
Abstract
Recently a noncanonical activity of autophagy proteins has been discovered that targets lipidation of microtubule-associated protein 1 light chain 3 (LC3) onto macroendocytic vacuoles, including macropinosomes, phagosomes, and entotic vacuoles. While this pathway is distinct from canonical autophagy, the mechanism of how these nonautophagic membranes are targeted for LC3 lipidation remains unclear. Here we present evidence that this pathway requires activity of the vacuolar-type H+-ATPase (V-ATPase) and is induced by osmotic imbalances within endolysosomal compartments. LC3 lipidation by this mechanism is induced by treatment of cells with the lysosomotropic agent chloroquine, and through exposure to the Heliobacter pylori pore-forming toxin VacA. These data add novel mechanistic insights into the regulation of noncanonical LC3 lipidation and its associated processes, including LC3-associated phagocytosis (LAP), and demonstrate that the widely and therapeutically used drug chloroquine, which is conventionally used to inhibit autophagy flux, is an inducer of LC3 lipidation.
Abbreviations:
- ATG, autophagy-related
- Baf, bafilomycin A1
- CALCOCO2/NDP52, calcium binding and coiled-coil domain 2
- ConA, concanamycin A
- CQ, chloroquine
- FYCO1, FYVE and coiled-coil domain containing 1
- GFP, green fluorescent protein
- LAMP1, lysosomal-associated membrane protein 1
- LAP, LC3-associated phagocytosis
- MAP1LC3/LC3, microtubule-associated protein 1 light chain 3
- MTOR, mechanistic target of rapamycin
- PIK3C3/VPS34, phosphatidylinositol 3-kinase
- catalytic subunit type 3
- PtdIns3P, phosphatidylinositol 3-phosphate
- PtdIns3K, phosphatidylinositol 3-kinase
- RB1CC1/FIP200, RB1-inducible coiled-coil 1
- SQSTM1/p62, sequestosome 1
- TEM, transmission electron microscopy
- TLR, toll-like receptor
- ULK1/2, unc-51 like autophagy activating kinase 1/2
- VacA, vacuolating toxin A
- V-ATPase, vacuolar-type H+-ATPase
Introduction
Autophagy (used here to refer to macroautophagy) is a conserved lysosomal degradation pathway activated by multiple cellular stresses, including nutrient deprivation.Citation1,2 During autophagy, intracellular macromolecules and organelles are sequestered within double-membrane autophagosomes, which fuse with lysosomes that degrade internalized cargo.Citation1,2 As autophagy removes damaged organelles and protein aggregates, and recycles essential nutrients, it contributes to maintaining cellular homeostasis and is an important cellular starvation response.Citation1,2
Recently a noncanonical activity of autophagy proteins has been discovered that is associated with macroendocytic engulfment processes.Citation3,4 Through this pathway, MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3) is lipidated onto a variety of single-membrane vacuoles, including phagosomes, macropinosomes, and entotic vacuoles and is important for innate immunity and vision.Citation3,5,6 Single-membrane LC3 lipidation requires components of the autophagy lipidation machinery (e.g., ATG5, ATG7), but is independent of the ULK1/2-ATG13-RB1CC1/FIP200 preinitiation complex and thus distinct from classical autophagy. However, the underlying mechanism regulating activation of this noncanonical pathway remains unclear. For LC3 lipidation onto phagosomes, called LC3-associated phagocytosis, or LAP, toll-like receptor (TLR) signalingCitation4 as well as NADPH oxidase activity and reactive oxygen speciesCitation7 are required. Whether these signals also mediate LC3 lipidation onto macropinosomes and entotic vacuoles, and the mechanism of how they activate LC3 lipidation, is unknown.Citation3
During canonical autophagy, the vacuolar-type H+-ATPase (V-ATPase) plays an important role in maintaining autophagy flux by acidifying endolysosomal compartments and thereby enabling the activity of lysosomal hydrolases. V-ATPase inhibitors (bafilomycin A1 (Baf) and concanamycin A (ConA)) raise pH and disrupt autophagy flux, leading to accumulation of autophagosomes in cells, measured as an increase in cleaved and lipidated LC3 (called LC3-II). Similarly, the weak base lysosomotropic amine chloroquine,Citation8-11 and the Na+/H+ ionophore monensinCitation12,13 also raise lysosomal pH and disrupt autophagy flux. These compounds are used interchangeably to inhibit autophagy in cultured cells,Citation14,15 but only chloroquine and its derivative hydroxychloroquine (HCQ) have been exploited for therapeutic use, as antimalarial drugs,Citation16 and potentially as anticancer compounds.Citation17
In this report we make the unexpected discovery that chloroquine and monensin induce LC3 lipidation onto endolysosomal compartments by activating a noncanonical pathway resembling LAP. We demonstrate that this process is driven by osmotic imbalances within endolysosomal compartments, which act in concert with V-ATPase activity to recruit the autophagy protein ATG5 and to lipidate LC3 onto vacuolar membranes. More broadly, we show that water influx is sufficient to induce single-membrane LC3 lipidation, and demonstrate that V-ATPase activity is indispensable for physiogical LC3 lipidation during LAP and entosis. Finally we find that pathophysiological LC3 lipidation is activated during exposure of cells to Heliobacter pylori VacA toxin through the same mechanism. Our data provide new mechanistic insight into single membrane, LAP-like LC3 lipidation, and identify a new disease relevant context for this process. Furthermore our findings demonstrate that lysosomotropic compounds used to inhibit canonical autophagy flux (chloroquine and monensin) are activators of parallel, endolysosomal LC3 lipidation.
Results
Chloroquine and monensin induce V-ATPase-dependent LC3 lipidation
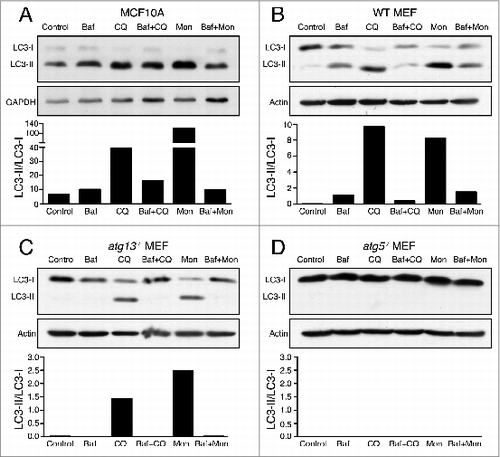
While examining autophagy flux in cells, we noticed that human mammary epithelial cells (MCF10A) and mouse embryo fibroblasts (MEF) treated with chloroquine exhibited a larger-fold accumulation of LC3-II than cells treated with Baf, despite similar effects on lysosomal pH ( and , Fig. S1A and S1B, Fig. S2A to D). Surprisingly, this effect of chloroquine was blocked by Baf pretreatment ( and ), suggesting that V-ATPase activity is required for a chloroquine-inducible LC3-II accumulation that is unrelated to autophagy flux. Like chloroquine, treatment of cells with monensin also induced a larger-fold accumulation of LC3-II than treatment with Baf, despite similar effects on lysosomal pH, and this increase in LC3-II accumulation was also Baf-inhibitable ( and , Fig. S1A and S1B). These data demonstrate a chloroquine and monensin-inducible LC3-II accumulation that is unrelated to autophagy flux and that requires V-ATPase activity.
