
Abstract
Antibody-targeted nanoparticles have the potential to significantly increase the therapeutic index of cytotoxic anti-cancer therapies by directing them to tumor cells. Using antibodies or their fragments requires careful engineering because multiple parameters, including affinity, internalization rate and stability, all need to be optimized. Here, we present a case study of the iterative engineering of a single chain variable fragment (scFv) for use as a targeting arm of a liposomal cytotoxic nanoparticle. We describe the effect of the orientation of variable domains, the length and composition of the interdomain protein linker that connects VH and VL, and stabilizing mutations in both the framework and complementarity-determining regions (CDRs) on the molecular properties of the scFv. We show that variable domain orientation can alter cross-reactivity to murine antigen while maintaining affinity to the human antigen. We demonstrate that tyrosine residues in the CDRs make diverse contributions to the binding affinity and biophysical properties, and that replacement of non-essential tyrosines can improve the stability and bioactivity of the scFv. Our studies demonstrate that a comprehensive engineering strategy may be required to identify a scFv with optimal characteristics for nanoparticle targeting.
Abbreviations
| scFv | = | single chain variable fragment |
| CDR | = | complementarity-determining region |
| EphA2 | = | ephrin type-A receptor 2 |
| FACS | = | fluorescence-activated cell sorting |
| DSF | = | differential scanning fluorimetry |
| HTP | = | high throughput |
| CLIA | = | chelated ligand-induced internalization assay |
Introduction
Nanoparticle-based drugs have advanced cancer therapy by delivering highly cytotoxic payloads to cancer cells while lowering systemic drug toxicity via sustained release.Citation1-Citation3 Nanoparticles serve as a carrier to concentrate the encapsulated drug into tumors, thus providing an optimal pharmacological coverage and extending the therapeutic index.Citation1-Citation3 Paclitaxel and doxorubicin, 2 widely used chemotherapeutic agents, have been incorporated into nanoparticle formulations.Citation4-Citation6 Albumin-conjugated paclitaxel (nab-paclitaxel; Abraxane®) has been approved by the Food and Drug Administration for the treatment of non-small-cell lung cancer,Citation7 as well as advanced breast cancerCitation8 and pancreatic cancer.Citation9 Pegylated liposomal doxorubicin (Doxil®) is used to treat many cancers including ovarian carcinomas,Citation6 AIDS-related Kaposi's sarcoma,Citation10 and refractory multiple myeloma.Citation11
The use of antibodies to target cytotoxic drugs can further increase the deposition of the drug into tumors that overexpress aberrant receptors while limiting the exposure of non-cancer cells. This targeted delivery concept has been illustrated by the success of antibody-drug conjugates (ADCs) such as brentuximab vedotin (Adcetris®, Seattle Genetics), an anti-CD30 antibody conjugated with monomethyl aurastatin,Citation12 and ado-trastuzumab emtansine (Kadcyla®, Roche), an anti-HER2 antibody conjugated with emtansine,Citation13 but has not been broadly applied to nanoparticles.
For nanoparticle targeting, scFvs can be advantageous over full-length antibodies because they have no effector function and do not alter the particle pharmacokinetics and biodistribution in Fc receptor mediated fashion.Citation14-Citation16 Broad medical application of scFvs has been limited, however, due to numerous biophysical challenges,Citation17 including the tendency of the molecules to aggregate and their low stability, especially at high concentrations. Moreover, robust conjugation of scFvs to lipid molecules requires incubation at temperatures greater than 60°C, and so high thermal stability of scFvs is highly desirable for nanoparticle targeting. Additionally, other biophysical properties must be optimized, including colloidal, serum, lipid phase and chemical denaturation stability. Finally, in order to be commercially manufactured, expression titers must be high and the scFvs selected should preferably bind to an affinity resin such as protein A.
To generate a ligand for nanoparticle targeting, antibodies or antibody fragments are selected for desired biological properties, including high affinity, cross-reactivity to other preclinical animal species, and internalization rates. scFvs are routinely isolated with in vitro display technologies using libraries that are derived from human repertoire (natural), computationally designed (synthetic), or are hybrid (semi-synthetic) in nature. Analysis of the amino acid distribution of antibodies from the human repertoire have revealed that tyrosine, glycine and serine are overrepresented in complementarity-determining regions (CDRs), Citation18 with tyrosine alone composing 10% of, and contributing up to 25% of, the antigen contact surface.Citation19-Citation21 Based on this statistical consideration, designed synthetic antibody libraries composed of a single frameworkCitation19 or multiple frameworksCitation22 often have a high frequency of tyrosines in the CDRs. While these tyrosines can facilitate antigen recognition, we have observed that they can also increase the instability of scFvs by providing a pleiotropic interaction surface for aggregate nucleation.
Here, we present a comprehensive engineering approach to optimize an anti-EphA2 scFv as a targeting ligand for a liposomal nanoparticle. EphA2 is a tyrosine kinase receptor that is selectively expressed on the surface of many solid tumors, but its targeting has shown only limited success in the clinic thus far.Citation23 Because nanoparticles provide more potent and better tunable delivery of chemotoxic agents toward tumors compared to ADCs, we sought to generate an EphA2-targeted nanoparticle for use as a potential cancer therapy. We isolated an anti-EphA2 scFv, clone 116, from a synthetic scFv library using yeast surface display. The initial hit had the essential functions of a targeting ligand: it internalized into cancer cell lines expressing human EphA2, it was cross-reactive to murine EphA2, and it could be readily conjugated to the lipid anchor and inserted efficiently into the liposomes. However, this clone did not have optimal thermostability and had the propensity to aggregate, which precluded its development into a therapeutic targeting agent. To improve the biophysical properties of this scFv while maintaining the affinity and epitope specificity, we explored the effect of orientation of the variable domains, interdomain protein linker composition, stabilizing mutations, and tyrosine replacements to the biophysical characteristics of clone 116. Ultimately, multiple approaches were required to engineer this scFv into a developable lead. Our studies suggest that a comprehensive optimization strategy is required to develop a scFv with optimal characteristics for nanoparticle targeting.
Results
Anti-EphA2 scFv lead identification and characterization
Anti-EphA2 binders were isolated from a synthetic, yeast-displayed scFv library through 5 rounds of selection consisting of 2 rounds of magnetic-activated cell sorting (MACS) and 3 rounds of quantitative fluorescent-activated cell sorting (FACS). To select specific and thermostable anti-EphA2 binders, progressively more stringent selection pressure was introduced into the last 2 rounds of FACS sorting. In the fourth round, the selected library was depleted with a mixture of biotinylated non-target proteins and streptavidin beads to remove non-specific binders. In the final round, to eliminate binders with low thermostability, the library was heat-shocked at 52.5°C for 5 minutes prior to antigen binding and sorting, and the binders were then picked.
To select scFvs with high thermal stability, these binders were further characterized using a more stringent thermal challenge assay. Specifically, yeast cells expressing individual scFvs were either heat shocked at 65°C or incubated at room temperature for 5 minutes prior to staining and FACS analysis. For every clone, we then calculated the thermal binding index defined as the ratio of the antigen binding signal of the heat shocked cells, compared to the signal of control cells that were not heat-shocked. Out of the 192 scFv binders that were characterized, approximately half showed residual binding between 60% and 110% (). Clones with residual binding of over 110% were deemed to be non-specific and eliminated from further analysis. DNA sequencing identified 42 unique thermostable clones, which were then characterized as soluble scFvs.
