
Abstract
During macroautophagy, conjugation of ATG12 to ATG5 is essential for LC3 lipidation and autophagosome formation. Additionally, ATG12 has ATG5-independent functions in diverse processes including mitochondrial fusion and mitochondrial-dependent apoptosis. In this study, we investigated the regulation of free ATG12. In stark contrast to the stable ATG12–ATG5 conjugate, we find that free ATG12 is highly unstable and rapidly degraded in a proteasome-dependent manner. Surprisingly, ATG12, itself a ubiquitin-like protein, is directly ubiquitinated and this promotes its proteasomal degradation. As a functional consequence of its turnover, accumulation of free ATG12 contributes to proteasome inhibitor-mediated apoptosis, a finding that may be clinically important given the use of proteasome inhibitors as anticancer agents. Collectively, our results reveal a novel interconnection between autophagy, proteasome activity, and cell death mediated by the ubiquitin-like properties of ATG12.
Abbreviations:
Introduction
Macroautophagy (hereafter termed autophagy) and proteasome-mediated degradation constitute the cell's 2 major means of protein turnover.Citation1,2 Proteasomal degradation typically allows the selective turnover of short-lived proteins whereas autophagy is often considered a process that favors degradation of long-lived proteins. Autophagy and proteasome-mediated degradation are highly interconnected, such that perturbation of one pathway can impact on the other.Citation3-6 Importantly, deregulation of either process likely underpins many diseases linked to defective protein degradation such as Alzheimer's disease.Citation7,8
At a mechanistic level, autophagy and proteasome-mediated degradation share several similarities. Ubiquitination is often required to target proteins for proteasomal degradation; this process requires step-wise transfer of ubiquitin onto the target protein by 3 classes of enzyme (E1, 2, 3).Citation9 In an analogous fashion, 2 converging ubiquitin-like conjugation pathways are essential for autophagy. In one pathway, the ubiquitin-like protein (UBL) ATG12 is transferred from an E1-like enzyme, ATG7 via ATG10 (E2-like) forming a covalent attachment with ATG5.Citation10 Separately, the UBL MAP1LC3B/LC3B (Atg8 in yeast) is transferred from ATG7 onto ATG3 (E2-like). Serving an E3-like function, the ATG12–ATG5 conjugate (together with ATG16L1) is required for LC3B conjugation to phosphatidylethanolamine that drives autophagosomal membrane expansion.Citation10
Besides its essential role in autophagy, the UBL ATG12 also carries out various autophagy-independent roles. These include induction of mitophagy, promotion of mitochondrial fusion and activation of mitochondrial apoptosis.Citation11-13 Importantly, ATG12 has been shown to mediate these functions independently of its ability to conjugate to ATG5. ATG12 conjugation to ATG3 promotes mitochondrial fusion and restricts mitochondrial mass whereas free ATG12 directly promotes mitochondrial apoptosis in a similar manner to proapoptotic BH3-only proteins.Citation12,13 Given these autophagy (and ATG5)-independent functions of ATG12, in this study we investigated the regulation of free ATG12.
Results
Free ATG12 is rapidly degraded in a proteasome-dependent manner
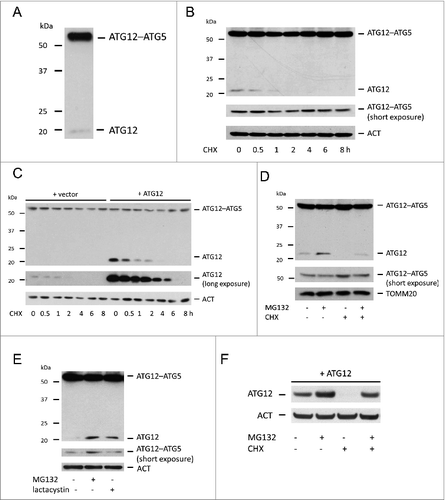
While investigating ATG12 regulated mitophagy we noted that the free form of ATG12 was expressed at very low levels compared with the ATG12–ATG5 conjugate (). We reasoned that this might either be due to efficient and complete conjugation of ATG12 to ATG5, instability of free ATG12 or a combination thereof. To address this, the stability of free ATG12 was examined following treatment of cells with the protein-translation inhibitor cycloheximide (CHX). Strikingly, endogenous ATG12 was rapidly depleted to below detectable levels following 2-h CHX treatment (). In a similar manner, ectopically expressed free ATG12 was also rapidly turned over, consistent with previous findings ().Citation14 Densitometric analysis revealed a half-life of 30 min for free ATG12 (Fig. S1). In contrast, levels of ATG12–ATG5 conjugate were unchanged following translational inhibition over an 8-h time-period (, , Fig. S1). Because the ubiquitin-proteasome system is the major degradative pathway for short-lived proteins, we next investigated whether free ATG12 was degraded in a proteasome-dependent manner. Treatment of cells with the proteasome inhibitor MG132 increased endogenous free ATG12 protein levels and completely prevented degradation of free ATG12 following CHX treatment (), demonstrating that free ATG12 is degraded in a proteasome-dependent manner. Supporting this, treatment of U2OS cells with an alternative proteasome inhibitor, lactacystin, also led to an accumulation of endogenous free ATG12 (). Consistent with ATG12 being degraded in a proteasome-dependent manner, MG132 treatment also increased levels of ectopically expressed free ATG12 and prevented degradation of free ATG12 following CHX treatment (). Taken together, these data demonstrate that the free form of ATG12 is highly unstable and degraded in a proteasome-dependent manner.