Figure 1. Chloroquine and monensin induce V-ATPase-dependent LC3 lipidation. (A to D) Representative western blots for LC3 and GAPDH on (A) MCF10A cells, (B) Wild-type MEFs (C), atg13−/− MEFs or (D) and atg5−/− MEFs treated with lysosome inhibitors bafilomycin A1 (Baf, 100 nM), chloroquine (CQ, 100 μM), monensin (Mon, 100 μM) for 1 h or with 15 min Baf pretreatment followed by CQ or Mon for 1 h. Ratios of lipidated LC3-II/unlipidated LC3-I were quantified and graphed. See Figure S2 for repeat protein gel blots and quantification. See also Figures S1 and S2.
Chloroquine and monensin activate endolysosomal LC3 lipidation in a V-ATPase-dependent manner
Recently noncanonical activities of autophagy pathway proteins have been reported, including the lipidation of LC3 onto nonautophagosomal membranes, including macroendocytic vacuoles, that occurs independently of the ULK1/2-ATG13-RB1CC1 preinitiation complex.Citation3 Interestingly, whereas the knockout of Atg13 abolished LC3-II accumulation induced by starvation and Baf (Fig. S1C), treatment with chloroquine and monensin still induced LC3-II accumulation in the absence of ATG13 (). As in wild-type cells, the accumulation of LC3-II induced by chloroquine and monensin in atg13−/− cells was Baf-inhibitable, demonstrating that the V-ATPase-dependent effects of chloroquine and monensin on LC3-II are genetically separable from canonical autophagy (). As expected, the knockout of Atg5, an essential component of the LC3 lipidation machinery, completely abolished LC3-II in control and inhibitor-treated cells ().
ATG13-independent LC3 lipidation has been reported to occur on single-membrane endolysosomal membranes during engulfment events such as LAP, macropinocytosis, and entosis. To explore whether LAP-like endolysosomal LC3 lipidation may underlie the effects of chloroquine and monensin, fluorescence imaging of GFP-LC3 was performed on drug-treated cells. Following chloroquine or monensin treatment, we noted that GFP-LC3 colocalized with LAMP1 on lysosomal structures in ring-like patterns, suggesting that LC3 is lipidated onto lysosomal membranes in a LAP-like manner (). This localization pattern of LC3 is different than that induced by Baf treatment, suggesting it does not arise from inhibition of autophagy flux (). Like LC3-II accumulation assessed by western blotting, the colocalization of LC3 and LAMP1 was inhibited by treatment with Baf, demonstrating dependence on V-ATPase activity ().
Figure 2. Chloroquine and monensin induce LC3 lipidation onto multiple lysosomal compartments dependent on V-ATPase activity. (A and B) Confocal images of GFP-LC3 and LAMP1 immunostaining of (A) lysosomes in MCF10A and (B) uncoated latex bead phagosomes in J774 macrophage following treatment with Baf (100 nM), CQ (100 mM) or Baf + CQ for 1 h. Arrow indicates GFP-LC3 lipidation onto a phagosome. Bar = 6 μm. (C and D) Images of GFP-LC3 and LAMP1 on entotic corpse vacuoles in MCF10A cells treated with Baf, CQ, Mon (100 μM) or Baf + CQ for 1 h. Arrows indicate GFP-LC3 lipidation onto vacuoles. Bar = 10 mm. (E) GFP-LC3 and ATP6V0D1 (V0D1) staining on entotic corpse vacuoles with our without CQ treatment. Bar = 10 μm. (F) GFP-LC3G120A and LAMP1 staining on entotic corpse vacuoles following CQ treatment. Bar = 10 μm. (G) GFP-LC3 and ATG5 immunostaining on entotic corpse vacuole (arrows) following CQ treatment. Bar = 10 μM. (H) (i) Electron microscopy of corpse containing cell-in-cell structure treated with CQ (100 μM), (ii) entotic corpse vacuole has a single membrane (arrows). See also Figure S3; Movie S1.
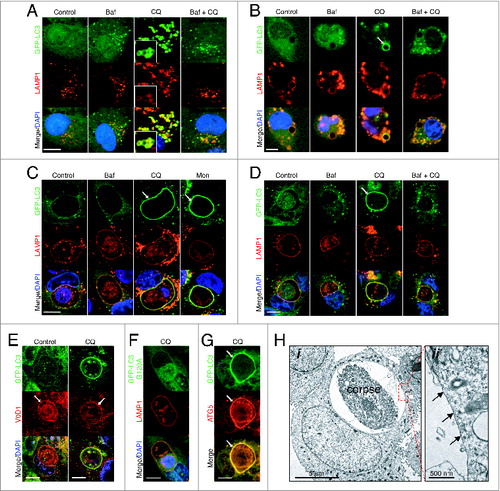
To further examine if chloroquine can induce LAP-like LC3 lipidation, we imaged phagosomes in macrophages to determine if LC3 would be lipidated onto these lysosomal compartments. Strikingly, treatment of macrophages harboring latex bead phagosomes with chloroquine, induced rapid localization of GFP-LC3 with LAMP1 in a ring-like pattern around latex beads, consistent with LAP. Recruitment of LC3 onto phagosomes was blocked by Baf treatment, demonstrating dependence on V-ATPase activity ( and Fig. S3A). Next we examined a different LAP-like activity that occurs during the live epithelial cell engulfment program entosis. Like phagosomes in macrophages, treatment with chloroquine or monensin induced rapid accumulation of GFP-LC3 at LAMP1-positive entotic vacuoles in MCF10A cells (; Movie S1). The colocalization of GFP-LC3 and LAMP1 on vacuoles was inhibited by treatment with Baf and also Con A ( and Fig. S3B to D), consistent with the vacuolar localization of V-ATPase as assessed by subunit ATP6V0D1 staining (). GFP-LC3 and LAMP1 colocalization required LC3 lipidation, as a nonconjugable mutant GFP-LC3G120A did not translocate upon chloroquine treatment (). Lipidation of LC3 was associated with the recruitment of ATG5 to the entotic vacuole (). Moreover, inspection of the membrane structure of entotic vacuoles after chloroquine treatment (which resulted in GFP-LC3 recruitment to 100% entotic corpse vacuoles, data not shown) by transmission electron microscopy revealed a single-membrane structure, consistent with the lipidation of LC3 onto single-membrane vacuoles (). By time-lapse microscopy we found no evidence of GFP-LC3 puncta fusing with the entotic vacuole during chloroquine treatment, and the accumulation of GFP-LC3 at the vacuole membrane was mirrored by a loss of cytosolic signal, as has been shown for GFP-LC3 lipidation during entosis (Fig. S3E).Citation18 These data are consistent with a model whereby chloroquine and monensin induce LAP-like LC3 lipidation onto phagosomes and entotic vacuoles, and lysosome compartments in cells, in a V-ATPase-dependent manner.
Chloroquine-mediated activation of endolysosomal LC3 lipidation occurs independent of PtdIns3P and autophagic receptor proteins
During canonical autophagy, PtdIns3P generated by the class III PtdIns3K PIK3C3/VPS34 is required for LC3 lipidation and autophagosome formation. To examine if PtdIns3P is involved in chloroquine-induced endolysosomal LC3 lipidation, we monitored PtdIns3P dynamics using cells expressing the 2xFYVE-mCherry reporter. Following chloroquine treatment, GFP-LC3 was lipidated onto entotic corpse vacuoles, but there was no evidence of recruitment of 2xFYVE-mCherry, suggesting that PtdIns3P is not generated at vacuole membranes in response to chloroquine (). Consistent with this, pretreatment with the PtdIns3K inhibitors LY290004 and wortmannin, at concentrations known to inhibit PIK3C3 and amino acid starvation-induced autophagy (Fig. S4A and S4B), completely inhibited the appearance of PtdIns3P puncta in cells, but did not inhibit GFP-LC3 lipidation following chloroquine treatment ( and , Fig. S4C). Together these data demonstrate the independence of chloroquine-induced LC3 lipidation from PtdIns3P generation.
