclonal antibody (generated in our lab, 1:5000 dilution), anti-HA mouse monoclonal antibody (Santa Cruz, 1:3000 dilution), and anti-α-tubulin mouse monoclonal antibody (Sigma Aldrich, 1:5000 dilution), respectively. An enhanced chemiluminescence system was used for blot analysis.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary_Files
Download Zip (3.4 MB)Acknowledgments
We thank Dr. Sanchez (Dartmouth Medical School) for the Δira1Δira2 double-knockout yeast strain (YMW208) and the CDC20 S52AS88A mutant clone, Dr. Mangoura (Biomedical Research Foundation of the Academy of Athens) for the neurofibromin CSRD/RasGAP domain and CTD clones, and Dr. Heitman (Duke University Medical Center) for the Ira1-3HA-tagged yeast strain (THY427a).
Supplemental Materials
Supplemental materials may be found here: http://dx.doi.org/10.4161/15384101.2015.945870
Additional information
Funding
References
- Cawthon RM, Weiss R, Xu GF, Viskochil D, Culver M, Stevens J, Robertson M, Dunn D, Gesteland R, O'Connell P, et al. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell 1990; 62:193-201; PMID:2114220; http://dx.doi.org/10.1016/0092-8674(90)90253-B
- Viskochil D, Buchberg AM, Xu G, Cawthon RM, Stevens J, Wolff RK, Culver M, Carey JC, Copeland NG, Jenkins NA, et al. Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell 1990; 62:187-92; PMID:1694727; http://dx.doi.org/10.1016/0092-8674(90)90252-A
- Wallace MR, Marchuk DA, Andersen LB, Letcher R, Odeh HM, Saulino AM, Fountain JW, Brereton A, Nicholson J, Mitchell AL, et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science 1990; 249:181-6; PMID:2134734; http://dx.doi.org/10.1126/science.2134734
- Bernards A, Haase VH, Murthy AE, Menon A, Hannigan GE, Gusella JF. Complete human NF1 cDNA sequence: two alternatively spliced mRNAs and absence of expression in a neuroblastoma line. DNA Cell Biol 1992; 11:727-34; PMID:1457041; http://dx.doi.org/10.1089/dna.1992.11.727
- Leondaritis G, Petrikkos L, Mangoura D. Regulation of the Ras-GTPase activating protein neurofibromin by C-tail phosphorylation: implications for protein kinase C/Ras/extracellular signal-regulated kinase 1/2 pathway signaling and neuronal differentiation. J Neurochem 2009; 109:573-83; PMID:19220708; http://dx.doi.org/10.1111/j.1471-4159.2009.05975.x
- Xu GF, Lin B, Tanaka K, Dunn D, Wood D, Gesteland R, White R, Weiss R, Tamanoi F. The catalytic domain of the neurofibromatosis type 1 gene product stimulates ras GTPase and complements ira mutants of S. cerevisiae. Cell 1990; 63:835-41; PMID:2121369; http://dx.doi.org/10.1016/0092-8674(90)90149-9
- Ballester R, Marchuk D, Boguski M, Saulino A, Letcher R, Wigler M, Collins F. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell 1990; 63:851-9; PMID:2121371; http://dx.doi.org/10.1016/0092-8674(90)90151-4
- Xu GF, O'Connell P, Viskochil D, Cawthon R, Robertson M, Culver M, Dunn D, Stevens J, Gesteland R, White R, et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 1990; 62:599-608; PMID:2116237; http://dx.doi.org/10.1016/0092-8674(90)90024-9
- Coleman ML, Marshall CJ, Olson MF. RAS and RHO GTPases in G1-phase cell-cycle regulation. Nat Rev Mol Cell Biol 2004; 5:355-66; PMID:15122349; http://dx.doi.org/10.1038/nrm1365
- Li Y, Bollag G, Clark R, Stevens J, Conroy L, Fults D, Ward K, Friedman E, Samowitz W, Robertson M, et al. Somatic mutations in the neurofibromatosis 1 gene in human tumors. Cell 1992; 69:275-81; PMID:1568247; http://dx.doi.org/10.1016/0092-8674(92)90408-5
- Fahsold R, Hoffmeyer S, Mischung C, Gille C, Ehlers C, Kucukceylan N, Abdel-Nour M, Gewies A, Peters H, Kaufmann D, et al. Minor lesion mutational spectrum of the entire NF1 gene does not explain its high mutability but points to a functional domain upstream of the GAP-related domain. Am J Hum Genet 2000; 66:790-818; PMID:10712197; http://dx.doi.org/10.1086/302809
- Origone P, De Luca A, Bellini C, Buccino A, Mingarelli R, Costabel S, La Rosa C, Garre C, Coviello DA, Ajmar F, et al. Ten novel mutations in the human neurofibromatosis type 1 (NF1) gene in Italian patients. Hum Mutat 2002; 20:74-5; PMID:12112660; http://dx.doi.org/10.1002/humu.9039
- Ars E, Kruyer H, Morell M, Pros E, Serra E, Ravella A, Estivill X, Lazaro C. Recurrent mutations in the NF1 gene are common among neurofibromatosis type 1 patients. J Med Genet 2003; 40:e82; PMID:12807981
- Gil R, Seeling JM. Characterization of Saccharomyces cerevisiae strains expressing ira1 mutant alleles modeled after disease-causing mutations in NF1. Mol Cell Biochem 1999; 202:109-8; PMID:10706001; http://dx.doi.org/10.1023/A:1007058427880
