
Abstract
Excessive ethanol exposure is detrimental to the brain. The developing brain is particularly vulnerable to ethanol such that prenatal ethanol exposure causes fetal alcohol spectrum disorders (FASD). Neuronal loss in the brain is the most devastating consequence and is associated with mental retardation and other behavioral deficits observed in FASD. Since alcohol consumption during pregnancy has not declined, it is imperative to elucidate the underlying mechanisms and develop effective therapeutic strategies. One cellular mechanism that acts as a protective response for the central nervous system (CNS) is autophagy. Autophagy regulates lysosomal turnover of organelles and proteins within cells, and is involved in cell differentiation, survival, metabolism, and immunity. We have recently shown that ethanol activates autophagy in the developing brain. The autophagic preconditioning alleviates ethanol-induced neuron apoptosis, whereas inhibition of autophagy potentiates ethanol-stimulated reactive oxygen species (ROS) and exacerbates ethanol-induced neuroapoptosis. The expression of genes encoding proteins required for autophagy in the CNS is developmentally regulated; their levels are much lower during an ethanol-sensitive period than during an ethanol-resistant period. Ethanol may stimulate autophagy through multiple mechanisms; these include induction of oxidative stress and endoplasmic reticulum stress, modulation of MTOR and AMPK signaling, alterations in BCL2 family proteins, and disruption of intracellular calcium (Ca2+) homeostasis. This review discusses the most recent evidence regarding the involvement of autophagy in ethanol-mediated neurotoxicity as well as the potential therapeutic approach of targeting autophagic pathways.
Abbreviations:
- AD, Alzheimer disease
- ALS, autophagy-lysosome system
- AMPK, adenosine 5′-monophosphate-activated protein kinase;
- ATG, autophagy-related
- CNS, central nervous system
- ER, endoplasmic reticulum
- FASD, fetal alcohol spectrum disorders
- FOXO3, forkhead box O3
- GSK3B, glycogen synthase kinase 3 β
- HD, Huntington disease, HNSCs, hippocampal neural stem cells
- LC3, microtubule-associated protein 1 light chain 3
- MTOR, mechanistic target of rapamycin (serine/threonine kinase)
- MTORC1, MTOR complex 1
- NOX, NADPH oxidase
- NFE2L2, nuclear factor, erythroid 2-like 2
- PD, Parkinson disease
- PI3K, class I phosphoinositide 3-kinase
- ROS, reactive oxygen species
- SQSTM1/p62, sequestosome 1
- TSC1/2, tuberous sclerosis 1/ 2
- UPR, unfolded protein response
Introduction
Heavy ethanol consumption produces profound damage to the brain, heart, liver, pancreas, lungs, bone, skeletal muscles, endocrine, and immune systems. The developing nervous system is particularly vulnerable to ethanol exposure. Prenatal exposure to ethanol causes fetal alcohol spectrum disorders. Fetal alcohol syndrome is the most severe form of FASD, characterized by mental retardation, malformation of the nervous system, deficits in craniofacial development, and growth deficiency.Citation1-5 The incidence of FASD in the United States is between 2% and 5%.Citation3 The lifetime cost of FASD is estimated to be approximately $2 million per case, and the total annual cost of FASD was estimated at $4 billion in 2004.Citation6 Neurodegeneration and the depletion of neurons in the brain, which contribute to some behavioral abnormalities observed in FASD, is the most devastating consequence of developmental exposure to ethanol.Citation5,7 Therefore, it is critical to elucidate the mechanisms of ethanol-induced neuroapoptosis in order to develop effective therapeutic approaches to ameliorate ethanol neurotoxicity.
Autophagy and Neuronal Survival
There are 2 major protein catabolism systems, namely, the ubiquitin-proteasome system (UPS) and the autophagy-lysosome system (ALS). Autophagy is a cellular catabolic process for amino acid/energy recycling and mitigation of metabolic stress.Citation8 Although some of these functions overlap with those of the UPS, ALS mainly regulates the turnover of long-lived proteins and the maintenance of amino acid pools in the condition of cellular stresses. The ALS is able to degrade entire organelles such as mitochondria, endoplasmic reticulum (ER), and peroxisomes.Citation8 This process is considered to be adaptive and plays an essential role in various cellular activities, including development, differentiation, survival, metabolism, and immunity.Citation9,10
Autophagy is activated by nutrient deprivation and other stress signals, such as exposure to toxic agents and metabolic stress.Citation11-14 Studies in the past decade have shown not only that autophagy provides cells with oxidizable substrates, but it can also offer protection against numerous pathologies such as cancer, neurodegeneration, cardiac disease, diabetes, and infections.Citation15,16 For the CNS, constitutive elimination of damaged proteins by basal autophagy is important, particularly to neurons.Citation17 The constitutive activation of autophagy is essential for neuronal cell survival.Citation12 Disruption in the normal flow of autophagic pathways has been shown in the aging process of the brain and in some neurodegenerative diseases, including Alzheimer (AD), Parkinson (PD), and Huntington (HD) diseases.Citation17 In some neuropathies, including AD, PD, HD, amyotrophic lateral sclerosis, and frontotemporal dementia, autophagy is beneficial due to its role in clearing toxic factors and enhancing cell survival particularly in neurons.Citation12,18-20 For example, impaired autophagy has been observed in several neurodegenerative diseases, such as AD models and PD, among others. Genetic activation of autophagy by overexpressing the autophagy-related (ATG) protein BECN1 promotes degradation of amyloid-β and reduces AD pathology in animal models.Citation21 Of potential clinical interest is that chemical stimulation of autophagy by rapamycin, or MTOR-independent autophagy inducers, such as lithium, valproic acid, carbamazepine, rilmenidine, or trehalose, can rescue the cellular toxicity and neurodegenerative phenotype in cellular and animal models of certain neurodegenerative disorders including AD, PD, and HD.Citation22
The role of autophagy in regulating cell survival, however, is still under debate. Since the activation of autophagy and cell death are frequently observed occurring at the same time, some scientists suggest that autophagy may be cytotoxic.Citation23-25 The terminology “autophagic cell death” or “type II cell death” has been used to describe cell death caused by autophagy.Citation23,24 For example, insulin withdrawal causes autophagic cell death of adult hippocampal neural stem cells (HNSCs) in culture. Insulin-starved HNSCs accumulate markers of autophagy, including increases in lipidated LC3 and BECN1, and knockdown of ATG7 suppresses autophagy, reducing cell death. Conversely, activation of autophagy by rapamycin increases death of insulin-deficient cells. Furthermore, even though the HNSCs have an intact apoptotic pathway, there is no evidence of caspase activation. This suggests a causative role of autophagy in HNSC death after insulin withdrawal.Citation24 It is possible that there is a mutual regulation of autophagy and apoptosis. For example, there is some evidence showing that autophagy is required for apoptosis.Citation24 Many pro-apoptotic signals induce autophagy and apoptosis simultaneously, suggesting that cell survival is determined by the balance of protective autophagy versus the cellular accumulation of toxins, such as damaged mitochondria.Citation12,26 The activation of apoptosis might inhibit autophagy by impairing some essential autophagy proteins such as BECN1.Citation12,26 However, activation of the autophagic pathway beyond a certain threshold may be toxic due to a collapse of cellular functions and cellular atrophy. The activation of autophagy may thus have dual functions. On the one hand, autophagy is a beneficial process to cope with metabolic stresses and immunological challenges. On the other hand, sustained activation of autophagy may induce pathogenic conditions by impairing protein recycling processes and depleting critical cellular constituents, resulting in cell death.Citation27