Figure 1. Measurement of thermostability and binding/internalization of anti-EphA2 scFvs 1A. Thermal challenge assay of scFvs on the surface of yeast. EphA2 binders from the final round of library selection were heat shocked prior to binding of EphA2-His6, and the antigen bound cells were evaluated for expression and EphA2 binding. The Y-axis represents the residual binding of the scFvs after incubation at 65°C for 5 minutes as measured by FACS. Mean fluorescent intensity (MFI) of antigen binding was normalized to MFI of expression. 1B. Chelated ligand-induced internalization assay (CLIA) detects the amount of scFv-conjugated liposomes on the cell surface or internalized. Soluble scFvs were conjugated to fluorescently labeled liposomes through a Ni (II)-activated nitrotriacetic (NTA) lipid, the fluorescent signal was measured and analyzed on FACS. Two scFvs (D2A6) and liposome without scFv were used as positive and negative control, respectively.
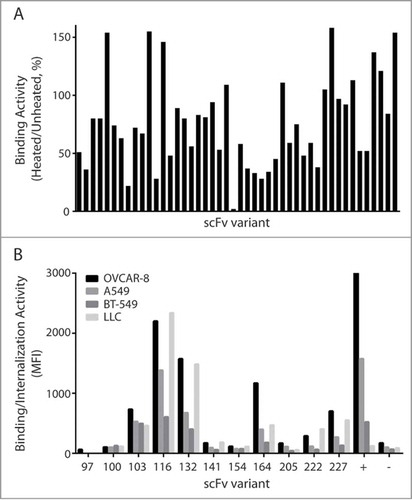
The hexahistidine tagged scFvs were expressed in S. cerevisiae and purified using a nickel resin. During purification, varying degrees of visible precipitation was observed for some clones and those with severe precipitation were not characterized further. We then measured the melting temperatures of the scFvs using differential scanning fluorimetry (DSF).Citation24 The scFvs had melting temperatures between 52.5°C to 65.5°C (Fig. S1). All of the characterized scFvs had melting temperatures equal to or higher than the screened temperature (52.5°C) of the yeast library, suggesting that thermal challenging the scFvs on the yeast surface prior to selection is an effective triage method for more thermostable antibodies.
Selection of internalizing antibodies
For a scFv to effectively target a liposomal nanoparticle, it needs to bind with high specificity and it must be capable of directing the internalization of the liposome. To screen for binding and internalization, we performed a high-throughput internalization screening assay,Citation25 which measures the amount of liposomal nanoparticles both on the cell surface and within the cell. Hexahistidine-tagged scFvs were conjugated to a fluorescently labeled liposome, added to cells expressing EphA2, and allowed to internalize for 4 hours. Fluorescence was measured both before and after dissociation of the bound antibodies by imidazole, allowing for quantification of the percentage of scFvs internalized. As shown in , 4 clones (103, 116, 132 and 164) demonstrated strong binding and internalization activity on cell lines expressing human or murine EphA2. Due to its superior internalization and melting temperature, clone 116 was selected as the scFv candidate best suited for further engineering for development into a therapeutic lead.
Engineering strategy for optimization of clone 116
The isolated scFv 116 (shown in Table S1) showed excellent cell binding and internalization activity in human and mouse cell lines expressing EphA2. However, its Tm of 57.5°C was lower than the desired 60°C for liposome conjugation and its partial precipitation during purification suggested that its stability was suboptimal. Before embarking on an engineering campaign, we first removed an aberrant N-linked glycosylation site on CDR-H1 by making a S30A mutation in the third position of NxS motif within CDR-H1. Homology modeling of 116 suggested that the sugar moiety was projected away from the antigen binding site, and, consistent with that model, the binding activity and cross reactivity were not affected after deglycosylation.
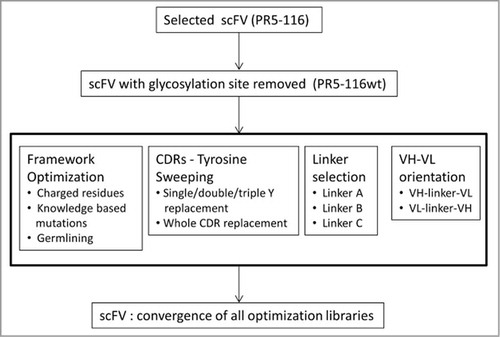
To improve the thermostability and solubility of 116 while maintaining its antigen binding, ability to internalize, and murine cross reactivity, a comprehensive engineering and optimization strategy was employed (). We designed a multitude of variants by switching variable domain orientation, optimizing the interdomain protein linkers, stabilizing the framework, CDR tyrosine “sweeping,” and the introduction of negatively-charged amino acids. All reengineered scFv variants were transiently expressed in mammalian cells and characterized in biophysical and functional assays.
Figure 2. Optimizing anti-EphA2 scFv 116 required several engineering strategies. The N-linked glycosylation site was removed from the parental clone prior to optimization. The iterative engineering approaches include (from left to right): (1) optimizing framework, (2) minimizing tyrosine content in the CDRs, (3) changing the interdomain protein linker, and (4) altering the VH/VL orientation. The best variants resulting from each approach were combined to yield lead candidates.
Initial engineering: stabilization of framework, interdomain protein linker optimization, and domain swapping
Framework optimization
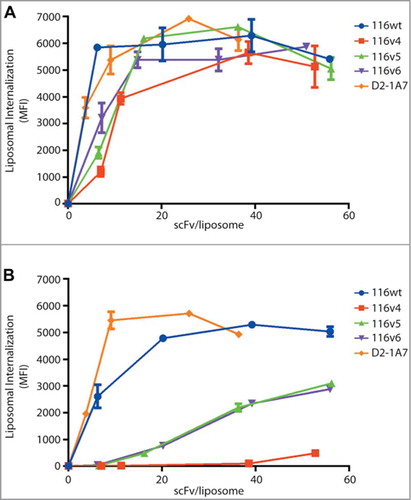
The framework of an antibody plays an important role in stability and affects the affinity by supporting the conformation of the CDRs. We hypothesized that introducing mutations into the framework may improve the biophysical properties of the scFv. The heavy chain is a member of the VH3 family (VH3-23), which is known to be stableCitation26 and capable of binding to protein A.Citation27,Citation28 Therefore, we only made one mutation, A49S, to make it more consistent with the VH3 consensus amino acid usage. However, we did extensive engineering to the light chain (VK1-39). We first added charge (S12E, S60D, and F83E) on the surface of the scFv to promote its stability.Citation29,Citation30 We also changed M4 to L and V58 to I to improve packing of the VL domain core.Citation31,Citation32 Finally, we ran the protein stability prediction algorithm Eris,Citation33 and changed R66 to K or to A. We introduced 3 mutations (VH-A49S, VL-R66K and VL-F83E) into the 116v4 framework and 7 mutations (VH-A49S, VL-M4L, VL-S12E, VL-V58I, VL-S60D, VL-R66A and VL-F83E) in the framework of 116v5. As a control, 116v6 had no additional mutations in the framework from the original wild-type clone, but it had both a different domain orientation and interdomain protein linker. The liposome conjugated variants showed comparable binding and internalization activity on human cell lines compared to 116wt clone (). Interestingly, the binding and internalization of these variants in murine cells line were significantly decreased (), indicating that their cross-reactivity to murine EphA2 was affected. Unexpectedly, changing the interdomain protein linker and orientation of the variable domains appeared to improve the melting temperature from 58°C to 63oC, but the expression was decreased 10-fold (). These unanticipated observations prompted us to investigate these engineering parameters more thoroughly.