Figure 3. Chloroquine-mediated LC3 recruitment is independent of PtdIns3P and autophagy receptor proteins. (A) Time-lapse microscopy of GFP-LC3 and 2xFYVE-mCherry on entotic corpse vacuoles following treatment with CQ (100 μM). Arrows indicate GFP-LC3 lipidation onto vacuole. Bar = 2 μm. (B) Confocal images of entotic corpse vacuoles treated with CQ (100 μM) with or without LY290004 (25 μM). Arrows indicate GFP-LC3 lipidation onto entotic vacuole, arrowheads indicate 2xFYVE-mCherry-positive vesicles. Bar = 2 μm. (C) Quantification of GFP-LC3 recruitment to LAMP1-positive entotic vacuoles with or without LY290004 (LY 25 μM) or wortmannin (WM, 200 nM); data are mean ± SEM from 3 independent experiments; NS, not significant. (D and E) Entotic corpse vacuoles treated with CQ (100 μM) for 1 h and immunostained for (D) SQSTM1 or (E) CALCOCO2. Arrows indicate autophagosomes with colocalized LC3 and SQSTM1 or CALCOCO2. Bar = 4 μm. See also Figure S4.
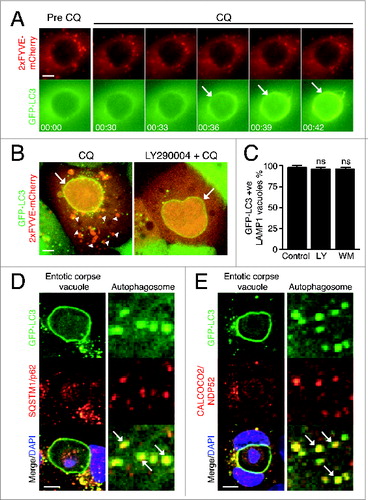
The autophagy receptor proteins SQSTM1/p62 and CALCOCO2/NDP52 are involved in targeting substrates to LC3-positive autophagosomes Citation19,20 and are also recruited to damaged membranes, which recruit LC3-positive vesicles.Citation21 To examine if these receptors might localize to vacuole membranes and mediate LC3 recruitment, we imaged their localization following chloroquine treatment. While SQSTM1 and CALCOCO2/NDP52 colocalized with autophagosomes in cells ( and ), these receptor proteins did not colocalize with GFP-LC3 on entotic vacuoles following chloroquine treatment ( and ). Together these findings demonstrate the independence of chloroquine-induced endolysosmal LC3 lipidation from PtdIns3P and autophagy receptor proteins.
Chloroquine-mediated activation of endolysosomal LC3 lipidation is osmotically regulated
In seeking to understand how chloroquine and monensin activate LAP-like LC3 lipidation, we considered the mechanism of action of these 2 compounds compared to Baf. Lysosomotropic agents such as chloroquine become protonated and entrapped within acidic compartments,Citation10 which alters the osmotic properties of lysosomes, promoting compartment swelling via water influx.Citation22 Similarly, by promoting the exchange of protons for osmotically active monovalent cations such as Na+, monensin alters the ionic balance and osmotic properties of intracellular compartments leading to water influx.Citation23 As raising lysosomal pH is insufficient to activate endolysosomal LC3 lipidation ( and and to ), we explored a possible role for osmotic imbalances and water flux.
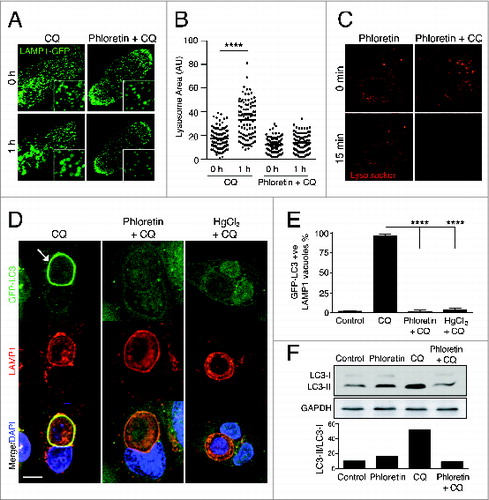
To inhibit water influx in our system we utilized 2 distinct aquaporin water channel inhibitors, mercury chlorideCitation24 and phloretin.Citation25 Treatment of cells with chloroquine induced lysosome swelling, measured by a significantly increased average size of LAMP1-GFP vesicles, which was inhibited in the presence of phloretin ( and ; Movie S2). The inhibition of lysosomal swelling by phloretin was not due to a failure to trap chloroquine inside of lysosomes, as chloroquine-mediated pH changes were not affected (). Importantly, chloroquine-induced endolysosomal LC3 lipidation, measured both by LC3 translocation to LAMP1-positive entotic vacuoles ( and ) and by LC3 protein gel blotting (), was inhibited by treatment of cells with either of the aquaporin channel inhibitors phloretin or mercury chloride. These data are consistent with a model where chloroquine-induced LC3 lipidation involves endolysosmal osmotic imbalance.
Figure 4. Chloroquine-mediated noncanonical LC3 lipidation is dependent on water flux. (A) Confocal images of LAMP1-GFP fluorescence in MCF10A cells before and after treatment with CQ (100 μM) with or without phloretin (180 μM). Insets show vesicles with LAMP1-GFP. (B) Quantification of LAMP1-GFP-labeled vesicle size; P < 0.01 **** by one-way ANOVA. (C) Confocal images of LysoTracker Red in MCF10A cells before and after treatment with phloretin with or without CQ for 15 min. (D) Confocal images of GFP-LC3 and LAMP1 immunostaining on entotic corpse vacuoles in MCF10A cells following treatment with CQ with or without phloretin (180 mM) or HgCl2 (15 mM). Arrow indicates GFP-LC3 lipidation onto a vacuole. Bar = 5 μm. (E) Quantification of GFP-LC3 lipidation onto LAMP1-positive entotic corpse vacuoles as in (D), data mean ± SEM from 3 independent experiments; P < 0.001 ****. (F) Western blot analysis of LC3 in MCF10A cells treated with phloretin, CQ or both for 1 h. Quantification of LC3-II/LC3-I graphed below. See also Movie S2.
Osmotic imbalances are sufficient to activate LC3 lipidation onto endolysosomal compartments
To explore the role of osmotic imbalances in endolysosomal LC3 lipidation more directly, intracellular compartment swelling was induced in the absence of chloroquine by placing cells under hypo-osmotic conditions. When MCF10A cells were placed in hypotonic medium, LAMP1-positive entotic vacuoles swelled and then rapidly recruited GFP-LC3 (; Movie S3). Lysosome size also increased under hypo-osmotic conditions, and there was an associated translocation of GFP-LC3 to LAMP1-positive lysosomes ( and ; Movie S4) along with an overall increase in lipidated LC3 (). Like MCF10A cells, wild-type and atg13−/− MEFs displayed GFP-LC3-positive swollen intracellular vacuoles under hypo-osmotic conditions (). These data demonstrate that the introduction of osmotic imbalance within lysosomal compartments, like chloroquine treatment, activates LAP-like LC3 lipidation.