Autophagic Protection Against Ethanol-Mediated Neuronal Damage
Since immature neurons are particularly sensitive to ethanol, a number of mechanisms have been suggested to explain ethanol-induced neurodegeneration in the developing brain. These potential mechanisms include interference with neurotrophic factor- and retinoid acid-mediated signaling, induction of oxidative stress, DNA damage and ER stress, increased proinflammatory proteins, disruption of the neurotransmitter system, and inhibition of translational regulation.Citation28-33 As mentioned above, autophagy can be activated by various types of cellular stress including deprivation of nutrients, hypoxia, oxidative stress, ER stress, DNA damage, and protein aggregates, as well as by the presence of intracellular pathogens.Citation16,34
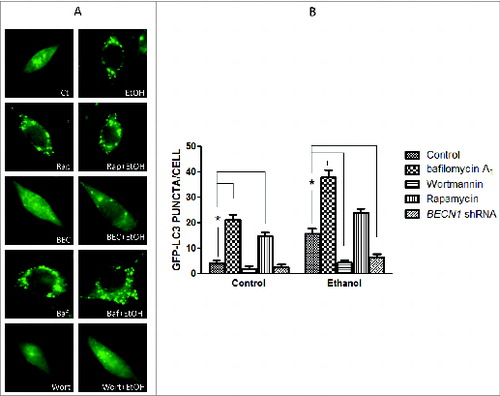
We have recently shown that ethanol activates autophagy in neurons in vitro and in the developing brain.Citation35 Autophagy involves the nucleation of the phagophore (the initial sequestering compartment), expansion and cargo sequestration, and subsequent maturation and degradation of autophagosomes. This dynamic flow is called “autophagic flux.”Citation36 LC3 lipidation or visualization of autophagosomes by electron microscopy has been frequently used to measure autophagic activity. However, an increase in LC3-II may be caused by either a stimulation of autophagosomal formation or inhibition of autophagosomal degradation, or may result from autophagy-independent mechanisms.Citation36 Therefore, both generation and degradation of autophagosomes need to be carefully evaluated when LC3-II is used to measure autophagy activity. Ethanol increases LC3 lipidation (), and reduces SQSTM1/p62 levels () in neuronal cells, which was accompanied by a significant increase of LC3 puncta (). At both 0.4% and 0.8%, ethanol is effective, but at 0.8% it produces a greater effect.Citation35 We therefore present the data using 0.8% to better illustrate the effect of ethanol, although we note that for in vitro studies, a concentration of 0.4% ethanol is considered physiologically relevant to heavy alcohol exposure in humans.Citation37 SQSTM1 levels are controlled by autophagy-dependent degradation. Bafilomycin A1, an inhibitor of the fusion of autophagosomes with the lysosome, prevents SQSTM1 degradation, further increasing the level of this protein (). Rapamycin, an activator of autophagy, enhances ethanol-mediated upregulation of LC3-lI Levels and the formation of LC3 puncta ( and ). In contrast, wortmannin, an inhibitor of initiation/maturation of autophagosomes, and shRNA for BECN1 block ethanol-increased LC3-II levels and LC3 puncta ( and ). Bafilomycin A1 alone also drastically increases LC3 puncta (). Taken together, these results indicate that ethanol enhances the development of autophagosomes rather than inhibiting autophagosome-lysosome fusion. Therefore, ethanol promotes autophagic flux in neurons.
Figure 1. Ethanol increases LC3 lipidation. SH-SY5Y neuroblastoma cells were treated with or without ethanol (0.8%) in the presence/absence of wortmannin (Wort: 10 μM), bafilomycin A1 (Baf: 10 nM), rapamycin (Rap: 10 nM) or BECN1 shRNA (BECN1). The protein samples were collected 8 h after the treatment. The levels of LC3 were examined with immunoblotting (top panel). Relative levels of LC3-II were quantified by densitometry and normalized to actin levels (bottom panel). * P < 0.05, **P < 0.01. (from ref. 35 with permission of the publisher).
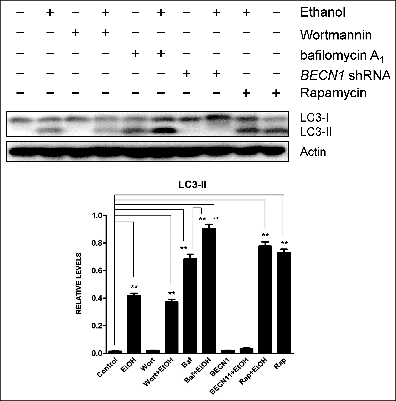
Figure 2. Ethanol decreases SQSTM1 levels. Notations are the same as in (from ref. 35 with permission of the publisher).
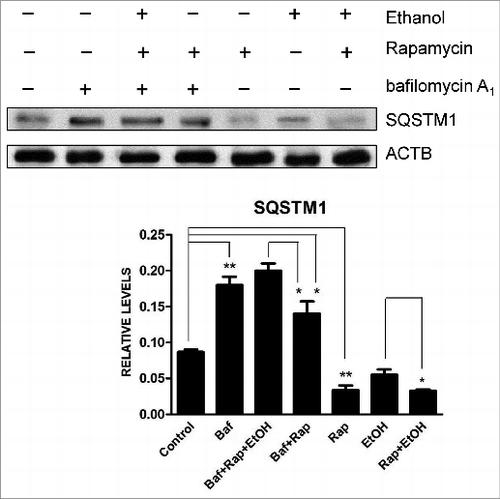
Figure 3. Ethanol increases LC3 puncta. (A) SH-SY5Y cells were transfected with a GFP-LC3 plasmid and exposed to ethanol (0.8%) in the presence/absence of bafilomycin A1 (Baf: 10 nM) or wortmannin (Wort: 10 μM) for 6 h. In some experimental groups, cells were cotransfected with BECN1 shRNA (BEC). The formation of GFP-LC3 puncta was examined under a fluorescence microscope 6 h after ethanol exposure. (B) GFP-LC3 puncta/cell was quantified. * P < 0.05. (from ref. 35 with permission of the publisher).
Ethanol causes damage to mitochondria resulting in the production of intracellular reactive oxygen species.Citation38-40 Because mitochondria are essential in regulating apoptosis, elimination of defective mitochondria by selective autophagy (mitophagy) is one of the mechanisms by which autophagy offers protection,Citation11,41 and ethanol increases mitophagy.Citation35 Ethanol-induced mitophagy is inhibited by BECN1 shRNA while it is increased by rapamycin. Rapamycin alleviated ethanol-mediated ROS accumulation and attenuates ethanol-induced neuroapoptosis in cultured neurons and in the developing brain, whereas wortmannin or knocking down BECN1 promotes ethanol-stimulated ROS accumulation and potentiated ethanol-induce neuroapoptosis. These results agree with a previous finding showing that in the context of liver cells, removal of defective mitochondria by autophagy is protective against mitochondrial release of pro-apoptotic factors and accumulation of toxic ROS.Citation42,43 Removing damaged mitochondria by autophagy not only promotes cell survival, but also may increase the threshold for the stress signals that are pro-apoptotic. Taken together, these findings indicate that neurons increase autophagic flux in response to ethanol neurotoxicity; enhanced autophagy is a protective mechanism that alleviates ethanol-mediated oxidative stress and neuronal death.
The protective role of autophagy against ethanol neurotoxicity is further supported by a study of hypoxic preconditioning. A modest hypoxic preconditioning (1% oxygen) can induce autophagy without affecting the viability of neuronal cells. This hypoxic preconditioning protects neuronal cells against ethanol-induced cell death.Citation44 Inhibition of autophagy blocks hypoxic preconditioning-mediated protection, whereas activation of autophagy enhances neuroprotection caused by hypoxic preconditioning.Citation41
During development, there is a temporal susceptibility to ethanol neurotoxicity. Using a mouse model of postnatal ethanol exposure, which is equivalent to human third trimester, it was found that ethanol treatment induces widespread neuroapoptosis in the brain of C57BL/6 mice at postnatal d 4 (PD4), but had little effect on PD12 mice.Citation45 We studied the expression pattern of genes involved in apoptosis, the pathways of the unfolded protein response (UPR) and autophagy in PD4 and PD12 mice. The expression of CASP3/caspase 3, a pro-apoptotic protein, is much higher in the ethanol-sensitive period, PD4, than in ethanol-resistant periods, corresponding to PD12; in contrast, the expression of genes related to UPR and autophagy are much higher on PD12 than PD4; these genes include Atg4, Atf6, Atg9, Atg10, Becn1, Bnip3, Cebpb, Ctsb, Ctsd, Ctss, Eif2ak3, Hspa5/Grp78, Ern1/Ire1α, Lamp1, Pik3c3, and Sqstm1.Citation42 The data suggest that the susceptibility of the developing nervous system to ethanol may be due to high expression of pro-apoptotic proteins and an immaturity of the stress mitigation systems such as UPR and autophagy.