Table 1. Sequence comparison and biophysical characterization of 116 and 116 variants
Figure 3. Variable domain or interdomain protein linker composition affect murine cross reactivity of anti-EphA2s. The chelated ligand-induced internalization assay was used to detect the amount of scFv-conjugated liposomes internalized on (A) human cancer cell line H-1993 and (B) murine cell line LLC-2. A benchmark antibody, 1A7, was used as positive control.
Domain swapping and interdomain protein linker composition variation
We then decided to test the domain orientation and interdomain protein linker composition separately in order to determine which parameter affected the stability and expression titers of the scFvs. To first test the effects of switching the orientation of the variable domains, we compared 116 in both orientations, VH-VL (116v7) and VL-VH (116v9) with the interdomain protein linker from the screened library. We found that the VL-VH orientation yielded 50 mg/L, while the VH-VL yielded 3.8 mg/L (). The melting temperatures were similar (65.4°C for 116v7 and 64°C for 116v9), suggesting that the altering the domain orientation of this scFv mainly affects expression.
We also tested different interdomain protein linkers that tether the variable regions together. Traditionally, these linkers compose low complexity amino acids (such as glycine and serine) to reduce potential immunogenicity.Citation34,Citation35 In this study, we compared the effect of 3 interdomain protein linkers on the biophysical properties and binding affinity of 116 variants (). Linker A was used in our library throughout the selections. Linker B is similar in composition to linker A, with a high percentage of glycines and serines, but it contains an additional 7 amino acids. Despite this similarity, we found that variants with linker A consistently had much higher expression (). We also tested a less hydrophilic linker, linker C. When comparing this linker (116v18) directly with the linker used in the library (116v7), we found that using linker C increased the melting temperature 4°C, but lowered expression 30-fold (). These results indicate that the choice of interdomain protein linker can affect both the thermal stability and expression yield of an engineered scFv irrespective of CDR composition.
Table 2. Composition and comparison of the interdomain protein linkers
Optimization of CDRs
CDR “tyrosine sweeping”
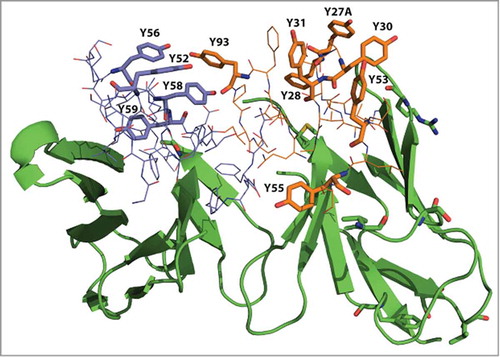
Tyrosine residues are often highly represented in the CDRs and play a critical role in antibody-antigen interaction by forming multiple types of macromolecular interactions.Citation36,Citation37 However, we have observed that abundant tyrosines in CDRs can promote instability of scFvs by providing an interaction surface for aggregates to nucleate. Sequence analysis of 116 revealed that 11 tyrosines are in the CDRs, representing 19.7% of their composition (). We then compared these tyrosines to a human consensus sequence we generated by compiling antibody repertoires from the National Center for Biotechnology Information (NCBI) protein sequence database, and noting the most common amino acid found at each position. Of these 11 tyrosines, only one, VH-Y59, is present in human consensus sequences at greater than 90% frequency, while the other 10 could have been acquired during the selection process. As shown in , these non-consensus tyrosine residues were located both in the light chain CDRs (Y27A, Y28, Y30, Y31, Y53 and Y55, Y93) and in the heavy chain CDR-H2 (Y52, Y56, Y58 and Y59). We hypothesized that the high number of tyrosines in clone 116 could be contributing to its overall instability.
Figure 4. Homology modeling of 116 shows abundance of tyrosines in the CDRs. Tyrosine residues in VL CDRs (Y27, Y28, Y30, Y31, Y53, Y55, Y93) are shown in orange and tyrosine residues in VH CDR2 (Y52, Y56, Y58 and Y59) are purple.
To investigate whether these tyrosine residues were essential to the antigen binding of clone 116, 2 dimensional “tyrosine sweeping” was carried out as shown in . Each tyrosine was individually mutated to the most frequently occurring amino acid based on our human consensus sequence. We also created double mutants to explore pairwise substitution effects. Over 60 scFv variants were made and the binding activity to human EphA2 was assessed on yeast surface by FACS analysis. Mutations in 4 positions, VH-Y52, VH-Y58, VL-Y30 and VL-Y31, significantly affected antigen binding activity, showing that these tyrosines were essential for EphA2 binding. Interestingly, the VH-Y56S mutation improved the antigen binding when combined with certain light chain variants including the wild type, VL-Y27S, and VL-Y55Q ().
Figure 5. Two dimensional tyrosine sweeping scheme. (1) Tyrosines of 116wt are mutated to corresponding consensus residues. (2) A consensus sequence clone was created first, then tyrosine were re-inserted into the CDRs.
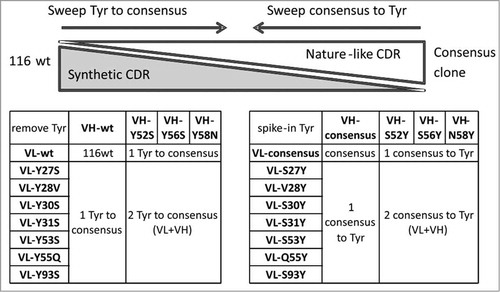
Figure 6. scFv variants with single or double tyrosine mutations to human consensus that retained binding activity to EphA2. Tyrosine sweeping-based identification of tyrosines that are essential for antigen binding. scFv variants with tyrosine to consensus mutations were expressed on yeast surface, the binding activity was measure by FACS.
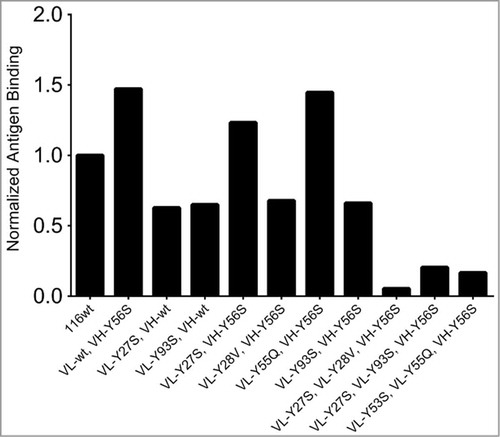
We next generated variants with triple and quadruple tyrosine mutations to the human consensus sequence. Variants with triple mutations lost over a 100-fold in binding activity () and the quadruple mutations completely lost antigen binding. These results show that it is only possible to remove a single tyrosine from each variable region without loss of at least some affinity. Three variants (VL-Y27AS+VH-Y56S, VL-Y55Q+VH-Y56S and VL-Y53S+VH-Y56S) (116v15, 116v16, 116v17) that still retained wild-type binding affinity were selected for characterization as soluble proteins. After expression, all 3 clones showed no visible precipitation during protein A purification. 116v17 was found to be the superior molecule with the highest level of expression (7.3 mg/L) and a melting temperature of 68.6°C.