Figure 5. Osmotic imbalances are sufficient to induce LC3 lipidation onto lysosomal compartments in a V-ATPase-dependent manner. (A and B) Confocal images of GFP-LC3 and LAMP1-RFP from time-lapse microscopy of (A) entotic corpse vacuoles or (B) lysosomes in MCF10A cells treated with hypotonic media. Arrow indicates GFP-LC3 lipidation onto entotic corpse vacuole. Bar = 2 μm. (C) Western blot analysis of LC3 in MCF10A cells cultured in control and hypotonic media for 1 h. Quantification of LC3-II/LC3-I graphed below. (D) Confocal images GFP-LC3 in wild-type, atg13−/− and atg5−/− MEFs cultured in control or hypotonic media for 30 min. Arrows indicate GFP-LC3 on vacuoles. Bar = 4 μm. (E) Quantification of LAMP1-GFP vesicle size in MCF10A cells under control or hypotonic conditions with or without Baf (100 nM); NS, not significant. (F) Quantification of hypo-osmotic induced LC3 lipidation onto LAMP1-positive entotic corpse vacuoles with or without Baf (100 nM). Data are mean ± SEM from 3 independent experiments; P < 0.01 *. (G) Western blot of LC3 in MCF10A cells in control or hypotonic media with or without Baf (100 nM). Quantification of LC3-II/LC3-I graphed below. See also Figure S5; Movie S3 and Movie S4.
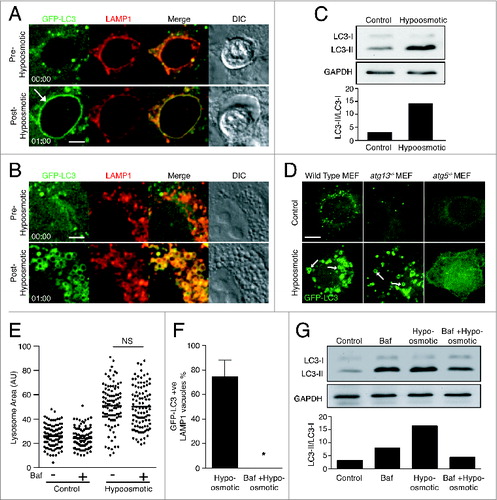
Osmotic activation of endolysosomal LC3 lipidation requires V-ATPase activity
We have shown that endolysosomal LC3 lipidation induced by chloroquine requires V-ATPase activity, as it is inhibited by Baf ( and ). This effect may be due to a failure to trap chloroquine in lysosomes in the absence of the V-ATPase-dependent proton gradient. The finding that hypo-osmotic conditions also activate noncanonical LC3 lipidation, similar to chloroquine, allowed for a more direct assessment of the role of the V-ATPase in endolysosmal LC3 lipidation. Interestingly, while hypo-osmotic conditions induced the swelling of multiple intracellular compartments, we noted that LC3 was lipidated only onto vacuoles that were acidic and LAMP1-positive. For example, entotic vacuoles housing live cells, which are not lysosomal compartments,Citation18 swelled dramatically under hypo-osmotic conditions, but did not recruit LC3 while entotic vacuoles housing dead cells, which are lysosomal vacuoles, did recruit LC3 (Fig. S5). V-ATPase inhibition by Baf treatment prior to exposure of cells to hypotonic medium did not inhibit water influx and lysosome swelling () but completely inhibited LC3 translocation to LAMP1-positive entotic vacuoles () and reduced overall LC3 lipidation (). These data demonstrate that osmotic imbalances act in concert with V-ATPase activity to induce LAP-like LC3 lipidation.
V-ATPase activity is required for LC3 lipidation during LAP and entosis
We next sought to determine whether V-ATPase activity is required to activate physiological examples of noncanonical LC3 lipidation such as LAP and entosis. During entosis, the internalized cell can die by either a nonapoptotic or an apoptotic program. In the latter case, LC3 is lipidated onto entotic vacuoles after cell death occurs.Citation18 Baf treatment has multiple effects on entosis. As previously reported, V-ATPase inhibition reduces the frequency of internalized cell death, while also switching the predominant mode of death from nonapoptotic to apoptotic ().Citation26 Furthermore, we show here that LC3 lipidation, which usually follows an apoptotic death, is completely blocked by Baf treatment ( and ), demonstrating a requirement for V-ATPase activity in this process.
Figure 6. LC3 recruitment during LAP and entosis is dependent on V-ATPase activity. (A) Quantification of entotic inner cell death over 20 h with or without Concanamycin A (ConA, 100 nM). Data are mean ± SEM of 3 separate experiments; P < 0.05 *. (B and C) Quantification of LC3 recruitment to entotic vacuole with or without ConA (100 μM) during (B) nonapoptotic or (C) apoptotic death of inner cells. Data are mean ± SEM of 3 separate experiments; P < 0.002 **; N/A, not applicable. (D and E) Quantification of GFP-LC3 recruitment to (D) zymosan phagosomes in RAW264.7 cells and (E) IgG-coated bead phagosomes in IFNG-treated RAW264.7 cells. Data are mean ± SEM of 3 separate experiments; P < 0.002 **. (F and G) Confocal time-lapse images of GFP-LC3 and 2xFYVE-mCherry in RAW264.7 cells during zymosan phagocytosis with or without Baf (100 μM). Arrows point to 2xFYVE recruitment and GFP-LC3 lipidation as indicated. Bar = 3 μm.
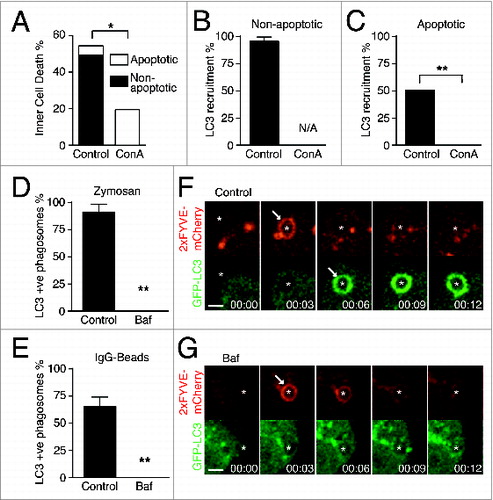
To examine the role of the V-ATPase during LAP, macrophages phagocytosing zymosan or IgG-coated beads were treated with Baf. Baf treatment blocked phagosomal LC3 lipidation ( and ), without inhibiting PtdIns3P formation that occurs upstream ( and ). These data demonstrate a previously undescribed role for V-ATPase activity in phagosome maturation, downstream of PtdIns3P generation and upstream of the lipidation of LC3. Together, these data uncover an essential role for V-ATPase activity in single-membrane LC3 lipidation during macroendocytic engulfment events.