- Harashima T, Anderson S, Yates JR, 3rd, Heitman J. The kelch proteins Gpb1 and Gpb2 inhibit Ras activity via association with the yeast RasGAP neurofibromin homologs Ira1 and Ira2. Mol Cell 2006; 22:819-30; PMID:16793550; http://dx.doi.org/10.1016/j.molcel.2006.05.011
- Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol 2007; 8:379-93; PMID:17426725; http://dx.doi.org/10.1038/nrm2163
- Li R, Murray AW. Feedback control of mitosis in budding yeast. Cell 1991; 66:519-31; PMID:1651172; http://dx.doi.org/10.1016/0092-8674(81)90015-5
- Hoyt MA, Totis L, Roberts BT. S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell 1991; 66:507-17; PMID:1651171; http://dx.doi.org/10.1016/0092-8674(81)90014-3
- Weiss E, Winey M. The Saccharomyces cerevisiae spindle pole body duplication gene MPS1 is part of a mitotic checkpoint. J Cell Biol 1996; 132:111-23; PMID:8567717; http://dx.doi.org/10.1083/jcb.132.1.111
- Cohen-Fix O, Peters JM, Kirschner MW, Koshland D. Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes Dev 1996; 10:3081-93; PMID:8985178; http://dx.doi.org/10.1101/gad.10.24.3081
- Yamamoto A, Guacci V, Koshland D. Pds1p, an inhibitor of anaphase in budding yeast, plays a critical role in the APC and checkpoint pathway(s). J Cell Biol 1996; 133:99-110; PMID:8601617; http://dx.doi.org/10.1083/jcb.133.1.99
- Visintin R, Prinz S, Amon A. CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science 1997; 278:460-3; PMID:9334304; http://dx.doi.org/10.1126/science.278.5337.460
- Schwab M, Neutzner M, Mocker D, Seufert W. Yeast Hct1 recognizes the mitotic cyclin Clb2 and other substrates of the ubiquitin ligase APC. EMBO J 2001; 20:5165-75; PMID:11566880; http://dx.doi.org/10.1093/emboj/20.18.5165
- Hilioti Z, Chung YS, Mochizuki Y, Hardy CF, Cohen-Fix O. The anaphase inhibitor Pds1 binds to the APC/C-associated protein Cdc20 in a destruction box-dependent manner. Curr Biol: CB 2001; 11:1347-52; PMID:11553328; http://dx.doi.org/10.1016/S0960-9822(01)00399-2
- Lau DT, Murray AW. Mad2 and Mad3 cooperate to arrest budding yeast in mitosis. Curr Biol: CB 2012; 22:180-90; PMID:22209528; http://dx.doi.org/10.1016/j.cub.2011.12.029
- Fraschini R, Formenti E, Lucchini G, Piatti S. Budding yeast Bub2 is localized at spindle pole bodies and activates the mitotic checkpoint via a different pathway from Mad2. J Cell Biol 1999; 145:979-91; PMID:10352016; http://dx.doi.org/10.1083/jcb.145.5.979
- Hardwick KG, Weiss E, Luca FC, Winey M, Murray AW. Activation of the budding yeast spindle assembly checkpoint without mitotic spindle disruption. Science 1996; 273:953-6; PMID:8688079; http://dx.doi.org/10.1126/science.273.5277.953
- Yu H. Regulation of APC-Cdc20 by the spindle checkpoint. Cur Opin Cell Biol 2002; 14:706-14; PMID:12473343
- Agarwal R, Tang Z, Yu H, Cohen-Fix O. Two distinct pathways for inhibiting pds1 ubiquitination in response to DNA damage. J Biol Chem 2003; 278:45027-33; PMID:12947083; http://dx.doi.org/10.1074/jbc.M306783200
- Searle JS, Schollaert KL, Wilkins BJ, Sanchez Y. The DNA damage checkpoint and PKA pathways converge on APC substrates and Cdc20 to regulate mitotic progression. Nat Cell Biol 2004; 6:138-45; PMID:14743219; http://dx.doi.org/10.1038/ncb1092
- Peeters T, Louwet W, Gelade R, Nauwelaers D, Thevelein JM, Versele M. Kelch-repeat proteins interacting with the Galpha protein Gpa2 bypass adenylate cyclase for direct regulation of protein kinase A in yeast. Proc Nat Acad Sci U S A 2006; 103:13034-9.
- Wang H, Liu D, Wang Y, Qin J, Elledge SJ. Pds1 phosphorylation in response to DNA damage is essential for its DNA damage checkpoint function. Genes Dev 2001; 15:1361-72; PMID:11390356; http://dx.doi.org/10.1101/gad.893201
- Longtine MS, McKenzie A3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 1998; 14:953-61; PMID:9717241; http://dx.doi.org/10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U
- Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno-Borchart A, Doenges G, Schwob E, Schiebel E, et al. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 2004; 21:947-62; PMID:15334558; http://dx.doi.org/10.1002/yea.1142
- Kim J, Jeong J, Song K. The C-terminus of Bfa1p in budding yeast is essential to induce mitotic arrest in response to diverse checkpoint-activating signals. Genes Cells 2004; 9:399-418; PMID:15147270; http://dx.doi.org/10.1111/j.1356-9597.2004.00731.x
- Sass P, Field J, Nikawa J, Toda T, Wigler M. Cloning and characterization of the high-affinity cAMP phosphodiesterase of Saccharomyces cerevisiae. Proc Nat Acad Sci U S A 1986; 83:9303-7.