Figure 1. Free ATG12 is rapidly degraded in a proteasome-dependent manner (A) Western blot detection of ATG12 in U2OS cells. (B) Endogenous ATG12 expression in U2OS cells following CHX treatment. (C) U2OS expressing empty vector or ATG12 were treated with CHX for various times and probed for ATG12 expression. (D) U2OS cells were treated were treated for 8 h as indicated with MG132 and/or CHX and probed for ATG12 expression. (E) U2OS were treated for 8 h with MG132 or lactacystin and examined for ATG12 expression. (F) U2OS expressing ATG12 were treated for 8 h with MG132 and/or CHX as indicated and cell lysates were probed for ATG12 expression. ACT or TOMM20 were used as a loading control.
Proteasomal degradation of free ATG12 is independent of autophagy
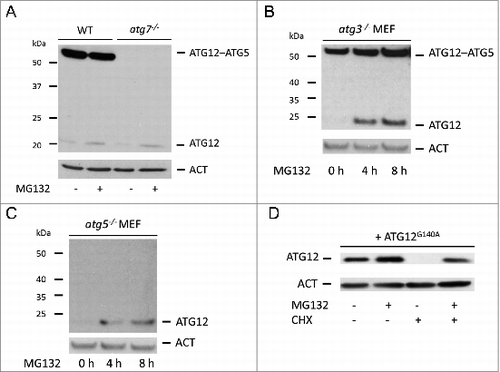
Autophagy requires ATG12 conjugation via its C-terminal glycine to ATG5 in a process that is catalyzed by ATG7 and ATG10. We next determined whether autophagy contributed to the degradation of free ATG12. Wild-type E1A and RAS-transformed murine embryonic fibroblasts (MEFs) expressed readily detectable ATG12–ATG5 conjugate but low amounts of free ATG12 (). As expected, ATG12–ATG5 conjugate was completely absent in Atg7-deficient MEFs, but, surprisingly, the level of free ATG12 was similar to wild-type MEFs even though Atg7-deficient MEFs expressed higher levels of Atg12 mRNA (, Fig. S2A). In both cases, MG132 treatment increased free ATG12 to the same extent (). Supporting this finding, proteasome inhibitor also increased ATG12 expression in SV40 immortalized MEFs deficient in Atg7 (Fig. S2B). These data demonstrate that ATG12 can be targeted for proteasome-dependent degradation independent of ATG7. To investigate whether other components of the autophagy pathway influenced ATG12 stability, we performed similar experiments in SV40 immortalized MEF deficient in Atg3 or Atg5 (, ). In both cases, MG132 treatment led to an increase in free ATG12 levels. ATG12 was also stabilized to a similar extent following proteasome-inhibitor treatment in cells following ATG5 knockdown by RNA interference (RNAi)(Fig. S2C). Finally, we analyzed the stability of ATG12 in which its C-terminal glycine was mutated to alanine (G140A) and is therefore unable to efficiently conjugate to ATG5.Citation15 Similar to ectopically expressed wild-type ATG12 (), ATG12G140A was highly unstable and degraded in a proteasome-dependent manner (). These results demonstrate that the rapid proteasomal degradation of free ATG12 neither requires the ATG12–ATG5 conjugation machinery nor autophagy.
Figure 2. Proteasomal degradation of free ATG12 protein occurs independent of autophagy (A) E1A and Ras-transformed WT or Atg7 knockout MEF were treated for 8 h with MG132 and cell lysates were probed for ATG12 expression. (B) Atg3 or (C) Atg5 knockout MEFs were treated with MG132 for 4 h and 8 h and analyzed for ATG12 expression. (D) U2OS cells expressing ATG12G140A were treated for 8 h with MG132 and/or CHX as indicated and lysates were examined for ATG12 expression. In all immunoblots, ACT was used as a loading control.