Short-term ethanol exposure induces a protective response of upregulation of autophagy in the developing brain. However, chronic ethanol exposure in adult mice activates MTOR and causes impairment of the autophagy-lysosome pathway in the brain.Citation46 This implies that long-term exposure to ethanol may damage this protective system and further jeopardize CNS neurons to subsequent ethanol exposure and other insults. In turn, this finding suggests that upregulation of autophagy may be beneficial not only for the damage caused by acute ethanol exposure in the developing brain but also for deficits in the adult brain induced by chronic exposure.
Indeed, the activation of autophagy protects against ethanol-induced liver damage. Autophagy is essential for liver health, and induction of autophagy is viewed as a promising approach for treating liver injury.Citation47-49 Ethanol induces autophagy in liver cells; suppression of autophagy exacerbates alcohol-induced apoptosis of liver cells.Citation50,51 Conversely, pharmacological promotion of autophagy alleviates steatosis and liver injury caused by ethanol exposure.Citation52,53 Therefore, in both the liver and developing brain, autophagy is a protective mechanism against alcohol-induced cellular damage. However, in some model systems, the activation of autophagy seems detrimental. For example, autophagy seems to have a negative effect in ethanol-induced cardiac defects and apoptosis because the activation of autophagy by rapamycin and AICAR exacerbates ethanol-induced cell damage in cardiomyocytes.Citation54 Patients or animals with alcoholic cirrhosis and hepatitis have severe muscle loss due to the degeneration of skeletal muscle protein. Furthermore, the degeneration of skeletal muscle protein and loss of skeletal muscle mass is mediated by ethanol-induced autophagy.Citation55 These examples suggest that the activation of autophagy may also contribute to the adverse effect of ethanol exposure in some situations.
Mechanisms Underlying Ethanol Activation of Autophagy in Neurons
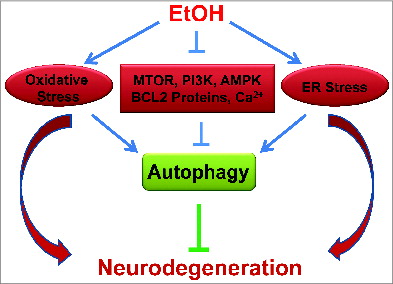
Many cellular mechanisms potentially contribute to ethanol-induced autophagy. Ethanol directly and indirectly induces many varied cellular consequences on factors such as ROS generation, MTOR and AMPK signaling, anti-apoptotic protein expression, and intracellular Ca2+ homeostasis (). Autophagy is a complex process that can be better understood by determining the altered roles of these various cellular factors. More specifically, through understanding and monitoring representative biological pathways the function of autophagy in the context of neurotoxicity can be better understood. These potential mechanisms are further addressed in the following section.
Figure 4. Autophagy as a potential protective response to ethanol neurotoxicity. Ethanol induces oxidative stress and ER stress in neurons of the developing brain, which results in neurodegeneration. Autophagy is activated in response to ethanol neurotoxicity. Several potential mechanisms contribute to ethanol-induced autophagy; these include the modulation of MTOR, PI3K-AKT, or AMPK signaling, alterations in BCL2 family proteins and disruption of intracellular calcium homeostasis. Ethanol-induced oxidative stress and ER stress may also trigger autophagy. There is considerable crosstalk among these signaling pathways during the activation of autophagy.
Oxidative stress
It is well established that the generation of reactive oxygen species, redox signaling and oxidative stress all stimulate autophagy.Citation56,57 ROS may mediate the balance between autophagy and apoptosis.Citation56 Free radicals regulate the activities of transcriptional factors, such as NFKB (nuclear factor of kappa light polypeptide gene enhancer in B-cells), TP53, and NFE2L2 (nuclear factor, erythroid 2-like 2), and affect various protein kinase cascades that are involved in the interaction between autophagy and apoptosis. Ethanol oxidation by ADH (alcohol dehydrogenase) and CYP2E1 (cytochrome P450, family 2, subfamily E, polypeptide1) produces ROS and causes oxidative stress in cells. In HepG2 liver cells, both ADH-generated acetaldehyde and CYP2E1-generated oxidants activates autophagy.Citation58
In neurons, ethanol-induced ROS mainly come from damaged mitochondria and the activation of NOX (NADPH oxidase).Citation38-40,59 NOX-derived production of ROS activates autophagy.Citation57 Antioxidants partially block ethanol-induced autophagic flux and mitophagy, suggesting that ROS is involved in ethanol activation of autophagy.Citation35
ER stress
The ER lumen is loaded with polypeptides at different stages of assembly. ER quality control systems monitor incorrect protein folding, stochastic errors, and off-pathway intermediates. As the accumulation of unfolded or misfolded proteins in the ER lumen overflows the capacity of the ER chaperone system, ER stress or UPR occurs. Induction of the UPR attempts to eliminate unfolded or misfolded proteins so that ER stress can be alleviated. The UPR is orchestrated by 3 ER transmembrane receptors, namely, EIF2AK3 (eukaryotic translation initiation factor 2-α kinase 3), ATF6 (activating transcription factor 6), and ERN1 (endoplasmic reticulum to nucleus signaling 1). In resting cells, all 3 UPR receptors are kept inactive through their association with an ER chaperone called HSPA5/Grp78 (heat shock 70 kDa protein 5 [glucose-regulated protein, 78 kDa]).
It is now known that ER stress can activate autophagy and there is considerable crosstalk between the UPR and autophagy.Citation52,61,62 There are multiple mechanisms underlying ER stress-induced autophagy. ER stress can activate autophagy by disrupting intracellular Ca2+ homeostasis or activating kinases and transcription factors that regulate autophagy; these include ERN1, MAPK/JNK, DDIT4/REDD1, AKT, DDIT3/CHOP, XBP1, DAPK1, and NFE2L1/NFE2L2.Citation52,61,62 Since ethanol induces ER stress in cultured neurons and in the brain,Citation31,33 it may trigger autophagy by inducing ER stress in neurons.
Class I PI3K and MTORC1
The pathways associated with the regulation of autophagy are very complex, involving more than 34 known proteins that assemble the autophagic machinery.Citation63 The best characterized regulator for autophagy is the mechanistic target of rapamycin complex 1 (MTORC1). MTORC1 is a protein complex that consists of a serine/threonine kinase, MTOR, a regulatory associated partner of MTOR (RPTOR) and MLST8 (MTOR associated protein, LST8 homolog [S. cerevisiae]). MTORC1 phosphorylates its substrates, EIF4EBP1 (eukaryotic translation initiation factor 4E binding protein 1) and the serine/threonine kinase RPS6KB/p70S6K (ribosomal protein S6 kinase, 70 kDa), and regulates their activity.Citation64 MTORC1 is regulated by the class I phosphoinositide 3-kinase (PI3K). PI3K phosphorylates AKT1 (v-akt murine thymoma viral oncogene homolog 1), which activates TSC1 and TSC2 (tuberous sclerosis 1/2). Other kinases, such as MAPK1/ERK2 (mitogen-activated protein kinase 1), MAPK3/ERK1, RSK1 (ribosomal protein S6 kinase, 90kDa, polypeptide 1), AMPK (adenosine 5’-monophosphate-activated protein kinase) and GSK3B (glycogen synthase kinase 3 β) also regulate the TSC1-TSC2 complex.Citation64 TSC1 and TSC2 form a functional complex with MTORC1 and negatively regulate its activity.Citation64 MTORC1 regulates autophagy through the modulation of specific proteins controlling the autophagic machinery; MTORC1 phosphorylates the mammalian homolog of yeast Atg13 (ATG13) and the mammalian Atg1 homologs ULK1/2 (unc-51 like autophagy activating kinase 1/2), inhibiting the activity of the ATG13-ULK1/2-RB1CC1/FIP200-ATG101 complex and preventing the activation of autophagy.Citation22,65 However, some evidence suggests that MTORC2 may also participate in the regulation of autophagy.Citation66
Ethanol inhibits the phosphorylation of EIF4EBP1 and RPS6KA, 2 main substrates of MTORC1, indicating that ethanol suppresses MTORC1 activity.Citation35 Rapamycin, a well-known inhibitor of MTORC1, and ethanol together produce greater dephosphorylation of RPS6KA and EIF4EBP1 than when cells are treated with either rapamycin or ethanol alone; this result is consistent with the finding that rapamycin potentiates ethanol-induced autophagy. Both rapamycin and the knockdown of MTOR by siRNA protect neurons against ethanol-induced apoptosis, further verifying the importance of the MTORC1 pathway in ethanol activation of autophagy and neurotoxicity.Citation35 Ethanol may regulate autophagy by MTORC1-dependent or -independent pathways. Further study is necessary to clearly establish whether ethanol-induced autophagy is mediated through the MTORC1 pathway; this can be achieved by determining whether a constitutive MTORC1 activation blocks ethanol-induced autophagy.