Finally, we investigated the role of tyrosines in CDR1 and CDR2 on both the heavy and light chain by changing the loops to the consensus sequence and adding the tyrosines back individually. Interestingly, we found that when the CDR1 or CDR2 were replaced with consensus sequences, these variants lost their binding completely. Although VL-CDR2 had low diversity in our library and was thought not to participate in the antigen binding, it did not tolerate the replacement with a consensus sequence. This suggested that the effect of VL-CDR2 on binding was not negligible and incorporation of targeted VL-CDR2 diversity into synthetic libraries was warranted. Further tyrosine spiked-back-in into consensus VL-CDR2 did not restore the binding ability, suggesting that the effect of tyrosine residues was conformation dependent and that tyrosine-rich loops of the same length adopted different conformation than canonical ones.
Effect of negatively charged residues on the biophysical properties of scFvs
It has been previously shown that adding negative charge, especially aspartate, to the loops of antibodies can improve their biophysical behaviors.Citation37,Citation38 Therefore, we created 6 scFv variants (116v20 - v25) by introducing negative charge within VH-CDR1 and VL-CDR2. To build on our earlier engineering insight, we also varied the linkers and replaced selected tyrosines with consensus amino acids. We found that the thermostability of many variants was improved by over 5 degrees (), but that the expression was variable, ranging from 1.4 mg/L to 10.4 mg/L. Our results suggest that increasing the negative charge in the CDRs generally result in an increase in thermostability, but improvements in expression need to be more empirically determined within the context of specific scFv sequences.
Recombination of the best variants to deliver scFvs that support robust liposome conjugation
After comparing properties of the variants generated through framework stabilization, varying the domain orientation, changing the interdomain protein linker, CDR tyrosine replacement and introduction of negatively charged amino acids into CDRs, we identified 4 scFvs (116v7, 116v17, 116v23 and 116v25) as potential leads for nanoparticle targeting. We then performed detailed kinetic analysis of their binding activity to human, cynomolgus monkey, and murine EphA2 (). We found that all our potential lead molecules had equivalent binding to the 3 antigens to the original 116 scFv, while 116v25 showed slightly improved antigen recognition in all 3 species. Consistent with our earlier findings, these scFvs all featured the same domain orientation (VL-VH) and intervening linker (linker A). However, they contained varying number of tyrosines in the CDRs and varying amounts of negative charge. These molecules were then scaled up to produce 10 mg of scFvs in a transient mammalian system and tested for their ability to be conjugated to liposomes, as well as for biophysical properties.
Table 3. Characterization of potential lead scFvs
Liposome conjugation of the selected scFvs
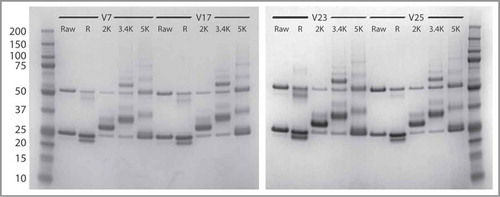
The selected scFvs were conjugated to a liposome using maleimide chemistry and polyethylene glycol (PEG) linkers of varying lengths (2K PEG, 3.4K PEG, and 5K PEG). The reactions were monitored for precipitation, and following an overnight incubation at room temperature, the conjugated liposomes were spun down, and the percent of precipitate was calculated for each scFv and linker combination. The conjugated liposomes were then run on SDS-PAGE to analyze the efficiency of the conjugation. As shown in , 116v7, 116v17, 116v23 and 116v25 were all successfully conjugated using the 5K PEG and 3.4K PEG linkers. However, there was a high level of precipitation (Table S2) when using the 2K PEG linker with all 4 scFvs. When using the 3.4K PEG linker, the amount of precipitation was variable. 116v17 did not precipitate at all, while 116v23 formed 20% precipitate. 116v7 and 116v25 had almost double the amount of precipitate, around 40%.
Figure 7. SDS PAGE analysis of scFv and covalently conjugated scFv-liposome (V7, V17, V23 and V25). There are 5 lanes for each variants: (1) raw: scFv; (2) R: reduced scFv; (3) 2K: scFv-Peg(2K)-DSPE conjugate; (4) 3.4K: scFv-Peg(3.4K)-DSPE conjugate; (5) 5K: scFv-Peg(5K)-DSPE conjugate.
Final selection of lead molecule
Our 4 lead scFvs, 116v7, 116v17, 116v23, and 116v25 had similar bioactivity as measured by binding and internalization on cells overexpressing EphA2. Additionally, kinetic analysis of binding to recombinant human, cynomolgus monkey, and murine EphA2 revealed their affinities were similar to the parental clone 116, ensuring we maintained both the affinity and cross reactivity of the antibodies. Therefore, we triaged our candidates based on their biophysical characteristics. Given our already significant screening of the scFvs based on purification stability, melting temperature, and expression, all of the selected clones had acceptable profiles for advancement. However, given the higher stability of 116v17 and 116v23 in the liposome conjugation studies, we decided that they were the best candidate anti-EphA2 scFvs for liposome nanoparticle targeting. Our 2 final scFvs, 116v17 and 117v23, kept the bioactivity of the original 116 clone while having much improved biophysical properties over the parental clone, including a melting temperature that was over 10 degrees higher, as well as no precipitation during the scFv purification process. One of these candidates, 116v17, did not precipitate as a liposome conjugate, thus meeting our predefined developability criteria for advancement as therapeutic lead for nanoliposomal targeting.
Discussion
scFvs generally require extensive engineering to be used as components of drugs due to their low intrinsic developability.Citation29,Citation39 To be useful as targeting ligands for nanoparticle delivery, scFvs need to internalize efficiently into antigen expressing cells to release the drug inside the cell, preferably cross-react to the target antigen in rodents and non-human primates to enable in vivo activity, pharmacokinetics and toxicological evaluation, and be highly thermostable to support liposome conjugation. In this study, we applied several engineering approaches to co-optimize these properties in anti-EphA2 scFvs. We introduced framework and CDR mutations, altered the interdomain protein linkers, and changed the orientation of the variable domains. Although scFvs can be found in both orientations, VH-VL is generally favored because the interdomain protein linker is further removed from CDR-H3, which is often a major contributor of antigen binding. However, in our case we found that VL-VH orientation in combination with a low complexity interdomain protein linker favors target binding, internalization capability, and expression of the scFv. Interestingly, in our study the 116v6 variant created in VH-VL orientation had poor cross-reactivity to murine EphA2 while the affinity to human EphA2 was mostly retained. This observation highlights the stress that even a long, low complexity interdomain protein linker can impose on the interface of the variable domains, possibly by introducing a paratope twist. We also observed that the best-expressing molecules featured original VL-VH orientation from the screened library. We also found, consistent with previously published results,Citation40 that adding negative charge in the CDRs can improve the physical stability of the molecule with one exception of 116V19. In our selected mutants, we found that the melting temperature was typically increased by 5 degrees. Although it is often stated that improving biophysical stability can lead to improved expression, we found that not to be the case for the clones we screened.
Regardless of the origin of the antibody, the abundance of tyrosine and serine residues in CDRs has been well documented. Tyrosine residues constitute 10% of the total CDR composition and up to 25% of the antigen contact surface.Citation41-Citation43 The tyrosine residues on the CDRs often play critical role in antigen-antibody binding affinity and specificity.Citation36,Citation42 However, due to their hydrophobic properties, there is concern about the solubility and specificity of scFvs if tyrosine residues are overly represented. This concern may be magnified when biased amino acid repertoire synthetic or semi-synthetic antibody libraries are used.