Direct ubiquitination of free ATG12 regulates its proteasomal degradation
The major means of targeting proteins for proteasomal degradation is by poly-ubiquitination. Therefore, we addressed whether ATG12 is directly ubiquitinated. Empty vector or ATG12 were coexpressed with His-tagged ubiquitin (His-UB) in 293T cells treated or not with MG132. Ubiquitinated proteins were isolated by Dynabead affinity isolation and probed with anti-ATG12 antibody (). Following ubiquitin affinity isolation, an ATG12 immunoreactive smear was detected, demonstrating that ATG12 is directly ubiquitinated. Furthermore, MG132 treatment increased the amount of ubiquitinated ATG12 and, as expected, led to a general increase in the level of protein ubiquitination (). We next assessed the contribution of ATG12 ubiquitination to its proteasome-mediated degradation. Ubiquitination most often occurs on substrate lysine residues, therefore we mutated all lysine residues in ATG12 to arginine (ATG12[K-]). First, we examined whether ATG12[K-] remained functionally active by stably expressing either WT ATG12 or ATG12[K-] in Atg12 knockout MEF. Cells were treated with the lysomotropic agent chloroquine to inhibit basal autophagy and assessed for ATG12–ATG5 conjugation and LC3 lipidation (). Expression of ATG12[K-] restored ATG12–ATG5 conjugate formation and LC3 lipidation to a similar extent as WT ATG12 in Atg12-deficient MEFs, thereby demonstrating that lysineless ATG12 retained functionality. Following this, we examined the effect of removing lysines upon ATG12 ubiquitination. His-tagged ubiquitin and WT or ATG12[K-] were coexpressed in cells that were treated or not with MG132 and ubiquitinated proteins were isolated by affinity isolation and probed for ATG12. Importantly, compared with WT ATG12, mutation of lysines in ATG12 completely abolished its ubiquitination (). This argues that ATG12 is ubiquitinated solely on lysine residues and not other possible acceptor residues such as its N terminus. We aimed to define which ATG12 lysine(s) were subject to ubiquitination by extensive mutagenesis of single or closely grouped lysine residues and assessing its effect upon ATG12 ubiquitination (Fig. S3A). All ATG12 lysine-mutants displayed similar levels of ubiquitination to WT ATG12, demonstrating that poly-ubiqutination of ATG12 can occur on multiple lysine residues (Fig. S3B). Using the ATG12[K-] mutant, we investigated the role of ATG12 ubiquitination upon its stability. Cells expressing either WT or ATG12[K-] were treated with CHX for 6 h and examined for ATG12 protein level by western blot. Whereas 90% of WT ATG12 was degraded with 6-h CHX treatment, 50% of ATG12[K-] remained, supporting a role for ubiquitination in proteasomal-degradation of ATG12 (). Nevertheless, we were intrigued that a significant pool of ATG12[K-] was still degraded even though it was not ubiquitinated. As such, we investigated whether lysineless ATG12 was still subject to proteasome-dependent degradation. Cells expressing ATG12[K-] were treated with CHX in the presence or absence of MG132. Interestingly, degradation of ATG12[K-] following CHX treatment could be inhibited by proteasome inhibition (). Together, these data show that ATG12 is directly ubiquitinated and that ATG12 can be targeted for proteasomal degradation by ubiquitin-dependent and -independent mechanisms.
Figure 3. Direct ubiquitination of free ATG12 regulates its proteasomal degradation (A) 293T cells expressing vector or ATG12 and His-ubiquitin were treated with MG132 as indicated. Cell lysates were subject to His-tag affinity isolation and immunoblotted for ATG12 and ubiquitin expression (B) Atg12 knockout MEFs, as well as MEFs stably expressing Atg12, were treated for 4 h with chloroquine, then examined for ATG12 and LC3B expression. (C) 293T cells expressing ATG12 or ATG12[K-] together with His-ubiquitin, were treated as indicated, subject to His-tag affinity isolation and probed for ATG12 and ubiquitin expression. (D) U2OS cells expressing ATG12 or ATG12[K-] and treated for 6 h with CHX and analyzed for ATG12 and TOMM20 expression, densitometric analysis was performed using ImageJ software, normalizing to TOMM20 levels. (E) U2OS cells expressing ATG12[K-] were treated with MG132 and/or CHX for 8 h as indicated and examined for ATG12 expression. ACT or TOMM20 were used as loading controls.