It has been previously shown that ethanol modulates MTORC1 activity in the brain.Citation67 Other evidence shows that ethanol suppresses MTOC1 activity in hepatocytes, skeletal muscles, and myocytes.Citation52,68-70 Ethanol-induced inhibition of MTORC1 activity in liver cells may be mediated by ROS.Citation52 However, chronic exposure to ethanol upregulates MTOR phosphorylation, leading to an inhibition of autophagic pathways in the brain.Citation46 The mechanisms of ethanol inhibition of MTORC1 are unclear. Ethanol induces ROS production and affects several upstream kinases of MTORC1, such as PI3K-AKT and GSK3B in neurons.Citation30,71-73 These potential mechanisms, particularly how ethanol-induced oxidative stress regulates MTORC1 signaling and autophagy, are worth further investigation.
AMPK
AMPK is a heterotrimeric protein kinase and is activated by alterations in the cellular AMP-to-ATP ratio. Upon activation, AMPK inhibits energy-requiring cellular activities and meanwhile stimulates catabolic pathways. AMPK plays a key role in cell survival during metabolic stress by maintaining metabolic homeostasis.Citation74 Recent studies indicate that AMPK plays a critical role in regulating autophagy in neurons.Citation75,76 AMPK is upstream of MTORC1 and the activation of AMPK inhibits MTORC1 activity through the phosphorylation of the MTORC1-associated proteins RPTOR and TSC2, thereby activating autophagy.Citation22 AMPK is activated by oxidative stress and also controls the redox-state and mitochondrial function.Citation74,77 AMPK is required for ROS-stimulated autophagy, which enhances cell survival in response to nutrient starvation, hypoxia, or ischemia.Citation56,77 Although it is commonly thought that activation of AMPK induces autophagy, some studies show that inhibition of AMPK stimulates autophagy.Citation78 Ethanol has been shown to modulate AMPK in neuronal cells as well as other cell types in vitro and in vivo.Citation74,79-83 Therefore, ethanol may activate autophagy by regulating AMPK activity in neurons.
In addition to inhibiting MTORC1 activity, AMPK regulates the activity of several transcription factors, such as CREB (cAMP response element binding protein), PPARG (peroxisome proliferator-activated receptor gamma), PPARA, and the FOXO (forkhead box O) family of transcription factors, as well as and NFE2L2.Citation84 Among these transcription factors, FOXO3 and NFE2L2 induce expression of genes regulating autophagy.Citation62,84 FOXO3 mediates ethanol-induced autophagy in primary hepatocytes and in the liver.Citation85 Overexpression of a dominant negative form of FOXO3 inhibits ethanol-induced autophagy-associated gene expression and enhances ethanol-induced cell death of hepatocytes. Similarly, the induction of FOXO3 nuclear localization activates autophagy and offers protection against ethanol-induced death of hepatocytes.Citation83 There is considerable interaction between ethanol and NFE2L2 in neuronal cells and other cell types.Citation86-89 Both FOXO3 and NFE2L2 are also regulated by oxidative stress.Citation62,90 Therefore, ethanol may also activate autophagy by modulating the activity of FOXO3 and NFE2L2.
BCL2 family proteins
It is well known that proteins of the BCL2 family play a critical role in the regulation of cell death/survival. Recent studies indicate that BCL2 family proteins have both a positive and negative regulatory role in autophagy.Citation91,92 Anti-apoptotic BCL2 family proteins, such as BCL2, BCL2L1/BCL-xL, and MCL1, inhibit autophagy by a direct association with BECN1. It seems that only the endoplasmic reticulum-BCL2 protein inhibits autophagy.Citation93,94 In addition to binding with BECN1, BCL2, and BCL2L1 also inhibit autophagy by modulating ER Ca2+ homeostasis, which plays an important role in the initiation of autophagy.Citation95,96 In contrast to anti-apoptotic BCL2 family proteins, pro-apoptotic BH3-only proteins, such as BNIP3, BNIP3L/NIX, BAD, BIK, PMAIP1/NOXA, BBC3/PUMA, and BCL2L11/BIM, generally induce autophagy through their binding to BCL2 and subsequent release of the inhibitory BCL2 from BECN1. However, the role of pro-apoptotic proteins BAX and BAK1 in autophagy is controversial. Both positive and negative regulation of autophagy by BAX and BAK1 have been reported.Citation52
Ethanol modulates BCL2 family proteins in neurons in vitro and in vivo.Citation73,97-99 Generally, ethanol inhibits the expression of anti-apoptotic BCL2 family proteins, but increases the expression of pro-apoptotic proteins, which may result in the activation of autophagy.
Calcium signaling
Intracellular Ca2+ is a ubiquitous second messenger regulating diverse cellular and physiological processes such as differentiation, survival, development, proliferation, muscle contraction, and secretion. Alterations in intracellular Ca2+ homeostasis could result in neuroapoptosis in the CNS. Homeostasis of intracellular Ca2+ is achieved in part by the buffering role of mitochondria and the ER. Recent studies indicate that intracellular Ca2+ signaling is involved in autophagy.Citation100,101 Although it has been suggested that autophagy may be activated by an increase in cytosolic Ca2+ concentration or a perturbed release from intra- or extracellular stores,Citation100 more compelling evidence has indicated that an increase in intracellular Ca2+ inhibits autophagic flux.Citation102-104 Both mitochondria and the ER play a critical role in Ca2+ homeostasis and regulation of autophagy.Citation96,100 Ethanol can damage mitochondria and cause ER stress, which may disrupt Ca2+ homeostasis.Citation31,38,39,105 Ethanol also disturbs intracellular Ca2+ homeostasis in neurons in vitro and in vivo.Citation106,107 Therefore, it is possible that ethanol may regulate autophagy by disrupting intracellular Ca2+ homeostasis in neurons.
Autophagy as a Potential Therapeutic Target for Treating Ethanol Neurotoxicity
Since the activation of autophagy appears protective against ethanol neurotoxicity, pharmacological manipulation of autophagy should be considered a method of potential therapeutic approaches. The most important and well-studied drug identified as an autophagy inducer is rapamycin, an MTORC1 inhibitor. Rapamycin is a FDA-approved drug and a widely used immune suppressor in clinical transplant patients. Rapamycin and its analogs (CCI-779, RAD001 and AP23573) have been used to activate autophagy and offer protection in a number of neurodegenerative models.Citation22,108 We have recently shown that rapamycin protects neurons against thiamine deficiency-induced neurodegenerationCitation109; in vitro studies indicate that rapamycin alleviates ethanol-induced death of neuronal cells by activating autophagy.Citation35,44 Similarly, rapamycin mitigates ethanol-induced liver pathogenesis.Citation52 In an animal model of ethanol-induced cardiac defect (in which autophagy plays a negative role), however, rapamycin exacerbates ethanol-induced cell injury in cardiomyocytes.Citation97 In addition to directly targeting MTOR, activation of autophagy by manipulating other pathways upstream of MTORC1, such as AMPK, PI3K-AKT, GSK3B, or TSC1/2 are alternative approaches.