We applied “tyrosine sweeping” in this study to explore the spectrum of sequences between human consensus and 116 that featured abundant tyrosines in CDRs. We investigated the contribution of CDR tyrosine residues to the antigen binding activity of scFv through tyrosine-to-consensus mutation, or through replacing whole CDR with consensus composition followed by spiking-in tyrosines. By sequence and homology analysis, 10 tyrosines on the CDRs were identified as non-consensus residues while one was highly conserved. Out of these 10 non-canonical CDR tyrosines, only 4 were essential for the antigen binding, and 6 tyrosines were non-essential, suggesting they are “passive," antigen-independent enrichment. Interestingly, when we combined nonessential tyrosine-to-consensus mutations, the binding activity to the antigen was improved, again suggesting that non-essential tyrosines were being selected in a nonspecific fashion.
Our studies suggest that abundant tyrosines can be detrimental to scFv biophysical properties. In another antibody discovery campaign for nanoparticle conjugation, we successfully compiled the concepts of tyrosine replacement from this work into a single step approach of batch scFv optimization. Therefore, the tyrosine sweeping strategy described in this report is antigen independent, and can be applied to optimization of other tyrosine-rich antibodies.
In summary, we have developed a comprehensive iterative engineering strategy to optimize scFvs for targeting of liposome nanoparticles. Our approach is distinct from the strategy used for the generation of targeting ligands for antibody-drug conjugates: it combines high-throughput liposome conjugation, internalization screening and biophysical analysis with rational molecular engineering. This engineering includes optimization of interdomain protein linker composition, variable domain orientation, framework stabilization and CDR mutagenesis toward human consensus. Using the described strategy, we successfully identified 4 developable anti-EphA2 scFv variants with the desired properties for nanoparticle targeting. Our 4 potential targeting scFvs varied in expression, melting temperature, and stability during conjugation, but were all superior to the original clone isolated from our library. Two, antibodies 116v17 and 116v23, showed improved stability during scFv purification and high melting temperature. Clone 116v17, which showed no precipitation during the liposome conjugation process, was nominated as the therapeutic lead for nanoliposomal targeting campaign. This study emphasizes the importance of high-throughput assays to test bioactivity and biophysical properties in order to screen hundreds of antibodies with small amounts of material. It also highlights a primary advantage of pursuing non-redundant focused engineering strategies in parallel, which gives us the ability to rapidly advance antibodies and their ligands from proof-of-concept molecules to viable therapeutic leads.
Materials and Methods:
Materials
Cell culture reagents
F17 media, L-glutamine, pluronic F-68, 5000X Sypro orange solution and pCEP4 vector, RPMI and 10% FBS were purchased from Invitrogen. Dextrose, galactose, raffinose, lithium acetate, salmon sperm DNA (ssDNA) and PEG3500 were obtained from Sigma. Linear Polyethylene imine (PEI, MW, 25 kDa) was acquired from Polysciences. All agar plates were from Teknova. Yeast extract, peptone, yeast nitrogen base and casamino acids were from VWR. RPMI-1640, DMEM and 1x PBS were from Lonza. PenStrep 100X, 0.25% Trypsin-EDTA, Hank's balanced Salt Solution (Ca2+, Mg+) and Hank's balanced Salt Solution (Ca-, Mg-) were from Gibco. The 96-well “V” shaped assay plate was from Axygen Scientific.
Molecular biology reagents
All PCR primers were custom synthesized by IDT. iProof High-Fidelity DNA Polymerase was purchased from Bio-Rad. Zymoprep yeast plasmid miniprep kits were purchased from Zymo Research and DNA MegaPrep kits were from Qiagen. PD-10 disposable columns (Sephadex G-25) were from GE Healthcare. All solutions were prepared in 18MΩ DI water.
Cell lines and other reagents
Human cancer cell lines (H-1993, A549, OVCAR-8) and mouse Lewis lung carcinoma cell line (LL-2) were ordered from ATCC. 293F cells and fluorescent lipid DiIC18(5)-DS (DiI5), M2 anti-Flag antibody, disodium phosphate, monosodium phosphate, Alexa Fluor 647 and 488 and goat-anti-human IgG (Fc specific) were purchased from Invitrogen. EphA2-His and anti-His6 mAb were purchased from R&D Systems. EZ-Link Sulfo-NHSLC-Biotin, Fluorescence Biotin Quantitation Kit and DTT were purchased from Pierce. Streptavidin MicroBeads and LS Columns (MACS columns) were purchased from Miltenyi. TSKgel SuperSW3000 column was purchased from Tosoh Bioscience. Hydrogenated soybean phosphatidylcholine (HSPC) and methoxy-PEG(2000)-distearoyl phosphatidylethanilamine (PEG-DSPE) were from Lipoid AG (Germany). Cholesterol (plant-derived) and maleimido-PEG(2000)-DSPE were from Avanti Polar Lipids (Alabama, USA). Nuclepore PCTE membranes with 100 nm pore size were from Whatman (USA). All chemicals were of analytical or greater purity.
Selection of EphA2 binders from yeast displayed scFv library
The methods of library growth, induction and selection were performed as described previously.Citation44 In the first 2 rounds of selection, the library was bound to biotinylated anti-EphA2-His6, and captured by Streptavidin MicroBeads using MACS cell separation system. The subsequent 2 rounds of selection were carried out using FACS sorting on the BD Aria system.
Affinity assessment of yeast surface display scFv using FACS
The induced yeast cells were incubated with titrated amounts of EphA2-His6. Cells were washed twice with FACS buffer (1x PBS, 0.5% BSA, pH7.4) to remove unbound antigen. The antigen bound cells were then incubated with 1 μg/mL Alexa647 labeled M2 anti-FLAG antibody and 1 μg/mL Alexa 488 labeled anti-His6 antibody for 30 minutes. After 2 washes, the cells were resuspended in FACS buffer and the fluorescent signals were measured using a BD FACS Calibur. The median fluorescent intensities (MFI) were determined using FlowJo software. Antigen binding MFI (anti-His6-Alexa 488) was normalized to expression MFI (anti-Flag-Alexa 647).
Assessment of thermal stability of selected scFvs on yeast surface using “Cook and Bind” thermal challenge assay
To select for scFvs with high thermal stability, yeast cells expressing individual scFvs were either heat shocked at 65°C or incubated at room temperature for 5 minutes, chilled on ice for 5 minutes, and then incubated with 150 nM EphA2-His6 for 2 hours. These antigen bound cells were stained and analyzed by FACS as described previously.Citation44 The thermal binding index for each clone was calculated as the ratio of the antigen binding signal of heat shocked cells to the signal of the control cells.
Expression and purification of soluble His6-tagged scFvs in yeast
The plasmid DNA of unique binders was digested to remove both the Flag-tag and fusion gene and co-transformed with a PCR fragment containing hexahistidine tag into yeast cells to induce ligation via homologous recombination. Transformed yeast colonies were grown in SD-CAA media, induced in YPG media for 2 days at 20°C shaker while shaking at 225 rpm, and the scFvs were purified using Protein A column.
Homology modeling of clone 116 and generation of human consensus sequence
Sequence and structural based analysis was performed as described previously.Citation44 First, a homology model was created using MolIDECitation45 with zero-gapped templates for the variable regions. The model was energy minimized using SCWRLCitation46-Citation48 and DeepView Citation49 and Eris.Citation33 All variants were inspected visually in PyMol for potential steric clashes.