![Figure 3. Direct ubiquitination of free ATG12 regulates its proteasomal degradation (A) 293T cells expressing vector or ATG12 and His-ubiquitin were treated with MG132 as indicated. Cell lysates were subject to His-tag affinity isolation and immunoblotted for ATG12 and ubiquitin expression (B) Atg12 knockout MEFs, as well as MEFs stably expressing Atg12, were treated for 4 h with chloroquine, then examined for ATG12 and LC3B expression. (C) 293T cells expressing ATG12 or ATG12[K-] together with His-ubiquitin, were treated as indicated, subject to His-tag affinity isolation and probed for ATG12 and ubiquitin expression. (D) U2OS cells expressing ATG12 or ATG12[K-] and treated for 6 h with CHX and analyzed for ATG12 and TOMM20 expression, densitometric analysis was performed using ImageJ software, normalizing to TOMM20 levels. (E) U2OS cells expressing ATG12[K-] were treated with MG132 and/or CHX for 8 h as indicated and examined for ATG12 expression. ACT or TOMM20 were used as loading controls.](/cms/asset/43b11ec1-d006-4ca0-b395-a23c9516f3f2/kaup_a_981914_f0003_b.gif)
Free ATG12 regulates proteasome inhibitor-mediated cell death
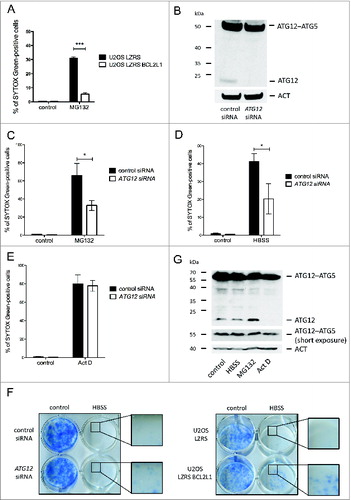
ATG12 has previously been implicated in mitochondrial-dependent apoptosis.Citation13 This, coupled with our finding that free ATG12 is turned over via the proteasome, led us to address whether ATG12 contributes to proteasome inhibitor-mediated toxicity. First, we examined whether proteasome inhibitor triggered cell death via the mitochondrial apoptotic pathway. Cells ectopically expressing antiapoptotic BCL2L1 were treated with MG132 and analyzed for cell death by uptake of the cell impermeable dye SYTOX Green or by ANXA5-propidium iodide staining and flow cytometry. BCL2L1 robustly protected against MG132-mediated killing, confirming that MG132 predominantly engages the mitochondrial apoptotic pathway (, Fig. S4A). Next, we utilized RNAi to knock down ATG12 in U2OS cells. Importantly, given the instability of free ATG12, RNAi allowed us to selectively downregulate free ATG12 with minimal effects upon levels of ATG12–ATG5 conjugate (). In accordance, no effect upon autophagy was observed following ATG12 RNAi (Fig. S4B). We examined the effect of depleting ATG12 upon proteasome inhibitor-mediated toxicity. U2OS cells treated with control or ATG12 RNAi were incubated with MG132 and monitored for cell death by uptake of the cell-impermeable dye SYTOX Green or by ANXA5-propidium iodide staining and flow cytometry. Consistently, RNAi knockdown of ATG12 protected against proteasome inhibitor-mediated toxicity (, Fig. S4C). Two individual siRNA oligos targeting ATG12 gave similar results (Fig. S4D, E). Extending these findings, we examined whether depletion of ATG12 could offer general protection against other prodeath stimuli including starvation (HBSS) and actinomycin D (Act D) treatment. Similar to proteasome inhibition, ectopic expression of antiapoptotic BCL2L1 effectively blocked cell death induced by HBSS starvation or Act D treatment demonstrating that these treatments kill via mitochondrial-dependent apoptosis (Fig. S4F–H). Interestingly, whereas ATG12 knockdown inhibited starvation induced apoptosis, it had no effect upon Act D-mediated apoptosis (, , Fig. S4I, J). Similar to ectopic BCL2L1 expression, ATG12 knockdown promoted long-term clonogenic survival following starvation in-line with a proapoptotic function for ATG12 residing upstream of the mitochondrial permeabilization (). The difference in requirement for ATG12 following divergent prodeath stimuli prompted us to investigate levels of free ATG12 following different treatments. Free ATG12 remained constant during starvation or, as before, increased following proteasome inhibitor treatment. In contrast, and in line with its ability to inhibit transcription, free ATG12 levels were rapidly depleted following Act D treatment ().
Figure 4. Free ATG12 promotes proteasome inhibitor-mediated cell death (A) U2OS cells stably expressing empty vector or BCL2L1 were treated with MG132 and cell viability was determined by SYTOX Green staining using an Incucyte Imager; data represents the mean +/− standard error of the mean (SEM) of 3 experiments at a representative time-point (24 h). (B) U2OS cells were assessed for ATG12 expression 2 d post-transfection with control or ATG12 siRNA. Cell viability of control or ATG12 siRNA-transfected U2OS cells, treated with MG132 (C) HBSS (D) or Act D (E) was determined by SYTOX Green staining using an Incucyte Imager; representative time-points shown (24 h MG132, 30 h HBSS, 24 h Act D). Graphs represent the mean +/− SEM of 4 experiments. (F) U2OS, transfected with control or ATG12 siRNA, or stably expressing vector or BCL2L1 were starved in HBSS. After 48 h of starvation, cells were washed, cultivated for 7 d in DMEM and colonies were stained with methylene blue. (G) U2OS cells were treated with HBSS (24 h), MG132 (16 h) or Act D (16 h) and examined for ATG12 expression. In all immunoblots, ACT was used as a loading control.