VacA toxin of Heliobacter pylori activates endolysosomal LC3 lipidation
In considering the mechanism of LC3 lipidation identified here, we hypothesized that any process that promotes osmotic imbalances within the endolysosomal system would have the potential to activate LAP-like LC3 lipidation. One physiologically relevant example is H. pylori infection, which is implicated in the pathogenesis of a variety of gastric diseases and promotes the appearance and swelling of large intracellular vacuoles derived from the endolysosomal system.Citation27 H. pylori secretes a virulence factor, vacuolating toxin A (VacA), which following endocytosis,Citation28 oligomerizes and inserts into endosomal membranes, where it functions as a selective anion channel, increasing intraluminal Cl− content.Citation29 To counteract the negative charge derived from Cl− influx and maintain electrogenic potential, V-ATPase proton pump activity is increased.Citation30 Membrane-permeant weak bases, such as ammonium chloride (NH4Cl), become protonated and accumulate within these proton-rich endolysosomal compartments, altering their osmotic properties, which in turn promote the influx of water.Citation31 The result is the appearance of large acidic single-membrane vacuoles that are enriched in late endosome and lysosome markers LAMP1 and RAB7.Citation32,33
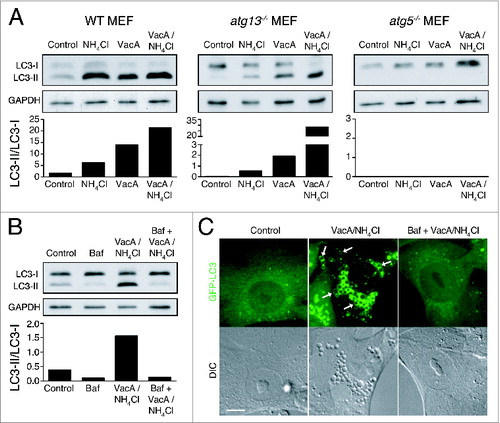
Considering that VacA alters osmotic properties and V-ATPase activity within endolysosomal compartments, we examined whether endolysosomal LC3 lipidation was also activated. Treatment of wild-type MEFs with VacA alone and in combination with NH4Cl, which promotes more efficient vacuolation, induced robust LC3 lipidation (). Strikingly, these treatments also promoted LC3 lipidation in atg13−/− MEFs, in a Baf-inhibitable, and ATG5-dependent manner, consistent with noncanonical, V-ATPase-dependent LC3 lipidation ( and ). Accordingly, GFP-LC3 recruited to VacA-induced vacuole membranes in both wild-type and atg13−/− MEFs (, data not shown). Together, these data demonstrate that VacA activates LAP-like lipidation of LC3 onto endolysosomal compartments.
Figure 7. VacA activates noncanonical LC3 lipidation. (A) Western blot analysis of LC3 in wild-type, atg13−/− and atg5−/− MEFs treated with NH4Cl (5 mM), VacA (10 μM) or both for 2 h. Quantification of LC3-II/LC3-I graphed below. (B) Western blot analysis of LC3 in atg13−/− MEFs treated with Baf (100 nM), NH4Cl (5 mM) + VacA (10 μM) or Baf + NH4Cl + VacA for 2 h. Quantification of LC3-II/LC3-I graphed below. (C) Confocal images of differential interference contrast and GFP-LC3 in atg13−/− MEFs treated with NH4Cl (5 mM) + VacA (10 μM) or Baf + NH4Cl + VacA for 2 h. Arrows indicate GFP-LC3 on vacuoles. Bar = 5 μm.
Discussion
Here we provide evidence for a novel mechanism controlling LC3 lipidation, involving V-ATPase activity and osmotic imbalances within endolysosomal compartments. We show that water influx into endolysosomal vacuoles induced by hypotonic medium, chloroquine treatment, and H. pylori VacA toxin activate this pathway, and provide evidence for the V-ATPase-dependency of LC3 lipidation during LAP and entosis.
The mechanism of LC3 lipidation identified here appears to occur independently of canonical autophagy based on a number of observations: (1) LC3 lipidation occurs under nutrient-replete conditions, where MTORC1 signaling is known to be active and autophagy is suppressed; (2) LC3 lipidation occurs in atg13−/− cells that are deficient for MTORC1-regulated autophagy; (3) PtdIns3K inhibitors that block autophagy have no effect on LC3 lipidation; (4) LC3 is lipidated onto single-membrane compartments as determined by TEM; (5) ATG5 is recruited to the lysosomal compartments where LC3 is lipidated; and (6) LC3 lipidation is V-ATPase-dependent unlike autophagosome formation that is induced by V-ATPase inhibition.Citation34 Altogether these data support a model where osmotic imbalances and the V-ATPase promote LC3 lipidation onto vacuole membranes in an autophagy-independent manner.
Our data uncover an important new activity for chloroquine and monensin as activators of noncanonical LC3 lipidation. These drugs are conventionally used as inhibitors of autophagic flux, but here we demonstrate a parallel induction of single-membrane endolysosomal LC3 lipidation. These findings support an emerging conceptCitation3 that the relative abundance of LC3-II in cells is not solely reflective of classical autophagy. Furthermore, our findings indicate that commonly used drugs have distinct, parallel activities on canonical and noncanonical autophagy processes. Chloroquine and monensin inhibit classical autophagic flux while simultaneously inducing single-membrane lipidation, while V-ATPase inhibitors (Baf, ConA) inhibit both autophagic flux and single-membrane LC3 lipidation.
The unexpected observations made with chloroquine led us to investigate a broader role for osmotic imbalance in controlling single-membrane LC3 recruitment. During LAP, signaling from a number of receptors including TLRs,Citation4,7 TIMD4/Tim4,Citation35 Fcγ receptors,Citation7,36 SLAM Citation37 and CLEC7A/Dectin1,Citation38 as well as activation of NADPH oxidase,Citation7,38 are implicated in promoting LC3 lipidation to the phagosome, although the mechanism of how they achieve this remains unclear. It is conceivable that these signals could feed into the pathway described here. Water influx occurs into phagosomes and is associated with bacterial killing potential.Citation39 NADPH oxidase and reactive oxygen species generation have the potential to alter phagosomal ionic balances,Citation40,41 which can influence osmotic properties, and the release of solutes from degrading cargo is proposed to alter the osmotic properties of phagosomes.Citation39 Multiple signals may converge to generate osmotic changes within phagosomes, which could activate LC3 lipidation in a V-ATPase-dependent manner. As LC3 lipidation precedes phagosome-lysosome fusion,Citation18,36 this model predicts a role for V-ATPase trafficking to phagosomes upstream of the appearance of other lysosomal markers, as has been observed in macrophages.Citation42 Of note, this mechanism appears to be distinct from LC3 recruitment to damaged endosomes or lysosomes, or pathogen-containing vacuoles, that involves targeting by the receptor proteins SQSTM1 or CALCOCO2/NDP52 Citation21,43,44 and is associated with the appearance of double-membrane structures.Citation43
We provide evidence that a bacterial virulence factor activates LC3 lipidation onto osmotically swollen lysosomal compartments in a V-ATPase-dependent manner. While previous work has provided evidence for VacA-dependent induction of autophagy,Citation45 our data suggest that most of the LC3 lipidation that occurs acutely upon treatment with VacA and NH4Cl occurs in an ATG13-independent manner, and is inhibited by Baf. As VacA-induced vacuoles serve as the intracellular niche for H. pylori, it will be of interest to determine what role LC3 lipidation plays in this context. The function of LC3 lipidated onto nonautophagosomal membranes remains poorly understood. It has been proposed that the presence of LC3 on vacuoles promotes lysosome fusion and the degradation of luminal contents,Citation4,18,35,36 or that LC3 regulates the inflammatory cytokine response or antigen presentation capacity of phagocytes.Citation35,36,38 However, LC3 lipidation may also delay phagosome maturation in some contexts to allow for more efficient antigen processing.Citation46 A clearer picture of the role of endolysosomal LC3 lipidation awaits identification of the molecular mechanism(s) whereby LC3 affects vacuole maturation. LC3-family proteins could regulate maturation by modulating the activity or localization of any of a number of binding partners,Citation47,48 or by facilitating membrane-membrane fusion.Citation3,49 Our finding here that treatment with lysosomotropic agents induces endolysosomal LC3 lipidation may provide a convenient system to uncover the changes that occur to endolysosomal compartments as a result of LC3 lipidation.