Tyrosine scanning
Databases of VH and VL protein sequences derived from NCBI Entrez Protein database were used to establish the individual amino acid frequencies at each residue position of natural antibody repertoire. The consensus analysis was performed in reference to sets of VH and VL consensus sequences defined by Kabat.Citation50 Each tyrosine residue in the CDR L1, L2, L3 or H2 of the initial scFv was identified and replaced with the amino acid that occurs most frequently in the natural antibody repertoire. Tyrosines that were found in a position more than 90% of the time were not mutated. The VH and VL variants were synthesized by IDT. The variable domains and interdomain protein linkers were amplified individually and assembled into full-length scFv by overlap PCR. The scFv PCR fragments containing designed mutations were co-transformed with XhoI and NheI digested yeast surface display vector into yeast cells. A few colonies of each transformed variants were grown and induced as described above. The antigen binding of these yeast surface displayed scFv variants were analyzed on FACS.
Expression and production of soluble scFvs in mammalian cells
All the re-engineered variants were expressed using transient transfection in 293F cells. The genes were synthesized at DNA 2.0 and subcloned into a modified pCEP4 vector containing the sequence “GGSGGC” at C-terminus for liposome conjugation. DNA was prepared according to Qiagen MegaPrep kit manual. For transient transfections, 293F cells were grown in cell culture media containing glutamine, pluronic F-68 and F17 media at 37°C, 5% CO2 to a density of 1.5–2.0 million/mL in a baffled shake flask and then transfected with 1 mg/L of DNA. After transfected cells were grown for 6 d, the culture media containing soluble scFv were harvested by centrifuging the cells at 5000 rpm, then filtered using an Akropak filter disc and re-filtered through a 0.22 μm vacuum filter. The soluble scFv was purified through a Protein A column (MAbSelect). Sucrose was added to the dialyzed protein to a final concentration of 5% to keep the scFvs soluble and stable.Citation35
Tm measurement by differential scanning fluorescence
The DSF assay was performed using IQ5 real time detection system (Bio-Rad). Briefly, for each measurement, 20 μL of the scFv solution containing 15 μM scFv protein and 1X Sypro Orange in 1X PBS was added to each well on a 96-well plate. The plate was heated from 20°C to 90°C at a rate of 1°C/min and the resulting fluorescence data collected. The data was transferred to GraphPad prism for analysis, and Tm was defined as the temperature of the maximal fluorescent value of the first derivative of the unfolding event.
High-throughput rapid scFv-assisted liposome cell uptake assay
To screen scFvs with good binding and internalization capability, a rapid high-throughput Chelated Ligand-induced Internalization Assay (CLIA)Citation25 was carried out as described below.
Conjugation of His6-tagged scFv onto fluorescent liposome
The scFv was conjugated to lipophilic fluorescent dye DiI18 (5)-DS containing liposomes through a Ni (II)-activated nitrotriacetic (NTA) lipid. The fluorescent liposome solution was prepared by diluting to 0.4 mM in HBSS then adding 0.1mM NiSO4 with vigorous mixing. His6-tagged scFv solution was prepared by diluting the scFv to a final concentration of 50 μg/mL in 100 μL of RPMI containing 10% FBS, Pen/Strep and glutamine on a 96-well “V” shaped plate. Using a multichannel pipette, 100 μL of the prepared fluorescent liposome solution was added to the 100 μL antibody solution per well on the plate and mixed by pipetting up and down several times.
scFv-mediated liposome internalization
Human cell lines (A-549, OVAR-8, BT-549) and murine cell line (LLC-2) were grown to 90% confluency. The culture media was removed, washed with Ca2+and Mg2+ free PBS and trypsinized with 0.25% Trypsin. The suspended cell solution was plated at 100 K/well in a 96-well “V” bottom shape polypropylene plate. The cells were washed with 1X PBS to remove any traces of EDTA that may interfere with the conjugate formation. After removal of PBS, 100 μL of the fluorescent liposome/antibody mixture was added into appropriate wells on the 96-well plate. The plate was covered with sealing tape and a piece of aluminum foil and incubated for 4 hrs at 37°C with 5% CO2 on a shaker to allow the scFv-assisted liposome binding to the cell surface target and subsequent internalization. After the liposome and antibody mixture was emptied to discard the unbound scFv-liposome conjugate, the attached cells were washed either with 200 μL/well of PBS to remove any unbound liposomes or with 200 μL/well of PBS containing 0.25 M imidazole (pH7.5) to remove any unbound liposome as well as the bound but not internalized liposomes. The fluorescent signals of these cells were analyzed by FACS.
Mammalian cell uptake of covalently conjugatedscFv-liposome
Covalently conjugated scFv-liposome and the mammalian cell uptake assay of these conjugates were carried out as follows:
Activation of terminal thiol of scFv proteins
Isolated scFv proteins were treated with 20 mM cysteine, 5 mM EDTA at 30°C for 1 hour to activate C-terminal cysteine thiol group. Free cysteine was removed by chromatography on PD-10 disposable columns (GE Healthcare, USA) in the conjugation buffer (5 mM citrate, 1 mM EDTA, 140 mM NaCl, pH 6.0). The reduced proteins contained 0.93 - 1.20 free thiol groups per molecule, as determined spectrophotometrically using Ellman's reagent.
Preparation and activation of liposomes
The chloroform solution containing HSPC, cholesterol, PEG-DSPE, and DiIC18(5)-DS mixed at the molar ratio of 3:2:0.24:0.009 was evaporated under reduced pressure to form a lipid film. The film was hydrated in a HEPES-buffered saline (5 mM HEPES, 144 mM NaCl, pH 6.5) at 65°C and 30 mM phospholipid, and the resulting multilamellar vesicles were repeatedly extruded under pressure through stacked 100-nm polycarbonate track-etched membranes (Nuclepore, Whatman, USA) using Lipex extruder (Northern Lipids, Canada). The liposome phospholipid concentration was determined spectrophotometrically after acid digestion using a modified molybdate method.Citation39 An mal-PEG-DSPE was inserted into the outer layer of the liposome membrane to provide an active group for scFv attachment by an incubation of the liposomes with mal-PEG-DSPE at 60°C for 20 min. The mal-PEG-DSPE and liposome mixture was quenched on ice and the liposomes were then purified from any traces of extraliposomal mal-PEG-DSPE by chromatography on Sepharose 4FF (GE Healthcare) in the conjugation buffer.
Covalent conjugation of scFv to liposome
The fractions containing activated liposome were collected and mixed with the activated scFv (reduced) at varying protein-phospholipid ratios to achieve at average 7.5, 15, 30, and 45 scFv per particle, as estimated using an averaged scFv molecular weight of 26 KD and 8 × 104 HSPC molecules per liposome. After overnight stirring at room temperature, the unreacted maleimide groups were blocked with 0.5 mM cysteine for 5 minutes and the scFv-liposome conjugates were purified by gel-chromatography on Sepharose 4FF, eluted in HEPES-buffered saline, pH 6.5. The amount of scFv molecules per liposome were determined by SDS-PAGE of the liposomes using Bio-Rad Mini-PROTEAN TGX gels and Coomassie G-250 staining according to the supplier's protocol, followed by the densitometry of the protein band. The sample buffer contains 4X SDS amount compared to regular 1X SDS, to achieve complete dissociation of the liposomes for release of conjugated scFvs. Free scFv proteins were run on the gel as reference standards. Control scFv-free liposomes were prepared identically, except that the protein solution was replaced by the conjugation buffer in the conjugation step.