Free ATG12 regulates proteasome inhibitor-mediated cell death independently of autophagy
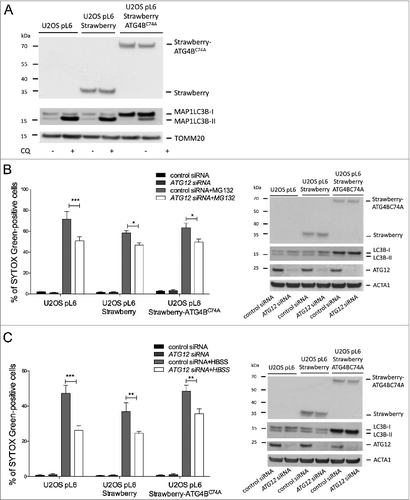
Ectopic expression of ATG12 has been shown to inhibit autophagy.Citation16,17 Therefore, we addressed whether the ability of free ATG12 to promote cell death was modulated by autophagy. For this purpose, we generated U2OS cells stably expressing a dominant-negative ATG4B mutant (C74A).Citation18 Verifying its dominant negative effect, the accumulation of lipidated LC3B following chloroquine treatment was prevented in ATG4BC74A -expressing cells (). We subjected ATG4BC74A or control cells to HBSS starvation, MG132 or Act D treatment. Inhibition of autophagy did not affect cell death induced by all 3 treatments (Fig. S5). Finally, we knocked down ATG12 in autophagy-deficient U2OS cells and subjected them to proteasome inhibitor or HBSS starvation. Knockdown of ATG12 equally protected autophagy- proficient and -deficient U2OS cells in response to both stimuli (, ), further arguing ATG12s proapoptotic effects are independent of effects on autophagy.
Figure 5. Free ATG12 promotes cell death independent of autophagy (A) U2OS cells stably expressing vector, Strawberry or Strawberry-ATG4BC74A were treated for 4 h with chloroquine and cell lysates were blotted for RFP and LC3B. U2OS cells stably expressing vector, Strawberry or Strawberry-ATG4BC74A were transfected with control or ATG12 siRNA. (B) Following MG132 treatment (24 h) or (C) HBSS starvation (48 h), cell viability was determined by SYTOX Green exclusion in an Incuyte Imager. Graphs show the mean +/− SEM of 3 (B) or 5 (C) experiments at representative time-points (24h MG132, 48 h HBSS). Western blots show cell lysates of U2OS cells stably expressing vector, Strawberry or Strawberry-ATG4BC74A, transfected with control or ATG12 siRNA, treated for 8 h with MG132 (B) or HBSS (C) and probed for RFP, LC3B, and ATG12. In all immunoblots, ACT or TOMM20 were used as a loading control.
Discussion
Here we investigated the regulation of ATG12 protein stability; unlike the ATG12–ATG5 conjugate, we find that free ATG12 is highly unstable. Free ATG12 was found to be directly ubiquitinated and targeted for proteasomal degradation in an autophagy-independent manner. Importantly, turnover of ATG12 regulates its cytotoxic function such that ATG12 contributes to proteasome inhibitor-mediated toxicity.
ATG12 is a member of the UBL protein family that comprises of approximately 20 diverse members.Citation19 Although sharing homology with ubiquitin, UBLs are typically neither ubiquitinated nor targeted for proteasomal degradation. In contrast, we find that the free form of ATG12 is directly ubiquitinated, most likely on lysine residues, and rapidly degraded via the proteasome. Mutation of individual lysine residues failed to effect ATG12 ubiquitination arguing that ATG12 can be ubiquitinated on multiple lysine residues. Supporting our findings, a recent proteome-wide, mass-spectrometry based analysis of ubiquitinated proteins has revealed several potential ubiquitin acceptor lysines in ATG12.Citation20 Besides ATG12, another UBL called ubiquitin D (UBD) has also been found to be directly ubiquitinated and targeted for proteasomal degradation.Citation21,22 Direct ubiquitination of UBD appears essential for its degradation. In the case of ATG12, direct ubiquitination promotes but is not required for its degradation since nonubiquitinated ATG12 is still targeted for proteasomal degradation (). Moreover, while UBD can target substrates for proteasomal degradation, this is not apparent with ATG12 given the highly stable nature of the ATG12–ATG5 conjugate that we and others have observed.Citation14 Why there is a profound difference in stability between free ATG12 and ATG12–ATG5 remains unclear. The possibility that ATG5 masks a destabilizing region in ATG12 seems unlikely given that disruption of the ATG12 and ATG5 binding interface does not appear to destabilize the ATG12–ATG5 conjugate.Citation16,23 Importantly, ATG12 proteasomal degradation neither requires autophagy nor an exposed C-terminal glycine (that conjugates to ATG5 or ATG3) since ATG12 instability persisted in various autophagy-deficient settings or following mutation of the ATG12 C-terminal glycine to an alanine (a modification that inhibits ATG12–ATG5 conjugation).Citation15
Free ATG12 has recently been shown to promote mitochondrial-dependent apoptosis.Citation13 Along similar lines, we find that free ATG12 contributes to proteasome inhibitor and HBSS starvation-mediated apoptosis. Interestingly, we find that disruption of the putative ATG12 BH3-domain effectively stabilizes it. The presence or upregulation of ATG12 may be sufficient to sensitize to cell death by antagonizing antiapoptotic BCL2 function. In contrast, ATG12 played no role in Act D-induced apoptosis. This differential contribution of ATG12 may be due to the rapid depletion of free ATG12 following transcriptional inhibition by Act D treatment relative to the other treatments. Importantly, the cytotoxic effect of ATG12 appears to be independent of any effect on autophagy.