Materials and Methods
Antibodies and reagents
The following antibodies were used: Anti-LC3A/B (Cell Signaling Technology, 4108), anti-human LAMP1 and anti-mouse LAMP1 (Becton Dickinson, 555798 and 553792), anti-GAPDH (SCBT, sc-25778), anti- SQSTM1/p62 (Becton Dickinson, 160832), anti-CALCOCO2/NDP52, anti-ATP6V0D1 (Abcam, ab68588 and ab56441). The following inhibitors and reagents were used at the indicated concentrations: Wortmannin (EMD, 12-338) 200 nM; LY290024 (EMD, 440202) 25 mM, monensin (Sigma, M5273) 100 mM, chloroquine (Sigma, C6628) 100 mM, bafilomycin A1 (EMD, 196000) 100 nM, concanamycin A (EMD, 344085) 100 nM.
Cell culture
MCF10A cells were cultured in DME/F12 (Invitrogen, 11320-033) + 5% horse serum (Invitrogen, 16050-122), 20 ng/ml EGF (Peprotech, AF-100-15), 10 μg/ml insulin (Sigma, I1882), 0.5 μg/ml hydrocortisone (Sigma, H0888), 100 ng/ml cholera toxin (Sigma, C8052), 50 U/ml penicillin, and pen/strep. Hypotonic medium was made by diluting MCF10A culture medium with ddH2O (20:80). Mouse embryonic fibroblast MEF cell lines, J774 and RAW264.7 mouse macrophages (ATCC) were cultured in DMEM + 10% fetal bovine serum (Atlanta Biologicals, S11595) with pen/strep.
Constructs and Amaxa nucleofection
The following constructs were used: pBabe-GFP-LC3,Citation18 pBabe-2xFYVE-mCherry,Citation18 N1-LAMP1-RFP,Citation18 pRetro-LAMP1-GFP.Citation18 Transient transfection was performed by electroporation using a Nucleofector II instrument (Lonza, Wakersville, MD, USA) and Lonza nucleofection kit V (Lonza, VCA-1003) following manufacturer's guidelines.
VacA treatment
Purified VacA toxin (kindly provided by Dr Tim Cover, Vanderbilt University, Nashville, TN) was acid activated by incubation with 200 mM HCl for 30 min before being diluted into tissue culture media at the desired final concentration.
Microscope image acquisition
For time-lapse microscopy, cells were grown on 35-mm glass-bottomed coverslip dishes (MatTek, P35G-1.5-14-C). For confocal time-lapse microscopy, imaging was performed with the Ultraview Vox spinning disc confocal system (Perkin Elmer, MA, USA) equipped with a Yokogawa CSU-X1 spinning disc head, and EMCCD camera (Hamamatsu, C9100-13, Hamamatsu Photonics K.K., Hamamatsu, Japan), and coupled with a Nikon Ti-E microscope using a 60× oil immersion 1.40 numerical aperture (NA) objective (Nikon, Melville, NY, USA). For widefield time-lapse microscopy, images were acquired using a coolSNAP HQ2 CCD camera (Photometrics, AZ, USA), coupled to a Nikon Ti-E microscope using a 20× 0.45NA objective. All imaging with live cells was performed within incubation chambers at 37°C and 5% CO2. Image acquisition and analysis was performed with Volocity software (Perkin Elmer, MA, USA) and Elements software (Nikon). All image processing (brightness and contrast) was performed on all pixels in each image.
For immunofluorescence, cells were fixed in ice-cold methanol at −20°C for 5 min. Samples were blocked in phosphate-buffered saline (Invitrogen, 10010023) + 5% BSA for 1 h at room temperature before incubation with primary antibodies in blocking media overnight at 4°C. Following phosphate-buffered saline washes, samples were incubated with Alexa Fluor tagged secondary antibodies (Invitrogen, A11036 and A11031) for 40 min at room temperature. DNA was stained using DAPI (Sigma, D8417) before mounting coverslips with ProLong antifade mounting media (Invitrogen, P36930). Confocal imaging was performed as above for time-lapse microscopy. All image processing (brightness and contrast) was performed on all pixels in each image. Images used in figures are representative of at least 3 separate repeats.
Electron microscopy
MCF10A cell-in-cell structures were treated with 100 mM chloroquine for 1 h and pelleted. GFP-LC3 recruitment was confirmed on all LAMP1-positive vacuoles by fluorescent microcopy. Samples were then fixed in 2.5% glutaraldehyde/2% paraformaldehyde in 0.075 M cacodylate buffer pH 7.5 for 1 h followed by rinsing in cacoldylate buffer and post fixation in 2% osmium tetroxide for 1 h. The samples were then rinsed in double-distilled water followed by dehydration in a graded series of alcohol 50%, 75% 95% through absolute alcohol and overnight in 1:1 propylene oxide/poly Bed 812 (Polysciences, 08791-500). Ultra thin sections were obtained with a Reichert Ultracut S microtome (Leica, Vienna, Austria). Sections were stained with uranyl acetate and lead citrate. Images were obtained using a JEOL 1200 EX transmission electron microscope (Peabody, MA, USA).
Phagocytosis and entosis assays
J774 or RAW264.7 cells expressing GFP-LC3 and 2xFYVE-mCherry were seeded onto glass-bottomed coverslip dishes for 2 d before zymosan (Sigma, Z4250) or uncoated 3-mm latex beads (Polysciences, Inc., PA, 17134-15) were added at a ratio of 10:1. IgG-coated latex beads were prepared by incubation overnight with 6 mg/ml human IgG (Sigma, I4506) in borate buffer and added to RAW264.7 cells pretreated with 200 U/ml IFNG (Peprotech, 315-05) for 2 d. Cells were monitored by confocal microscopy and images taken every 5 min.
To quantify entotic cell fate, MCF10A cells expressing GFP-LC3 were seeded overnight on 35-mm glass-bottomed dishes (MatTek, P35G-1.5-14-C) and cell-in-cell structures were imaged by time-lapse microscopy the next day in the presence or absence of 100 nM concanamycin. Fluorescent and differential interference contrast images were acquired every 4 min for 20 h using a Nikon Ti-E inverted microscope attached to a CoolSNAP CCD camera (Photometrics, AZ, USA). Images were captured by NIS Elements software (Nikon). Only live internalized cells at the start of time-lapse were quantified for cell fate. Internalized cells were scored for apoptotic or nonapoptotic death and for the recruitment of GFP-LC3 to the entotic vacuole.
Measurement of lysosome size
MCF10A expressing LAMP1-GFP were imaged by confocal microscopy. The mean area of 100 individual LAMP1-GFP vesicles from 5 cells were measured using Image J software before and after treatment as indicated.