Cell uptake of scFv-conjugated liposomes
Cultured human lung cancer cell line H-1993 and LL-2 mouse Lewis lung carcinoma cells were grown in RPMI containing 10% FBS, 2 mM glutamine and pep/strep and harvested by trypsinization. The cells were suspended in the growth medium and incubated with the liposomes (25 μM phospholipid) at 37°C for 4 hours. The cells were pelleted by centrifugation, re-suspended in PBS, and mean cell fluorescence intensity (MFI) of the DiI5 label was determined using FACScalibur (BD bioscience). The fluorescence signal is the sum of surface-bound and internalized nanoparticles. The MFI of the cells incubated with blank liposomes (no conjugated scFv) was 7.99 ± 1.21 for H-1993 cells and 11.07 ± 1.15 for LL-2 cells.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental_Files.zip
Download Zip (459.5 KB)Acknowledgments
We thank Professor Sachdev S Sidhu, The University ofToronto, for his assistance of generating synthetic antibody library.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Drummond DC, Meyer O, Hong K, Kirpotin DB, Papahadjopoulos D. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol Rev 1999; 51:691-743; PMID:10581328
- Drummond DC, Noble CO, Hayes ME, Park JW, Kirpotin DB. Pharmacokinetics and in vivo drug release rates in liposomal nanocarrier development. J Pharm Sci 2008; 97:4696-740; PMID:18351638
- Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev 2013; 65:36-48; PMID:23036225; http://dx.doi.org/10.1016/j.addr.2012.09.037
- Sparano JA, Malik U, Rajdev L, Sarta C, Hopkins U, Wolff AC. Phase I trial of pegylated liposomal doxorubicin and docetaxel in advanced breast cancer. J Clin Oncol 2001; 19:3117-25; PMID:11408509
- Gaitanis A, Staal S. Liposomal doxorubicin and nab-paclitaxel: nanoparticle cancer chemotherapy in current clinical use. Methods in Mol Biol 2010; 624:385-92; PMID:20217610; http://dx.doi.org/10.1007/978-1-60761-609-2_26
- Gordon AN, Fleagle JT, Guthrie D, Parkin DE, Gore ME, Lacave AJ. Recurrent epithelial ovarian carcinoma: a randomized phase III study of pegylated liposomal doxorubicin versus topotecan. J Clin Oncol 2001; 19:3312-22; PMID:11454878
- Socinski MA, Bondarenko I, Karaseva NA, Anatoly M. Makhson AM, Vynnychenko I, Okamoto I, Hon JK, Hirsh V, Bhar P, et al. Weekly nab-paclitaxel in combination with carboplatin versus solvent-based paclitaxel plus carboplatin as first-line therapy in patients with advanced non-small cell lung cancer: final results of a phase III trial. J Clin Oncol 2012; 30:2055-62; PMID:22547591; http://dx.doi.org/10.1200/JCO.2011.39.5848
- Gradishar WJ, Tjulandin S, Davidson N, Shaw H, Desai N, Bhar P, Hawkins M, O’Shaughnessy J. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J Clin Oncol 2005; 23:7794-803; PMID:16172456; http://dx.doi.org/10.1200/JCO.2005.04.937
- Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013; 369:1691-703; PMID:24131140; http://dx.doi.org/10.1056/NEJMoa1304369
- Gill PS1, Wernz J, Scadden DT, Cohen P, Mukwaya GM, von Roenn JH, Jacobs M, Kempin S, Silverberg I, Gonzales G, et al. Randomized phase III trial of liposomal daunorubicin versus doxorubicin, bleomycin, and vincristine in AIDS-related Kaposi's sarcoma. J Clin Oncol 1996; 14:2353-64; PMID:8708728
- Orlowski RZ, Nagler A, Sonneveld P, Bladé J, Hajek R, Spencer A, Miguel JS, Robak T, Dmoszynska A, Horvath N, et al. Phase III study of pegylated liposomal doxorubicin plus bortezomib compared with bortezomib alone in relapsed or refractory multiple myeloma: combination therapy improves time to progression. J Clin Oncol 2007; 25:3892-901; PMID:17679727; http://dx.doi.org/10.1200/JCO.2006.10.5460
- Pro B, Advani R, Brice P, Bartlett NL, Rosenblatt JD, Illidge T, Matous J, Ramchandren R, Fanale M, Connors JM, et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study. J Clin Oncol 2012; 30:2190-6; PMID:22614995; http://dx.doi.org/10.1200/JCO.2011.38.0402
- Krop IE, Kim SB, González-Martín A, LoRusso PM, Ferrero JM, Smitt M, Yu R, Leung AC, Wildiers H. Trastuzumab emtansine versus treatment of physician's choice for pretreated HER2-positive advanced breast cancer (TH3RESA): a randomised, open-label, phase 3 trial. Lancet Oncol 2014; 15:689-99; PMID:24793816; http://dx.doi.org/10.1016/S1470-2045(14)70178-0
- Noble CO, Kirpotin DB, Hayes ME, Mamot C, Hong K, Park JW, Benz CC, Marks JD, Drummond DC. Development of ligand-targeted liposomes for cancer therapy. Expert Opin Ther Targets 2004; 8:335-53; PMID:15268628; http://dx.doi.org/10.1517/14728222.8.4.335
- Koning GA, Morselt HWM, Gorter A, Allen TM, Zalipsky S, Scherphof GL, Kamps JAAM. Interaction of differently designed immunoliposomes with colon cancer cells and Kupffer cells. An in vitro comparison. Pharm Res 2003; 20:1249-57; PMID:12948023; http://dx.doi.org/10.1023/A:1025009300562
- Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong K, Nielsen UB, Marks JD, Benz CC, Park JW. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res 2006; 66:6732-40; PMID:16818648; http://dx.doi.org/10.1158/0008-5472.CAN-05-4199
- Wang T, Duan Y. Probing the stability-limiting regions of an antibody single-chain variable fragment: a molecular dynamics simulation study. PEDS 2011; 24:649-57; PMID:21729946
- Zemlin M, Klinger M, Link J, Zemlin C, Bauer K, Engler JA, Schroeder HW Jr, Kirkham PM. Expressed murine and human CDR-H3 intervals of equal length exhibit distinct repertoires that differ in their amino acid composition and predicted range of structures. J Mol Biol 2003; 334:733-49; PMID:14636599; http://dx.doi.org/10.1016/j.jmb.2003.10.007
- Birtalan S, Zhang Y, Fellouse FA, Shao L, Schaefer G, Sidhu SS. The intrinsic contributions of tyrosine, serine, glycine and arginine to the affinity and specificity of antibodies. J Mol Biol 2008; 377:1518-28; PMID:18336836; http://dx.doi.org/10.1016/j.jmb.2008.01.093
- Mian IS, Bradwell, AR, Olson AJ. Structure, function and properties of antibody binding sites. J Mol Biol 1991; 217:133-51; PMID:1988675; http://dx.doi.org/10.1016/0022-2836(91)90617-F
- Wilkinson RA, Piscitelli C, Teintze M, Cavacini LA, Posner MR, Lawrence CM. Structure of the Fab fragment of F105, a broadly reactive anti-human immunodeficiency virus (HIV) antibody that recognizes the CD4 binding site of HIV type 1 gp120. J Virology 2005; vol:13060-69; PMID:16189008; http://dx.doi.org/10.1128/JVI.79.20.13060-13069.2005