In summary, our findings develop an emerging paradigm that UBLs themselves can be directly modified by ubiquitin and targeted for proteasomal degradation. Moreover, these results provide a new ATG12-mediated link between autophagy and proteasome activity. Regulation of ATG12 degradation has the potential to impact on both its expanding repertoire of autophagy-independent functions as well as the clinical efficacy of proteasome inhibitors utilized in cancer treatments.
Materials and Methods
Cells
All cell lines were maintained in DMEM high glucose medium supplemented with 10% fetal calf serum, 2 mM glutamine, 1 mM sodium pyruvate, 50 μM mercaptoethanol, and penicillin/streptomycin. Atg7-deficient MEFs have been previously described.Citation24 Atg12 knockout MEFs were derived from atg12−/- animals that will be described elsewhere (J.D., manuscript in preparation). Drs. Masaaki Komatsu (Tokyo Metropolitan Institute of Medical Science) and Mathew Albert (Pasteur Institute) kindly provided Atg3- and Atg5-deficient MEFs, respectively.
Retroviral transduction
Phoenix Ecotropic cells (0.5 × 106 in a 10 cm dish) were transfected with LZRS zeo, LZRS FLAG-BCL2L1, pLenti6puro (pL6) Strawberry-ATG4BC74A or empty vector controls using Lipofectamine 2000 (Invitrogen, 11668-019) according to the manufacturer's instructions. Two days later virus-containing supernatant was harvested, filtered, and used to infect U2OS cells in the presence of 1 μg/ml polybrene. Two days postinfection, stably expressing cells were selected by growth in 200 ug/ml zeocin (Invitrogen, R250-01) or 1 ug/ml puromycin (Sigma, P8833), respectively.
Western blotting
Cell lysates were prepared using NP-40 lysis buffer (1% NP-40 [Sigma, I8896], 1 mM EDTA, 150 mM NaCl, 50 mM Tris pH 7.4, 1 mM phenylmethylsulfonyl fluoride [Sigma, 93482], Complete Protease Inhibitors [Roche, 11697498001]). Protein content was determined by Bio-Rad assay (Bio-Rad, 500-0006), proteins were separated by SDS-PAGE and blotted onto nitrocellulose. Membranes were probed with anti-ACT (actin) (MP Biomedicals, 08691001, 1/10000), anti-ATG12 (Cell Signaling Technology, 4180, 1/1000), anti-UB/ubiquitin (Santa Cruz Biotechnology, sc-8017, 1/1000), anti-TOMM20 (Santa Cruz Biotechnology, sc-11415, 1/1000), anti-RFP (Rockland, 600-901-379, 1/1000), anti-LC3B (Cell Signaling Technology, 2775, 1/1000) antibodies followed by incubation with the appropriate HRP conjugated secondary antibody (GE Healthcare, NA934V, NXA931) and detection of immunoreactive proteins by ECL.
Cell-based ubiquitination assay
293T cells (1 × 106) were transfected with 2.5 μg His-ubiquitinCitation25 and 2.5 μg pcDNA3 (Invitrogen, V790-20), pcDNA3 ATG12 or pcDNA3 ATG12[K-] using GeneJuice (Novagen, 70967) according to the manufacturer's protocol. Forty-eight h after transfection, cells were harvested in phosphate-buffered saline (PBS; Thermo Scientific, BR0014G) and pellets were resuspended in UBA buffer (6 M guanidinium HCl, 300 mM NaCl, 50 mM NaH2PO4 pH 8.0, 100 μg/ml N-ethylmaleimide [Sigma, E3876]). Cell lysates were incubated overnight with His-tag Dynabeads (Novex Life Technologies, 1003D) rotating at 4°C. The next day samples were subjected to consecutive washes in UBA, UBB (1:1 mix of UBA and UBC), UBC buffer (300 mM NaCl, 50 mM Na2PO4 pH 8.0) and PBS. For SDS-Page LDS sample buffer (Novex Life Technologies, NP0007) was added containing 50 mM imidazole.