Although rapamycin has been used clinically for immune suppression, it is unclear how it will affect the human CNS. In addition to the induction of autophagy, rapamycin may have potential side effects because MTORC1 signaling affects multiple pathways, such as protein translation, cell growth, and metabolism.Citation110,111 Chronic stimulation of autophagy may impose additional risks, because sustained autophagy may result in depletion of healthy mitochondria, causing neuronal death. The discovery of MTOR-independent autophagy enhancers may thus provide an alternative method for stimulating this process. A number of studies of chemical screening have identified chemicals and small molecule enhancers that regulate autophagy independent of MTOR, including some FDA-approved drugs.Citation22 It is worthwhile to test these chemicals for their induction of autophagy in the CNS and potential neuroprotection against ethanol neurotoxicity. For example, various mood-stabilizing drugs such as lithium, carbamazepine, L-690,330, and sodium valproate, could activate autophagy through the suppression of IMPA (inositol monophosphatase).Citation63,109 Lithium-induced autophagy seems to mediate its protective function in the neurodegeneration see in the PD model.Citation112,113 In fact, lithium alleviates ethanol-induced neuroapoptosis in cultured neurons and developing brain.Citation7,32,73 Although these drugs may be beneficial by activating autophagy in the brain, the potential teratogenic effects need to be carefully evaluated. GSK3B is an important regulator of brain development and these drugs alter the activity of this kinase.Citation114
A potentially more effective approach to induce autophagy is the application of a cocktail of autophagy activators. Combination therapy for enhancing autophagy with lower concentrations of each chemical inducer may be safer for chronic drug administration and may lessen side effects. Additive effects in enhancing autophagy can be achieved through simultaneous stimulation by MTOR-dependent and MTOR-independent routes. For example, combined treatment with lithium and rapamycin causes greater induction of autophagy compared to the effects of each individual compound.Citation22 Similar additive effects for activating autophagy have been shown by using rapamycin with other MTOR-independent autophagy enhancers, such as trehalose, calpastatin, or small molecule enhancers.Citation22
Future Directions
Several critical questions remain regarding the role of autophagy in ethanol neurotoxicity. These include: 1) During brain development, there is a spatiotemporal susceptibility to ethanol neurotoxicity. Is there a correlation between this selective vulnerability and the status of autophagy activation? What are the factors that mediate the region and cell type specificity of autophagic activation in the brain? 2) What are the primary molecular targets of ethanol that initiate autophagy? Are these targets region-specific and developmentally regulated in the brain? 3) Recent attempts targeting autophagy in the CNS bring tremendous excitement for the development of novel therapeutic approaches. What is the most effective paradigm of manipulation of autophagy in the brain? How can the potential for detrimental side effects be limited? What combination of autophagy inducers will be most effective? Taken together, the studies carried out to date suggest that autophagy will undoubtedly be an attractive target for future therapeutic efforts. A better understanding of neuronal autophagy shows promise in providing avenues for future therapy against ethanol neurotoxicity.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgment
I would like to thank Ashley Comer and Jacqueline Frank for reading this manuscript.
Funding
This research is supported by grants from National Institutes of Health (NIH) (AA019693 and AA015407).
References
- Sampson PD, Streissguth AP, Bookstein FL, Little RE, Clarren SK, Dehaene P, Hanson JW, Graham JM Jr. Incidence of fetal alcohol syndrome and prevalence of alcohol-related neurodevelopmental disorder. Teratology 1997; 56(5):317-26; PMID:9451756; http://dx.doi.org/10.1002/(SICI)1096-9926(199711)56:5%3c317::AID-TERA5%3e3.0.CO;2-U
- Sampson PD, Streissguth AP, Bookstein FL, Barr HM. On categorizations in analyses of alcohol teratogenesis. Environ Health Perspect 2000; 108 Suppl 3:421-8; PMID:10852839; http://dx.doi.org/10.1289/ehp.00108s3421
- May PA, Gossage JP, Kalberg WO, Robinson LK, Buckley D, Manning M, Hoyme HE. Prevalence and epidemiologic characteristics of FASD from various research methods with an emphasis on recent in-school studies. Dev Disabil Res Rev 2009; 15(3):176-92; PMID:19731384; http://dx.doi.org/10.1002/ddrr.68
- May PA, Blankenship J, Marais AS, Gossage JP, Kalberg WO, Joubert B, Cloete M, Barnard R, De Vries M, Hasken J, et al. Maternal alcohol consumption producing fetal alcohol spectrum disorders (FASD): quantity, frequency, and timing of drinking. Drug Alcohol Depend 2013; 133(2):502-12; PMID:23932841; http://dx.doi.org/10.1016/j.drugalcdep.2013.07.013
- Riley EP, Infante MA, Warren KR. Fetal alcohol spectrum disorders: an overview. Neuropsychol Rev 2011; 21(2):73-80; PMID:21499711; http://dx.doi.org/10.1007/s11065-011-9166-x
- Lupton C, Burd L, Harwood R. Cost of fetal alcohol spectrum disorders. Am J Med Genet C Semin Med Genet 2004; 127C(1):42-50; PMID:15095471; http://dx.doi.org/10.1002/ajmg.c.30015
- Luo J. GSK3beta in ethanol neurotoxicity. Mol Neurobiol 2009; 40(2):108-21; PMID:19507062; http://dx.doi.org/10.1007/s12035-009-8075-y
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132(1):27-42; PMID:18191218; http://dx.doi.org/10.1016/j.cell.2007.12.018
- Wirawan E, Vanden Berghe T, Lippens S, Agostinis P, Vandenabeele P. Autophagy: for better or for worse. Cell Res 2012; 22(1):43-61; PMID:21912435; http://dx.doi.org/10.1038/cr.2011.152
- Lu H, Li G, Liu L, Feng L, Wang X, Jin H. Regulation and function of mitophagy in development and cancer. Autophagy 2013; 9(11):1720-36; PMID:24091872; http://dx.doi.org/10.4161/auto.26550
- Kim I, Lemasters JJ. Mitochondrial degradation by autophagy (mitophagy) in GFP-LC3 transgenic hepatocytes during nutrient deprivation. Am J Physiol Cell Physiol 2011; 300(2):C308-17; PMID:21106691; http://dx.doi.org/10.1152/ajpcell.00056.2010
- Lee JA. Neuronal autophagy: a housekeeper or a fighter in neuronal cell survival? Exp Neurobiol 2012; 21(1):1-8; PMID:22438673; http://dx.doi.org/10.5607/en.2012.21.1.1
- Shen HM, Codogno P. Autophagy is a survival force via suppression of necrotic cell death. Exp Cell Res 2012; 318(11):1304-8; PMID:22366289; http://dx.doi.org/10.1016/j.yexcr.2012.02.006
- Loos B, Engelbrecht AM, Lockshin RA, Klionsky DJ, Zakeri Z. The variability of autophagy and cell death susceptibility: Unanswered questions. Autophagy 2013; 9(9):1270-85; PMID:23846383; http://dx.doi.org/10.4161/auto.25560
- Sridhar S, Botbol Y, Macian F, Cuervo AM. Autophagy and disease: always two sides to a problem. J Pathol 2012; 226(2):255-73; PMID:21990109; http://dx.doi.org/10.1002/path.3025
- Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res 2014; 24(1):24-41; PMID:24366339; http://dx.doi.org/10.1038/cr.2013.168
- Xilouri M, Stefanis L. Autophagy in the central nervous system: implications for neurodegenerative disorders. CNS Neurol Disord Drug Targets 2010; 9(6):701-19; PMID:20942791; http://dx.doi.org/10.2174/187152710793237421
- Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol 2010; 221(1):3-12; PMID:20225336; http://dx.doi.org/10.1002/path.2697
- Rodríguez-Muela N, Boya P. Axonal damage, autophagy and neuronal survival. Autophagy 2012; 8(2):286-8; http://dx.doi.org/10.4161/auto.8.2.18982
- Giordano S, Darley-Usmar V, Zhang J. Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox Biol 2014; 2:82-90; PMID:24494187; http://dx.doi.org/10.1016/j.redox.2013.12.013
- Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest 2008 118(6):2190-9; PMID:18497889
- Sarkar S. Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: autophagydysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem Soc Trans 2013; 41(5):1103-30; PMID:24059496; http://dx.doi.org/10.1042/BST20130134
- Galluzzi L, Vicencio JM, Kepp O, Tasdemir E, Maiuri MC, Kroemer G. To die or not to die: that is the autophagic question. Curr Mol Med 2008; 8(2):78-91; PMID:18336289; http://dx.doi.org/10.2174/156652408783769616
- Denton D, Nicolson S, Kumar S. Cell death by autophagy: facts and apparent artefacts. Cell Death Differ 2012; 19(1):87-95; PMID:22052193; http://dx.doi.org/10.1038/cdd.2011.146
- Puyal J, Ginet V, Grishchuk Y, Truttmann AC, Clarke PG. Neuronal autophagy as a mediator of life and death: contrasting roles in chronic neurodegenerative and acute neural disorders. Neuroscientist 2012; 18(3):224-3; PMID:21525331; http://dx.doi.org/10.1177/1073858411404948
- Luo S, Rubinsztein DC. BCL2L11/BIM: a novel molecular link between autophagy and apoptosis. Autophagy 2013; 9(1):104-5; PMID:23064249; http://dx.doi.org/10.4161/auto.22399
- Liang C. Negative regulation of autophagy. Cell Death Differ 2010; 17(12):1807-15; PMID:20865012; http://dx.doi.org/10.1038/cdd.2010.115
- Cherian PP, Schenker S, Henderson GI. Ethanol-mediated DNA damage and PARP-1 apoptotic responses in cultured fetal cortical neurons. Alcohol Clin Exp Res 2008; 32(11):1884-92; PMID:18717656
- Crews FT, Nixon K. Mechanisms of neurodegeneration and regeneration in alcoholism. Alcohol Alcohol 2009; 44(2):115-27; PMID:18940959; http://dx.doi.org/10.1093/alcalc/agn079
- Brocardo PS, Gil-Mohapel J, Christie BR. The role of oxidative stress in fetal alcohol spectrum disorders. Brain Res Rev 2011; 67(1-2):209-25; PMID:21315761; http://dx.doi.org/10.1016/j.brainresrev.2011.02.001
- Ke Z, Wang X, Liu Y, Fan Z, Chen G, Xu M, Bower KA, Frank JA, Li M, Fang S, Shi X, Luo J. Ethanol induces endoplasmic reticulum stress in the developing brain. Alcohol Clin Exp Res 2011; 35(9):1574-83; PMID:21599712
- Luo J. Lithium-mediated protection against ethanol neurotoxicity. Front Neurosci 2010; 4:41; PMID:20661453
- Luo J. Mechanisms of ethanol-induced death of cerebellar granule cells. Cerebellum 2012; 11(1):145-54; PMID:20927663; http://dx.doi.org/10.1007/s12311-010-0219-0
- Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40(2):280-93; PMID:20965422; http://dx.doi.org/10.1016/j.molcel.2010.09.023
- Chen G, Ke Z, Xu M, Liao M, Wang X, Qi Y, Zhang T, Frank JA, Bower KA, Shi X, Luo J. Autophagy is a protective response to ethanol neurotoxicity. Autophagy 2012; 8(11):1577-89; PMID:22874567; http://dx.doi.org/10.4161/auto.21376
- Matus S, Valenzuela V, Hetz C. A new method to measure autophagy flux in the nervous system. Autophagy 2014; 10(4):710-714; PMID:24717689; http://dx.doi.org/10.4161/auto.28434
- Luo J, Lindstrom CL, Donahue A, Miller MW. Differential effects of ethanol on the expression of cyclo-oxygenase in cultured cortical astrocytes and neurons. J. Neurochem 2001; 76:1354-1363; PMID:11238720; http://dx.doi.org/10.1046/j.1471-4159.2001.00129.x
- Ramachandran V, Watts LT, Maffi SK, Chen J, Schenker S, Henderson G. Ethanol-induced oxidative stress precedes mitochondrially mediated apoptotic death of cultured fetal cortical neurons. J Neurosci Res 2003; 74(4):577-88; PMID:14598302; http://dx.doi.org/10.1002/jnr.10767
- Haorah J, Ramirez SH, Floreani N, Gorantla S, Morsey B, Persidsky Y. Mechanism of alcohol-induced oxidative stress and neuronal injury. Free Radic Biol Med 2008; 45(11):1542-50; PMID:18845238; http://dx.doi.org/10.1016/j.freeradbiomed.2008.08.030
- Wang X, Ke Z, Chen G, Xu M, Bower KA, Frank JA, Zhang Z, Shi X, Luo J. Cdc42 dependent activation of NADPH oxidase is involved in ethanol-induced neuronal oxidative stress. PLoS One 2012; 7(5):e38075; PMID:22662267; http://dx.doi.org/10.1371/journal.pone.0038075
- Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys 2007; 462(2):245-53; PMID:17475204; http://dx.doi.org/10.1016/j.abb.2007.03.034
- Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 2005; 8(1):3-5; PMID:15798367; http://dx.doi.org/10.1089/rej.2005.8.3
- Mizushima N. Autophagy: process and function. Genes Dev 2007; 21(22):2861-73; PMID:18006683; http://dx.doi.org/10.1101/gad.1599207
- Wang H, Bower KA, Frank JA, Xu M, Luo J. Hypoxic preconditioning alleviates ethanol neurotoxicity: the involvement of autophagy. Neurotox Res 2013; 24(4):472-7; PMID:23568540; http://dx.doi.org/10.1007/s12640-013-9390-7
- Alimov A, Wang H, Liu M, Frank JA, Xu M, Ou X, Luo J. Expression of autophagy and UPR genes in the developing brain during ethanol-sensitive and resistant periods. Metab Brain Dis 2013; 28(4):667-76; PMID:23979425; http://dx.doi.org/10.1007/s11011-013-9430-2
- Pla A, Pascual M, Renau-Piqueras J, Guerri C. TLR4 mediates the impairment of ubiquitin-proteasome and autophagy-lysosome pathways induced by ethanol treatment in brain. Cell Death Dis 2014; PMID:24556681; http://dx.doi.org/10.1038/cddis.2014.46
- Komatsu M. Liver autophagy: physiology and pathology. J Biochem 2012; 152(1):5-15; PMID:22661765; http://dx.doi.org/10.1093/jb/mvs059
- Czaja MJ, Ding WX, Donohue TM Jr, Friedman SL, Kim JS, Komatsu M, Lemasters JJ, Lemoine A, Lin JD, Ou JH, et al. Functions of autophagy in normal and diseased liver. Autophagy 2013; 9(8):1131-58; PMID:23774882; http://dx.doi.org/10.4161/auto.25063
- Ding WX. Induction of autophagy, a promising approach for treating liver injury. Hepatology 2014; 59(1):340-3; PMID:23775596; http://dx.doi.org/10.1002/hep.26572
- Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, Lu B, Stolz DB, Clemens DL, Yin XM. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology 2010; 139(5):1740-52; PMID:20659474; http://dx.doi.org/10.1053/j.gastro.2010.07.041
- Dolganiuc A, Thomes PG, Ding WX, Lemasters JJ, Donohue TM Jr. Autophagy in alcohol-induced liver diseases. Alcohol Clin Exp Res 2012; 36(8):1301-8; PMID:22551004; http://dx.doi.org/10.1111/j.1530-0277.2012.01742.x
- Ding WX, Manley S, Ni HM. The emerging role of autophagy in alcoholic liver disease. Exp Biol Med (Maywood) 2011; 236(5):546-56; PMID:21478210