- Prassler J, Thiel S, Pracht C, Polzer A, Peters S, Bauer M, Nörenberg S, Stark Y, Kölln J, Popp A, et al. HuCAL PLATINUM, a synthetic Fab library optimized for sequence diversity and superior performance in mammalian expression systems. J Mol Biol 2011; 413:261-78; PMID:21856311; http://dx.doi.org/10.1016/j.jmb.2011.08.012
- Barrett NA, Austen FK. Innate cells and T helper 2 cell immunity in airway inflammation. Immunity 2009; 31:425-37; PMID:19766085; http://dx.doi.org/10.1016/j.immuni.2009.08.014
- Lavinder JJ, Hari SB, Sullivan BJ, Thomas J, Magliery TJ. High-throughput thermal scanning: a general, rapid dye-binding thermal shift screen for protein engineering. J Am Chem Soc 2009; 131:3794-95; PMID:19292479; http://dx.doi.org/10.1021/ja8049063
- Nielsen UB, Kirpotin DB, Pickering EM, Drummond DC, Marks JD. A novel assay for monitoring internalization of nanocarrier coupled antibodies. BMC Immunol 2006; 7:24; PMID:17014727; http://dx.doi.org/10.1186/1471-2172-7-24
- Ewert S1, Huber T, Honegger A, Plückthun A. Biophysical properties of human antibody variable domains. J Mol Biol 2003; 325:531-53; PMID:12498801; http://dx.doi.org/10.1016/S0022-2836(02)01237-8
- Starovasnik MA1, O’Connell MP, Fairbrother WJ, Kelley RF. Antibody variable region binding by Staphylococcal protein A: thermodynamic analysis and location of the Fv binding site on E-domain. Protein Sci 1999; 8:1423-31; PMID:10422830; http://dx.doi.org/10.1110/ps.8.7.1423
- Roben PW1, Salem AN, Silverman GJ. VH3 family antibodies bind domain D of staphylococcal protein A. J Immunol 1995;154:6437-45; PMID:7759880
- Wörn A, Plückthun A. Stability engineering of antibody single-chain Fv fragments. J Mol Biol 2001; 305:989-1010; PMID:11162109; http://dx.doi.org/10.1006/jmbi.2000.4265
- Jordan JL, Arndt JW, Hanf K, Li G, Hall J, Demarest S, Huang F, Wu X, Miller B, Glaser S, et al. Structural understanding of stabilization patterns in engineered bispecific Ig-like antibody molecules. Proteins 2009;77:832-41; PMID:19626705; http://dx.doi.org/10.1002/prot.22502
- Benhar I, Pastan I. Identification of residues that stabilize the single-chain Fv of monoclonal antibodies B3. J Biol Chem 1995; 270:23373-80; PMID:7559495; http://dx.doi.org/10.1074/jbc.270.40.23373
- McConnell AD, Spasojevich V, Macomber JL, Krapf IP, Chen A, Sheffer JC, Berkebile A, Horlick RA, Neben S, King DJ, et al. An integrated approach to extreme thermostabilization and affinity maturation of an antibody. Protein Eng Des Sel 2013; 26:151-64; PMID:23173178; http://dx.doi.org/10.1093/protein/gzs090
- Yin S, Ding F, Dokholyan NV. Eris: an automated estimator of protein stability. Nat Methods 2007; 4:466-7; PMID:17538626; http://dx.doi.org/10.1038/nmeth0607-466
- Alfthan K, Takkinen K, Sizmann D, Söderlund H, Teeri TT. Properties of a single-chain antibody containing different linker peptides. Protein Eng 1995; 8:725-31; PMID:8577701; http://dx.doi.org/10.1093/protein/8.7.725
- Huston JS1, Levinson D, Mudgett-Hunter M, Tai MS, Novotný J, Margolies MN, Ridge RJ, Bruccoleri RE, Haber E, Crea R, et al. Protein engineering of antibody binding sites: recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. PNAS USA 1988; 85:5879-83; PMID:3045807; http://dx.doi.org/10.1073/pnas.85.16.5879
- Fellouse FA, Wiesmann C, Sidhu SS. Synthetic antibodies from a four-aminoacid code: a dominant role for tyrosine in antigen recognition. PNAS USA 2004; 101:12467-72; PMID:15306681; http://dx.doi.org/10.1073/pnas.0401786101
- Perchiacca JM, Lee CC, Tessie PM. Optimal charged mutations in the complementarity-determining regions that prevent domain antibody aggregation are dependent on the antibody scaffold. PEDS 2014; 27:29-39; PMID:24398633
- Dudgeon K, Rouet R, Kokmeijer I, Schofield P, Stolp J, Langley D, Stock D, Christ D. General strategy for the generation of human antibody variable domains with increased aggregation resistance. PNAS USA 2012; 109:10879-84; PMID:22745168; http://dx.doi.org/10.1073/pnas.1202866109
- Weaver-Feldhaus JM, Miller KD, Feldhaus MJ, Siegel RW. Directed evolution for the development of conformation-specific affinity reagents using yeast display. PEDS 2005; 18: 527-36; PMID:16186140
- Perchiacca JM, Lee CC, Tessier PM. Optimal charged mutations in the complementarity-determining regions that prevent domain antibody aggregation are dependent on the antibody scaffold. Protein Eng Des Sel 2014; 27:29-39; PMID:24398633; http://dx.doi.org/10.1093/protein/gzt058
- Mian IS, Bradwell AR, und Olson AJ. Structure, function and properties of antibody binding sites. J Mol Biol 1991; 217:133-51; PMID:1988675; http://dx.doi.org/10.1016/0022-2836(91)90617-F
- Wilkinson RA, Piscitelli C, Teintze M, Cavacini LA, Posner MR, Lawrence CM. Structure of the Fab fragment of F105, a broadly reactive anti-human immunodeficiency virus (HIV) antibody that recognizes the CD4 binding site of HIV type 1 gp120. J Virol 2005; 79:13060-9; PMID:16189008; http://dx.doi.org/10.1128/JVI.79.20.13060-13069.2005
- Collis AV, Brouwer AP, Martin AC. Analysis of the antigen combining site: correlations between length and sequence composition of the hypervariable loops and the nature of the antigen. J Mol Biol 2003; 325:337-54; PMID:12488099; http://dx.doi.org/10.1016/S0022-2836(02)01222-6
- Xu L, Kohli N, Rennard R, Jiao Y, Razlog M, Zhang K, Baum J, Johnson B, Tang J, Schoeberl B, et al. Rapid optimization and prototyping for therapeutic antibody-like molecules. MABS 2013; 5:237-54; PMID:23392215; http://dx.doi.org/10.4161/mabs.23363
- Wang Q, Canutescu AA, Dunbrack RL. SCWRL and MolIDE: computer programs for side-chain conformation prediction and homology modeling. Nat Protoc 2008; 3:1832-47; PMID:18989261; http://dx.doi.org/10.1038/nprot.2008.184
- Canutescu AA, Dunbrack RL. MollDE: a homology modeling framework you can click with. Bioinformatics 2005; 21:2914-16; PMID:15845657; http://dx.doi.org/10.1093/bioinformatics/bti438
- Canutescu AA, Dunbrack RL. Cyclic coordinate descent: a robotics algorithm for protein loop closure. Protein Sci 2003; 12:963-72; PMID:12717019; http://dx.doi.org/10.1110/ps.0242703
- Canutescu AA, Shelenkov AA, Dunbrack RL. A graph-theory algorithm for rapid protein side-chain prediction. Protein Sci 2003; 12:2001-14; PMID:12930999; http://dx.doi.org/10.1110/ps.03154503
- Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 1997; 18:2714-23; PMID:9504803; http://dx.doi.org/10.1002/elps.1150181505
- Wu TT, Kabat EA, Bilofsky H. Some sequence similarities among cloned mouse DNA segments that code for lambda and kappa light chains of immunoglobulins. PNAS USA 1979; 76:4617-21; PMID:116235; http://dx.doi.org/10.1073/pnas.76.9.4617