Plasmids and site-directed mutagenesis
ATG12 was cloned from pMT2 myc-ATG12 via BamHI and EcoRI into pcDNA3 by standard cloning techniques. Site-directed mutagenesis was performed using Phusion site-directed mutagenesis kit (Thermo Scientific, F-541) according to the manufacturer's instructions.
siRNA transfection
U2OS (1.5 × 105) cells were transfected with 100 nM nontargeting control (Thermo Scientific, D-001210-02-05) siGENOME human Atg12 SMARTpool siRNA (Thermo Scientific, M-010212-02-0005), a mixture of 3 different Stealth RNAi oligos against Atg5 (Invitrogen, HSS114103, HSS114104, HSS190366) or the individual RNAi duplexes using Lipofectamine 2000 (Invitrogen, 11668027) according to the manufacturer's protocol. Twenty-four h post-transfection, cells were analyzed by immunoblotting or subjected to cell death or autophagic flux assays.
Treatments and cell death assays
For protein stability assays cells were treated with 1mg/ml cycloheximide (Sigma, C7698-1G), 10 μM MG132 (Calbiochem, 474790) or 10 μM lactaystin (Enzo, BML-PI104-1000) for 8 h unless otherwise indicated. Before starvation with HBSS (Gibco, 14025-050), cells were washed for 4 times with PBS. Actinomycin D (Calbiochem, 114666) was used at a concentration of 1 μM. Cell viability was determined using an Incucyte FLR imaging system (Essen Bioscience, Ann Arbor, MI). Cells were plated in medium containing 30 nM SYTOX Green (Invitrogen, S7020). Cells were treated as described, imaged every 30 min over a period of 3 d, and analyzed using Incucyte image analysis software (Essen Bioscience). For quantification, the SytoxGreen fluorescence was normalized to the confluency factor of the respective well and the percentage of SYTOX Green-positive cells was calculated using the maximum SYTOX Green fluorescence at 100% cell death. Alternatively, cell viability was analyzed by flow cytometry using FACSCalibur (BD Biosciences, San Jose, CA). For this purpose, cells were harvested following 24 h of treatment as indicated and stained with Alexa Fluor 647-ANXA5/annexin V (BioLegend, 640911) and 1 μg/ml propidium iodide according to the manufacturer's protocols. Analysis was performed using Cellquest Pro software (BD Biosciences).
Autophagic flux assays
To determine autophagic flux U2OS cells were treated for 8 h with 20 μM leupeptin (Tocris, 1167) and for 4 h with 20 mM NH4Cl or 100 nM bafilomycin A1 (LC Laboratories, B-1080). Chloroquine (Sigma, C6628) was used at a concentration of 50 uM for a duration of 4 h.
Real-time PCR
RNA was isolated from E1A wt or Atg7 knockout MEFs using QIAGEN RNeasy Mini Kit (Qiagen, 74104). 2 μg RNA per sample were used for cDNA synthesis and PCR with DyNAmo SYBR Green 2-step qRT-PCR kit (Thermo Scientific, F430-L). PCR fragments were amplified 3 min at 95°C, followed by 40 cycles of 20 sec at 95°C, 30 sec at 57°C, 30 sec at 72°C and final 5 min at 72°C. The following primers were used: Acta1 (actin, α 1, skeletal muscle) forward (CTAAGGCCAACCGTGAAAAG), Acta1 reverse (ACCAGAGGCATACAGGGACA), Atg12 forward (AACAAAGAAATGGGCTGTGG), Atg12 reverse (TTGCAGTAATGCAGGACCAG).
Statistical analysis
For comparison of multiple groups, the 2-way Analysis of Variance (ANOVA) followed by the Bonferroni post-test was used. Analyses were performed using Prism 5.0 software (GraphPad).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary_Figure_Legends.docx
Download MS Word (16.5 KB)Supplemental_Figures.pdf
Download PDF (3.1 MB)Acknowledgments
We thank Drs. Masaaki Komatsu and Mathew Albert for providing cells.
Funding
This work was supported by funding from the Royal Society, BBSRC grant BB/K008374/1 and a Marie-Curie Career Integration Grant (S.W.G.T). S.W.G.T is a Royal Society University Research Fellow. Generation of Atg12-deficient MEFs was supported by NIH R01CA126792 to J.D.