- Lin CW, Zhang H, Li M, Xiong X, Chen X, Chen X, Dong XC, Yin XM. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol 2013; 58(5):993-9; PMID:23339953; http://dx.doi.org/10.1016/j.jhep.2013.01.011
- Ge W, Guo R, Ren J. AMP-dependent kinase and autophagic flux are involved in aldehyde dehydrogenase-2-induced protection against cardiac toxicity of ethanol. Free Radic Biol Med 2011; 51(9):1736-48; PMID:21871561; http://dx.doi.org/10.1016/j.freeradbiomed.2011.08.002
- Thapaliya S, Runkana A, McMullen MR, Nagy LE, McDonald C, Naga Prasad SV, Dasarathy S. Alcohol-induced autophagy contributes to loss in skeletal muscle mass. Autophagy 2014; 10(4):677-90; PMID:24492484; http://dx.doi.org/10.4161/auto.27918
- Kaminskyy VO, Zhivotovsky B. Free Radicals in Cross Talk Between Autophagy and Apoptosis. Antioxid Redox Signal 2014; 21(1):86-102; PMID:24359220; http://dx.doi.org/10.1089/ars.2013.5746
- Navarro-Yepes J, Burns M, Anandhan A, Khalimonchuk O, Del Razo LM, Quintanilla-Vega B, Pappa A, Panayiotidis MI, Franco R. Oxidative Stress, Redox Signaling, and Autophagy: Cell Death Versus Survival. Antioxid Redox Signal 2014; 21(1):66-85; PMID:24483238; http://dx.doi.org/10.1089/ars.2014.5837
- Thomes PG, Ehlers RA, Trambly CS, Clemens DL, Fox HS, Tuma DJ, Donohue TM. Multilevel regulation of autophagosome content by ethanol oxidation in HepG2 cells. Autophagy 2013; 9(1):63-73; PMID:23090141; http://dx.doi.org/10.4161/auto.22490
- Chen G, Luo J. Anthocyanins: are they beneficial in treating ethanol neurotoxicity? Neurotox Res 2010; 17(1):91-101; PMID:19590929; http://dx.doi.org/10.1007/s12640-009-9083-4
- Sciarretta S, Volpe M, Sadoshima J. NOX4 regulates autophagy during energy deprivation. Autophagy 2014; 10(4):699-701; PMID:24492492; http://dx.doi.org/10.4161/auto.27955
- Deegan S, Saveljeva S, Gorman AM, Samali A. Stress-induced self-cannibalism: on the regulation of autophagy by endoplasmic reticulum stress. Cell Mol Life Sci 2013; 70(14):2425-41; PMID:23052213; http://dx.doi.org/10.1007/s00018-012-1173-4
- Digaleh H, Kiaei M, Khodagholi F. Nrf2 and Nrf1 signaling and ER stress crosstalk: implication for proteasomal degradation and autophagy. Cell Mol Life Sci 2013; 70(24):4681-94; PMID:23800989; http://dx.doi.org/10.1007/s00018-013-1409-y
- Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem 2011; 80:125-56; PMID:21548784; http://dx.doi.org/10.1146/annurev-biochem-052709-094552
- Maiese K, Chong ZZ, Shang YC, Wang S. mTOR: on target for novel therapeutic strategies in the nervous system. Trends Mol Med 2013; 19(1):51-60; PMID:23265840; http://dx.doi.org/10.1016/j.molmed.2012.11.001
- Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009; 20(7):1992-2003; PMID:19225151; http://dx.doi.org/10.1091/mbc.E08-12-1249
- Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 2007; 6(6):458-71; PMID:18054315; http://dx.doi.org/10.1016/j.cmet.2007.11.001
- Gutala R, Wang J, Kadapakkam S, Hwang Y, Ticku M, Li MD. Microarray analysis of ethanol-treated cortical neurons reveals disruption of genes related to the ubiquitin-proteasome pathway and protein synthesis. Alcohol Clin Exp Res 2004; 28(12):1779-88; PMID:15608593; http://dx.doi.org/10.1097/01.ALC.0000148117.17707.B4
- Lang CH, Frost RA, Deshpande N, Kumar V, Vary TC, Jefferson LS, Kimball SR. Alcohol impairs leucine-mediated phosphorylation of 4E-BP1, S6K1, eIF4G, and mTOR in skeletal muscle. Am J Physiol Endocrinol Metab 2003; 285(6):E1205-15; PMID:12944322
- Vary TC, Deiter G, Goodman SA. Acute alcohol intoxication enhances myocardial eIF4G phosphorylation despite reducing mTOR signaling. Am J Physiol Heart Circ Physiol 2005; 288(1):H121-8; PMID:15388509; http://dx.doi.org/10.1152/ajpheart.00440.2004
- Hong-Brown LQ, Brown CR, Huber DS, Lang CH. Alcohol and indinavir adversely affect protein synthesis and phosphorylation of MAPK and mTOR signaling pathways in C2C12 myocytes. Alcohol Clin Exp Res 2006; 30(8):1297-307; PMID:16899032; http://dx.doi.org/10.1111/j.1530-0277.2006.00157.x
- Li Z, Ding M, Thiele CJ, Luo J. Ethanol inhibits brain-derived neurotrophic factor-mediated intracellular signaling and activator protein-1 activation in cerebellar granule neurons. Neuroscience 2004; 126(1):149-62; PMID:15145081; http://dx.doi.org/10.1016/j.neuroscience.2004.03.028
- Cozzoli DK, Goulding SP, Zhang PW, Xiao B, Hu JH, Ary AW, Obara I, Rahn A, Abou-Ziab H, Tyrrel B, etal. Binge drinking upregulates accumbens mGluR5-Homer2-PI3K signaling: functional implications for alcoholism. J Neurosci 2009; 29(27):8655-68; PMID:19587272; http://dx.doi.org/10.1523/JNEUROSCI.5900-08.2009
- Liu Y, Chen G, Ma C, Bower KA, Xu M, Fan Z, Shi X, Ke ZJ, Luo J. Overexpression of glycogen synthase kinase 3beta sensitizes neuronal cells to ethanol toxicity. J Neurosci Res 2009; 87(12):2793-802; PMID:19382207; http://dx.doi.org/10.1002/jnr.22098
- Sid B, Verrax J, Calderon PB. Role of AMPK activation in oxidative cell damage: Implications for alcohol-induced liver disease. Biochem Pharmacol 2013; 86(2):200-9; PMID:23688501; http://dx.doi.org/10.1016/j.bcp.2013.05.007
- Meijer AJ, Codogno P. AMP-activated protein kinase and autophagy. Autophagy 2007; 3(3):238-40; PMID:17224623; http://dx.doi.org/10.4161/auto.3710
- Poels J, Spasić MR, Callaerts P, Norga KK. Expanding roles for AMP-activated protein kinase in neuronal survival and autophagy. Bioessays 2009; 31(9):944-52; PMID:19644919; http://dx.doi.org/10.1002/bies.200900003
- Huang Q, Wu YT, Tan HL, Ong CN, Shen HM. A novel function of poly(ADP-ribose) polymerase-1 in modulation of autophagy and necrosis under oxidative stress. Cell Death Differ 2009; 16(2):264-77; PMID:18974775; http://dx.doi.org/10.1038/cdd.2008.151
- Vucicevic L, Misirkic M, Janjetovic K, Vilimanovich U, Sudar E, Isenovic E, Prica M, Harhaji-Trajkovic L, Kravic-Stevovic T, Bumbasirevic V, et al. Compound C induces protective autophagy in cancer cells through AMPK inhibition-independent blockade of Akt/mTOR pathway. Autophagy 2011; 7(1):40-50; PMID:20980833; http://dx.doi.org/10.4161/auto.7.1.13883
- You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW. The role of AMP activated protein kinase in the action of ethanol in the liver. Gastroenterology 2004; 127(6):1798-808; PMID:15578517; http://dx.doi.org/10.1053/j.gastro.2004.09.049
- Saito M, Chakraborty G, Mao RF, Wang R, Cooper TB, Vadasz C, Saito M. Ethanol alters lipid profiles and phosphorylation status of AMP-activated protein kinase in the neonatal mouse brain. J Neurochem 2007; 103(3):1208-18; PMID:17683484; http://dx.doi.org/10.1111/j.1471-4159.2007.04836.x
- Noh BK, Lee JK, Jun HJ, Lee JH, Jia Y, Hoang MH, Kim JW, Park KH, Lee SJ. Restoration of autophagy by puerarin in ethanol-treated hepatocytes via the activation of AMP-activated protein kinase. Biochem Biophys Res Commun 2011; 414(2):361-6; PMID:21964292; http://dx.doi.org/10.1016/j.bbrc.2011.09.077
- Guo R, Ren J. Deficiency in AMPK attenuates ethanol-induced cardiac contractile dysfunction through inhibition of autophagosome formation. Cardiovasc Res 2012; 94(3):480-91; PMID:22451512; http://dx.doi.org/10.1093/cvr/cvs127