Western blotting
Cells were scraped into ice-cold RIPA (150 mM NaCl, 50 mM Tris-HCl, 1% TritonX100, 0.1% SDS, 0.1% sodium deoxycholate) buffer and lysed for 10 min on ice. Lysates were centrifuged for 12 min at 4°C, supernatants were then separated on 15% polyacrylamide SDS-PAGE gels and transferred to a polyvinyldifluoride membrane. The membrane was blocked in TBS-T (50 mM Tris-Cl, pH 7.6, 150 mM NaCl, 1% Tween20) + 5% BSA (Sigma, A7906) and incubated overnight at 4°C with primary antibodies diluted blocking buffer. Blots were incubated with horseradish peroxidase conjugated to secondary antibodies (Cell Signaling Technology, 7074S and 7076S) and protein detected using enhanced chemiluminescence (Invitrogen, WP20005). Densitometry analysis was carried out using ImageJ software (NIH). Blots used in figures are representative of at least 3 separate repeats.
Statistics
The indicated P values were obtained using the Student t test, utilizing Graphpad Prism software.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Movie_S4.mov
Download QuickTime Video (1.3 MB)Movie_S3.mov
Download QuickTime Video (1,003.8 KB)Movie_S2.mov
Download QuickTime Video (7.4 MB)Movie_S1.mov
Download QuickTime Video (1.7 MB)Supplemental_file.pdf
Download PDF (357.8 KB)Acknowledgments
We thank members of the Overholtzer lab for helpful discussions, reagents, and for reading the manuscript. We also thank Nina Lampen of the Memorial Sloan Kettering Cancer Center Electron Microscopy Facility for processing of EM samples. We thank Dr. Tim Cover, Vanderbilt University TN, for providing us with purified VacA toxin and protocols.
Funding
This work was supported by a National Cancer Institute grant RO1CA154649, National Institutes of Health grants RO1CA166413 and 1F32CA162691, and by a Cancer Research UK fellowship C47718/A16337.
References
- Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. New Engl J Med 2013; 368:651-62; PMID:23406030; http://dx.doi.org/10.1056/NEJMra1205406
- Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res 2014; 24:24-41; PMID:24366339; http://dx.doi.org/10.1038/cr.2013.16
- Florey O, Overholtzer M. Autophagy proteins in macroendocytic engulfment. Trends Cell Biol 2012; 22:374-80; PMID:22608991; http://dx.doi.org/10.1016/j.tcb.2012.04.005
- Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007; 450:1253-7; PMID:18097414; http://dx.doi.org/10.1038/nature06421
- Kim JY, Zhao H, Martinez J, Doggett TA, Kolesnikov AV, Tang PH, Ablonczy Z, Chan CC, Zhou Z, Green DR, et al. Noncanonical autophagy promotes the visual cycle. Cell 2013; 154:365-76; PMID:23870125; http://dx.doi.org/10.1016/j.cell.2013.06.012
- Mehta P, Henault J, Kolbeck R, Sanjuan MA. Noncanonical autophagy: one small step for LC3, one giant leap for immunity. Curr Opin Immunol 2014; 26:69-75; PMID:24556403; http://dx.doi.org/10.1016/j.coi.2013.10.012
- Huang J, Canadien V, Lam GY, Steinberg BE, Dinauer MC, Magalhaes MA, Glogauer M, Grinstein S, Brumell JH. Activation of antibacterial autophagy by NADPH oxidases. Proc Nat Acad Sci U S A 2009; 106:6226-31; PMID:19339495; http://dx.doi.org/10.1073/pnas.0811045106
- Bowman EJ, Siebers A, Altendorf K. Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc Nat Acad Sci U S A 1988; 85:7972-6; PMID:2973058; http://dx.doi.org/10.1073/pnas.85.21.7972
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313-26; PMID:20144757; http://dx.doi.org/10.1016/j.cell.2010.01.028
- Poole B, Ohkuma S. Effect of weak bases on the intralysosomal pH in mouse peritoneal macrophages. J Cell Biol 1981; 90:665-9; PMID:6169733; http://dx.doi.org/10.1083/jcb.90.3.665
- Seglen PO, Grinde B, Solheim AE. Inhibition of the lysosomal pathway of protein degradation in isolated rat hepatocytes by ammonia, methylamine, chloroquine and leupeptin. Eur J Biochem 1979; 95:215-25; PMID:456353; http://dx.doi.org/10.1111/j.1432-1033.1979.tb12956.x
- Grinde B. Effect of carboxylic ionophores on lysosomal protein degradation in rat hepatocytes. Exp Cell Res 1983; 149:27-35; PMID:6641796; http://dx.doi.org/10.1016/0014-4827(83)90377-4
- Nakazato K, Hatano Y. Monensin-mediated antiport of Na+ and H+ across liposome membrane. Biochim Biophys Acta 1991; 1064:103-10; PMID:1851038; http://dx.doi.org/10.1016/0005-2736(91)90416-6
- Barth S, Glick D, Macleod KF. Autophagy: assays and artifacts. J Pathol 2010; 221:117-24; PMID:20225337; http://dx.doi.org/10.1002/path.2694
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/10.4161/auto.19496
- O’Neill PM, Bray PG, Hawley SR, Ward SA, Park BK. 4-Aminoquinolines–past, present, and future: a chemical perspective. Pharmacol Ther 1998; 77:29-58; PMID:9500158; http://dx.doi.org/10.1016/S0163-7258(97)00084-3
- Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, DiPaola RS, Lotze MT, White E. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res 2011; 17:654-66; PMID:21325294; http://dx.doi.org/10.1158/1078-0432.CCR-10-2634
- Florey O, Kim SE, Sandoval CP, Haynes CM, Overholtzer M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat Cell Biol 2011; 13:1335-43; PMID:22002674; http://dx.doi.org/10.1038/ncb2363
- Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011; 7:279-96; PMID:21189453; http://dx.doi.org/10.4161/auto.7.3.14487
- Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol 2009; 10:1215-21; PMID:19820708; http://dx.doi.org/10.1038/ni.1800
- Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 2012; 482:414-8; PMID:22246324; http://dx.doi.org/10.1038/nature10744
- Marceau F, Bawolak MT, Lodge R, Bouthillier J, Gagne-Henley A, Gaudreault RC, Morissette G. Cation trapping by cellular acidic compartments: beyond the concept of lysosomotropic drugs. Toxicol Appl Pharmacol 2012; 259:1-12; PMID:22198553; http://dx.doi.org/10.1016/j.taap.2011.12.004
- Mollenhauer HH, Morre DJ, Rowe LD. Alteration of intracellular traffic by monensin; mechanism, specificity and relationship to toxicity. Biochim Biophys Acta 1990; 1031:225-46; PMID:2160275; http://dx.doi.org/10.1016/0304-4157(90)90008-Z
- Savage DF, Stroud RM. Structural basis of aquaporin inhibition by mercury. J Mol Biol 2007; 368:607-17; PMID:17376483; http://dx.doi.org/10.1016/j.jmb.2007.02.070