References
- Mizushima N. Autophagy in protein and organelle turnover. Cold Spring Harbor Symp Quant Biol 2011; 76:397-402; PMID:21813637; http://dx.doi.org/10.1101/sqb.2011.76.011023
- Kleiger G, Mayor T. Perilous journey: a tour of the ubiquitin-proteasome system. Trends Cell Biol 2014; 24:352-9; PMID:24457024; http://dx.doi.org/10.1016/j.tcb.2013.12.003
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007; 447:859-63; PMID:17568747; http://dx.doi.org/10.1038/nature05853
- Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol 2007; 171:513-24; PMID:17620365; http://dx.doi.org/10.2353/ajpath.2007.070188
- Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell 2009; 33:517-27; PMID:19250912; http://dx.doi.org/10.1016/j.molcel.2009.01.021
- Qiao L, Zhang J. Inhibition of lysosomal functions reduces proteasomal activity. Neurosci Lett 2009; 456:15-9; PMID:19429125; http://dx.doi.org/10.1016/j.neulet.2009.03.085
- Harris H, Rubinsztein DC. Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol 2012; 8:108-17; PMID:22187000; http://dx.doi.org/10.1038/nrneurol.2011.200
- Ihara Y, Morishima-Kawashima M, Nixon R. The ubiquitin-proteasome system and the autophagic-lysosomal system in Alzheimer disease. Cold Spring Harbor Perspect Med 2012; 2:a006361; PMID:22908190; http://dx.doi.org/10.1101/cshperspect.a006361
- Schulman BA, Harper JW. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat Rev Mol Cell Biol 2009; 10:319-31; PMID:19352404; http://dx.doi.org/10.1038/nrm2673
- Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep 2008; 9:859-64; PMID:18704115; http://dx.doi.org/10.1038/embor.2008.163
- Colell A, Ricci JE, Tait S, Milasta S, Maurer U, Bouchier-Hayes L, Fitzgerald P, Guio-Carrion A, Waterhouse NJ, Li CW, et al. GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell 2007; 129:983-97; PMID:17540177; http://dx.doi.org/10.1016/j.cell.2007.03.045
- Radoshevich L, Murrow L, Chen N, Fernandez E, Roy S, Fung C, Debnath J. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell 2010; 142:590-600; PMID:20723759; http://dx.doi.org/10.1016/j.cell.2010.07.018
- Rubinstein AD, Eisenstein M, Ber Y, Bialik S, Kimchi A. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol Cell 2011; 44:698-709; PMID:22152474; http://dx.doi.org/10.1016/j.molcel.2011.10.014
- Sou YS, Waguri S, Iwata J, Ueno T, Fujimura T, Hara T, Sawada N, Yamada A, Mizushima N, Uchiyama Y, et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol Biol Cell 2008; 19:4762-75; PMID:18768753; http://dx.doi.org/10.1091/mbc.E08-03-0309
- Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y. A protein conjugation system essential for autophagy. Nature 1998; 395:395-8; PMID:9759731; http://dx.doi.org/10.1038/26506
- Otomo C, Metlagel Z, Takaesu G, Otomo T. Structure of the human ATG12∼ATG5 conjugate required for LC3 lipidation in autophagy. Nat Struct Mol Biol 2013; 20:59-66; PMID:23202584; http://dx.doi.org/10.1038/nsmb.2431
- Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell 2008; 19:2092-100; PMID:18321988; http://dx.doi.org/10.1091/mbc.E07-12-1257
- Fujita N, Hayashi-Nishino M, Fukumoto H, Omori H, Yamamoto A, Noda T, Yoshimori T. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell 2008; 19:4651-9; PMID:18768752; http://dx.doi.org/10.1091/mbc.E08-03-0312
- van der Veen AG, Ploegh HL. Ubiquitin-like proteins. Ann Rev Biochem 2012; 81:323-57; PMID:22404627; http://dx.doi.org/10.1146/annurev-biochem-093010-153308
- Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell 2011; 44:325-40; PMID:21906983; http://dx.doi.org/10.1016/j.molcel.2011.08.025
- Hipp MS, Kalveram B, Raasi S, Groettrup M, Schmidtke G. FAT10, a ubiquitin-independent signal for proteasomal degradation. Mol Cell Biol 2005; 25:3483-91; PMID:15831455; http://dx.doi.org/10.1128/MCB.25.9.3483-3491.2005
- Buchsbaum S, Bercovich B, Ciechanover A. FAT10 is a proteasomal degradation signal that is itself regulated by ubiquitination. Mol Biol Cell 2012; 23:225-32; PMID:22072791; http://dx.doi.org/10.1091/mbc.E11-07-0609
- Noda NN, Fujioka Y, Hanada T, Ohsumi Y, Inagaki F. Structure of the Atg12-Atg5 conjugate reveals a platform for stimulating Atg8-PE conjugation. EMBO Rep 2013; 14:206-11; PMID:23238393; http://dx.doi.org/10.1038/embor.2012.208
- Taherbhoy AM, Tait SW, Kaiser SE, Williams AH, Deng A, Nourse A, Hammel M, Kurinov I, Rock CO, Green DR, et al. Atg8 transfer from Atg7 to Atg3: a distinctive E1-E2 architecture and mechanism in the autophagy pathway. Mol Cell 2011; 44:451-61; PMID:22055190; http://dx.doi.org/10.1016/j.molcel.2011.08.034
- Hock AK, Vigneron AM, Carter S, Ludwig RL, Vousden KH. Regulation of p53 stability and function by the deubiquitinating enzyme USP42. EMBO J 2011; 30:4921-30; PMID:22085928; http://dx.doi.org/10.1038/emboj.2011.419