- Nepal S, Park PH. Activation of autophagy by globular adiponectin attenuates ethanol-induced apoptosis in HepG2 cells: involvement of AMPK/FoxO3A axis. Biochim Biophys Acta 2013; 1833(10):2111-25; PMID:23688633; http://dx.doi.org/10.1016/j.bbamcr.2013.05.013
- Cantó C, Auwerx J. AMP-activated protein kinase and its downstream transcriptional pathways. Cell Mol Life Sci 2010; 67(20):3407-23; http://dx.doi.org/10.1007/s00018-010-0454-z
- Ni HM, Du K, You M, Ding WX. Critical role of FoxO3a in alcohol-induced autophagy and hepatotoxicity. Am J Pathol 2013; 183(6):1815-25; PMID:24095927; http://dx.doi.org/10.1016/j.ajpath.2013.08.011
- Wu KC, Liu J, Klaassen CD. Role of Nrf2 in preventing ethanol-induced oxidative stress and lipid accumulation. Toxicol Appl Pharmacol 2012; 262(3):321-9; PMID:22627062; http://dx.doi.org/10.1016/j.taap.2012.05.010
- Lu Y, Zhang XH, Cederbaum AI. Ethanol induction of CYP2A5: role of CYP2E1-ROS-Nrf2 pathway. Toxicol Sci 2012; 128(2):427-38; PMID:22552773; http://dx.doi.org/10.1093/toxsci/kfs164
- Jin M, Kumar A, Kumar S. Ethanol-mediated regulation of cytochrome P450 2A6 expression in monocytes: role of oxidative stress-mediated PKC/MEK/Nrf2 pathway. PLoS One 2012; 7(4):e35505; PMID:22530035; http://dx.doi.org/10.1371/journal.pone.0035505
- Chen X, Liu J, Chen SY. Over-expression of Nrf2 diminishes ethanol-induced oxidative stress and apoptosis in neural crest cells by inducing an antioxidant response. Reprod Toxicol 2013; 42:102-9; PMID:23994065; http://dx.doi.org/10.1016/j.reprotox.2013.08.003
- Storz P. Forkhead homeobox type O transcription factors in the responses to oxidative stress. Antioxid Redox Signal 2011; 14(4):593-605; PMID:20618067; http://dx.doi.org/10.1089/ars.2010.3405
- Decuypere JP, Parys JB, Bultynck G. Regulation of the autophagic bcl-2/beclin 1 interaction. Cells 2012; 1(3):284-312; PMID:24710477; http://dx.doi.org/10.3390/cells1030284
- Hardwick JM, Soane L. Multiple functions of BCL-2 family proteins. Cold Spring Harb Perspect Biol 2013; 5(2); PMID:23378584; http://dx.doi.org/10.1101/cshperspect.a008722
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122(6):927-39; PMID:16179260; http://dx.doi.org/10.1016/j.cell.2005.07.002
- Chang NC, Nguyen M, Germain M, Shore GC. Antagonism of Beclin 1-dependent autophagy by BCL-2 at the endoplasmic reticulum requires NAF-1. EMBO J 2010; 29(3):606-18; PMID:20010695; http://dx.doi.org/10.1038/emboj.2009.369
- Brady NR, Hamacher-Brady A, Yuan H, Gottlieb RA. The autophagic response to nutrient deprivation in the hl-1 cardiac myocyte is modulated by Bcl-2 and sarco/endoplasmic reticulum calcium stores. FEBS J 2007; 274(12):3184-97; PMID:17540004; http://dx.doi.org/10.1111/j.1742-4658.2007.05849.x
- Høyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell 2007; 25(2):193-205; PMID:17244528; http://dx.doi.org/10.1016/j.molcel.2006.12.009
- Ge Y, Belcher SM, Pierce DR, Light KE. Altered expression of Bcl2, Bad and Bax mRNA occurs in the rat cerebellum within hours after ethanol exposure on postnatal day 4 but not on postnatal day 9. Brain Res Mol Brain Res 2004; 129(1-2):124-34; PMID:15469889; http://dx.doi.org/10.1016/j.molbrainres.2004.06.034
- Heaton MB, Paiva M, Siler-Marsiglio K. Ethanol influences on Bax translocation, mitochondrial membrane potential, and reactive oxygen species generation are modulated by vitamin E and brain-derived neurotrophic factor. Alcohol Clin Exp Res 2011; 35(6):1122-33; PMID:21332533; http://dx.doi.org/10.1111/j.1530-0277.2011.01445.x
- Ali Shah S, Ullah I, Lee HY, Kim MO. Anthocyanins protect against ethanol-induced neuronal apoptosis via GABAB1 receptors intracellular signaling in prenatal rat hippocampal neurons. Mol Neurobiol 2013; 48(1):257-69; PMID:23645118; http://dx.doi.org/10.1007/s12035-013-8458-y
- East DA, Campanella M. Ca2+ in quality control: an unresolved riddle critical to autophagy and mitophagy. Autophagy 2013; 9(11):1710-9; PMID:24121708; http://dx.doi.org/10.4161/auto.25367
- Ureshino RP, Rocha KK, Lopes GS, Bincoletto C, Smaili SS. Calcium Signaling Alterations, Oxidative Stress, and Autophagy in Aging. Antioxid Redox Signal 2014; 21(1):123-37; PMID:24512092; http://dx.doi.org/10.1089/ars.2013.5777
- Gordon PB, Holen I, Fosse M, Røtnes JS, Seglen PO. Dependence of hepatocytic autophagy on intracellularly sequestered calcium. J Biol Chem 1993; 268(35):26107-12; PMID:8253727
- Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, Jahreiss L, Fleming A, Pask D, Goldsmith P, et al. Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol 2008; 4(5):295-305; PMID:18391949; http://dx.doi.org/10.1038/nchembio.79
- Ganley IG, Wong PM, Gammoh N, Jiang X. Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol Cell 2011; 42(6):731-43; PMID:21700220; http://dx.doi.org/10.1016/j.molcel.2011.04.024
- Kaplowitz N, Ji C. Unfolding new mechanisms of alcoholic liver disease in the endoplasmic reticulum. J Gastroenterol Hepatol 2006; Suppl 3:S7-9; http://dx.doi.org/10.1111/j.1440-1746.2006.04581.x
- Webb B, Walker DW, Heaton MB. Nerve growth factor and chronic ethanol treatment alter calcium homeostasis in developing rat septal neurons. Brain Res Dev Brain Res 2003; 143(1):57-71; PMID:12763581; http://dx.doi.org/10.1016/S0165-3806(03)00100-7
- Kouzoukas DE, Li G, Takapoo M, Moninger T, Bhalla RC, Pantazis NJ. Intracellular calcium plays a critical role in the alcohol-mediated death of cerebellar granule neurons. J Neurochem 2013; 124(3):323-35; PMID:23121601; http://dx.doi.org/10.1111/jnc.12076
- Tan CC, Yu JT, Tan MS, Jiang T, Zhu XC, Tan L. Autophagy in aging and neurodegenerative diseases: implications for pathogenesis and therapy. Neurobiol Aging 2014; 35(5):941-57; PMID:24360503; http://dx.doi.org/10.1016/j.neurobiolaging.2013.11.019
- Meng Y, Yong Y, Yang G, Ding H, Fan Z, Tang Y, Luo J, Ke ZJ. Autophagy alleviates neurodegeneration caused by mild impairment of oxidative metabolism. J Neurochem 2013; 126(6):805-18; PMID:23586593; http://dx.doi.org/10.1111/jnc.12268
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149(2):274-93; PMID:22500797; http://dx.doi.org/10.1016/j.cell.2012.03.017
- Polak P, Hall MN. mTOR and the control of whole body metabolism. Curr Opin Cell Biol 2009; 21(2):209-18; PMID:19261457; http://dx.doi.org/10.1016/j.ceb.2009.01.024
- Xiong N, Jia M, Chen C, Xiong J, Zhang Z, Huang J, Hou L, Yang H, Cao X, Liang Z, et al. Potential autophagy enhancers attenuate rotenone-induced toxicity in SH-SY5Y. Neuroscience 2011; 199:292-302; PMID:22056603; http://dx.doi.org/10.1016/j.neuroscience.2011.10.031
- Shimada K, Motoi Y, Ishiguro K, Kambe T, Matsumoto SE, Itaya M, Kunichika M, Mori H, Shinohara A, Chiba M, et al. Long-term oral lithium treatment attenuates motor disturbance in tauopathy model mice: implications of autophagy promotion. Neurobiol Dis 2012; 46(1):101-8; PMID:22249108; http://dx.doi.org/10.1016/j.nbd.2011.12.050
- Luo J. The role of GSK3beta in the development of the central nervous system. Front. Biol 2012; 7(3):212-220; http://dx.doi.org/10.1007/s11515-012-1222-2