- Schorn C, Frey B, Lauber K, Janko C, Strysio M, Keppeler H, Gaipl US, Voll RE, Springer E, Munoz LE, et al. Sodium overload and water influx activate the NALP3 inflammasome. J Biol Chem 2011; 286:35-41; PMID:21051542; http://dx.doi.org/10.1074/jbc.M110.139048
- Overholtzer M, Mailleux AA, Mouneimne G, Normand G, Schnitt SJ, King RW, Cibas ES, Brugge JS. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell 2007; 131:966-79; PMID:18045538; http://dx.doi.org/10.1016/j.cell.2007.10.040
- Leunk RD, Johnson PT, David BC, Kraft WG, Morgan DR. Cytotoxic activity in broth-culture filtrates of Campylobacter pylori. J Med Microbiol 1988; 26:93-9; PMID:3385767; http://dx.doi.org/10.1099/00222615-26-2-93
- Garner JA, Cover TL. Binding and internalization of the Helicobacter pylori vacuolating cytotoxin by epithelial cells. Infect Immun 1996; 64:4197-203; PMID:8926088
- Szabo I, Brutsche S, Tombola F, Moschioni M, Satin B, Telford JL, Rappuoli R, Montecucco C, Papini E, Zoratti M. Formation of anion-selective channels in the cell plasma membrane by the toxin VacA of Helicobacter pylori is required for its biological activity. EMBO J 1999; 18:5517-27; PMID:10523296; http://dx.doi.org/10.1093/emboj/18.20.5517
- Genisset C, Puhar A, Calore F, de Bernard M, Dell’Antone P, Montecucco C. The concerted action of the Helicobacter pylori cytotoxin VacA and of the v-ATPase proton pump induces swelling of isolated endosomes. Cell Microbiol 2007; 9:1481-90; PMID:17253977; http://dx.doi.org/10.1111/j.1462-5822.2006.00886.x
- Morbiato L, Tombola F, Campello S, Del Giudice G, Rappuoli R, Zoratti M, Papini E. Vacuolation induced by VacA toxin of Helicobacter pylori requires the intracellular accumulation of membrane permeant bases, Cl(-) and water. FEBS Lett 2001; 508:479-83; PMID:11728476; http://dx.doi.org/10.1016/S0014-5793(01)03133-7
- Molinari M, Galli C, Norais N, Telford JL, Rappuoli R, Luzio JP, Montecucco C. Vacuoles induced by Helicobacter pylori toxin contain both late endosomal and lysosomal markers. J Biol Chem 1997; 272:25339-44; PMID:9312153; http://dx.doi.org/10.1074/jbc.272.40.25339
- Ricci V, Sommi P, Fiocca R, Romano M, Solcia E, Ventura U. Helicobacter pylori vacuolating toxin accumulates within the endosomal-vacuolar compartment of cultured gastric cells and potentiates the vacuolating activity of ammonia. J Pathol 1997; 183:453-9; PMID:9496263; http://dx.doi.org/10.1002/(SICI)1096-9896(199712)183:4%3c453::AID-PATH950%3e3.0.CO;2-2
- Li M, Khambu B, Zhang H, Kang JH, Chen X, Chen D, Vollmer L, Liu PQ, Vogt A, Yin XM. Suppression of lysosome function induces autophagy via a feedback down-regulation of MTOR complex 1 (MTORC1) activity. J Biol Chem 2013; 288:35769-80; PMID:24174532; http://dx.doi.org/10.1074/jbc.M113.511212
- Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, Hengartner MO, Green DR. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Nat Acad Sci U S A 2011; 108:17396-401; PMID:21969579; http://dx.doi.org/10.1073/pnas.1113421108
- Henault J, Martinez J, Riggs JM, Tian J, Mehta P, Clarke L, Sasai M, Latz E, Brinkmann MM, Iwasaki A, et al. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity 2012; 37:986-97; PMID:23219390; http://dx.doi.org/10.1016/j.immuni.2012.09.014
- Berger SB, Romero X, Ma C, Wang G, Faubion WA, Liao G, Compeer E, Keszei M, Rameh L, Wang N, et al. SLAM is a microbial sensor that regulates bacterial phagosome functions in macrophages. Nat Immunol 2010; 11:920-7; PMID:20818396; http://dx.doi.org/10.1038/ni.1931
- Ma J, Becker C, Lowell CA, Underhill DM. Dectin-1-triggered recruitment of light chain 3 protein to phagosomes facilitates major histocompatibility complex class II presentation of fungal-derived antigens. J Biol Chem 2012; 287:34149-56; PMID:22902620; http://dx.doi.org/10.1074/jbc.M112.382812
- Reeves EP, Lu H, Jacobs HL, Messina CG, Bolsover S, Gabella G, Potma EO, Warley A, Roes J, Segal AW. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature 2002; 416:291-7; PMID:11907569; http://dx.doi.org/10.1038/416291a
- Lamb FS, Moreland JG, Miller FJ Jr. Electrophysiology of reactive oxygen production in signaling endosomes. Antioxid Redox Signal 2009; 11:1335-47; PMID:19207039; http://dx.doi.org/10.1089/ars.2008.2448
- Segal AW. The function of the NADPH oxidase of phagocytes and its relationship to other NOXs in plants, invertebrates, and mammals. Int J Biochem Cell Biol 2008; 40:604-18; PMID:18036868; http://dx.doi.org/10.1016/j.biocel.2007.10.003
- Sun-Wada GH, Tabata H, Kawamura N, Aoyama M, Wada Y. Direct recruitment of H+-ATPase from lysosomes for phagosomal acidification. J Cell Sci 2009; 122:2504-13; PMID:19549681; http://dx.doi.org/10.1242/jcs.050443
- Maejima I, Takahashi A, Omori H, Kimura T, Takabatake Y, Saitoh T, Yamamoto A, Hamasaki M, Noda T, Isaka Y, et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J 2013; 32:2336-47; PMID:23921551; http://dx.doi.org/10.1038/emboj.2013.171
- Hung YH, Chen LM, Yang JY, Yang WY. Spatiotemporally controlled induction of autophagy-mediated lysosome turnover. Nat Commun 2013; 4:2111; PMID:23817530; http://dx.doi.org/10.1038/ncomms3111
- Terebiznik MR, Raju D, Vazquez CL, Torbricki K, Kulkarni R, Blanke SR, Yoshimori T, Colombo MI, Jones NL. Effect of Helicobacter pylori's vacuolating cytotoxin on the autophagy pathway in gastric epithelial cells. Autophagy 2009; 5:370-9; PMID:19164948; http://dx.doi.org/10.4161/auto.5.3.7663
- Romao S, Gasser N, Becker AC, Guhl B, Bajagic M, Vanoaica D, Ziegler U, Roesler J, Dengjel J, Reichenbach J, et al. Autophagy proteins stabilize pathogen-containing phagosomes for prolonged MHC II antigen processing. J Cell Biol 2013; 203:757-66; PMID:24322427; http://dx.doi.org/10.1083/jcb.201308173
- Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature 2010; 466:68-76; PMID:20562859; http://dx.doi.org/10.1038/nature09204
- Ma J, Becker C, Reyes C, Underhill DM. Cutting edge: FYCO1 recruitment to dectin-1 phagosomes is accelerated by light chain 3 protein and regulates phagosome maturation and reactive oxygen production. J Immunol 2014; 192:1356-60; PMID:24442442; http://dx.doi.org/10.4049/jimmunol.1302835
- Weidberg H, Shpilka T, Shvets E, Abada A, Shimron F, Elazar Z. LC3 and GATE-16 N termini mediate membrane fusion processes required for autophagosome biogenesis. Dev Cell 2011; 20:444-54; PMID:21497758; http://dx.doi.org/10.1016/j.devcel.2011.02.006