
Abstract
Cytotoxic T lymphocytes (CTLs) play a central role in antitumor immunity. We utilized B16 melanoma cells expressing the fluorescent ubiquitination-based cell cycle indicator B16-fucci implanted in host mice and adoptively transferred with pmel-1-TCR transgenic T cells to demonstrate that tumor growth reduction is largely dependent on interferon γ-mediated cell cycle arrest rather than the cytotoxic killing of tumor cells by CTLs.
Adaptive immunity and interferon γ (IFNγ) mediate cancer immunosurveillance and immunoediting.Citation1 Adoptive cytotoxic T lymphocyte (CTL) transfer (ACT) therapy exploits cells with the potential to recognize and kill tumor cells and induce tumor regression.Citation2 To develop effective immunotherapy, we conducted gene expression analysis of the whole tumor after ACT into tumor-bearing mice. Antigen-specific CTLs infiltrated into the tumor as early as day 1, peaked on day 3 to 5 and suppressed tumor growth, while tumors grew progressively in untreated mice.Citation3,4 Histological analysis demonstrated that the number of CTLs in the tumor was far lower than the number of tumor cells on day 3 after ACT; we hypothesized that this small number of CTLs infiltrating the tumor would insufficient to prevent its growth exclusively by direct cell-to-cell cytotoxicity. Indeed, only small areas of necrosis or apoptosis were observed after ACT.
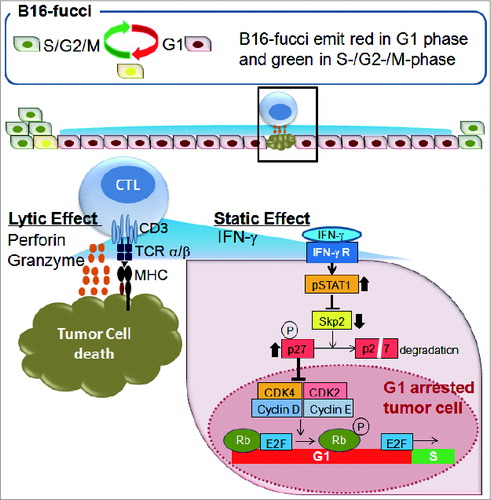
Gene expression analysis revealed that genes related to CD8+ T cells, the MHC Class I pathway, IFNγ signaling, cytotoxic effector molecules, and others were upregulated in tumors from ACT-treated mice. In contrast, genes related to the cell cycle, such as Skp2, E2f2, Ccnf, Mki67 and Wee1, were downregulated. Therefore, to examine the cell cycle regulation of malignant cells in the presence of antitumor CTLs, we developed a new mouse model using B16 melanoma cells expressing a fluorescent ubiquitination-based cell cycle indicator (B16-fucci) and infusions of transgenic T cells expressing premelanosome protein-specific T cell receptor (pmel-1-TCR).Citation5,6 In the S, G2, and M phases of the cell cycle, B16-fucci cells fluoresce green, in the G1 phase red, and during the G1/S transition, green and red merge, and nuclei fluoresce yellow. The majority of B16-fucci tumor cells inoculated subcutaneously into C57BL/6 mice were in the S/G2/M phase in the growing tumor and fluoresced green. After CTL transfer, most tumor cells promptly turned red, although the presence only of spotty apoptotic or necrotic areas accompanied the diffuse infiltration of CTLs. These results suggest that only a minority of tumor cells was recognized and killed by CTLs infiltrating the tumor; nonetheless, the majority of tumor cells underwent cell cycle arrest in G1 without direct contact with CTLs ().
Figure 1. CTLs regulate tumor growth via cytostatic effects rather than cytotoxicity. B16 melanoma cells expressing a fluorescent ubiquitination-based cell cycle indicator (B16-fucci) fluoresce green in the S, G2, and M phases of the cell cycle, red in the G1 phase and yellow during the G1/S transition. Cytotoxic T lymphocytes (CTLs) infiltrate the tumor, where they recognize and directly kill a portion of the cancer cells. At the same time, interferon γ (IFNγ) secreted by CTLs induces wide spread G1 cell cycle arrest of additional tumor cells through the IFNγ receptor, phosphorylation of Stat1, downregulation of Skp2 and accumulation of its target p27 cyclin-dependent kinase inhibitor. The inhibition of tumor growth mediated by tumor-reactive CTLs is thus largely dependent on IFNγ-mediated cell cycle arrest rather than the cytolytic killing of tumor cells.
Treating ACT mice with IFNγ neutralizing antibody (Ab) abrogated the inhibitory effects of CTLs on tumor growth. Tumors fluoresced green again when signal transducer and activator of transcription 1 (Stat1) phosphorylation and expression of IFNγ inducible genes, such as MIG (Cxcl9), IP10 (Cxcl10), or I-TAC (Cxcl11), was prevented by IFNγ blockade. However, expression of the lytic molecules perforin, granzyme B or FasL was unaffected. To examine the direct effects of IFNγ on B16 tumor cell growth, we generated B16-fucci tumor cells with an IFNγ receptor lacking the intracellular component (B16-fucciΔIC).Citation7 Exposure to IFNγ completely inhibited the proliferation of B16-fucci cells and arrested them in G1 phase, but had no effect on B16-fucciΔIC cells. Thus, CTLs in vivo or IFNγ treatment in vitro induced G1 cell cycle arrest, a response that was blocked with an antibody in vivo or in B16-fucci cells with defective IFNγ receptors. These results therefore indicate that tumor growth reduction mediated by ACT is largely dependent on IFNγ-mediated cell cycle arrest rather than the direct killing of tumor cells.
IFNγ is known to inhibit cell proliferation. In ACT-treated B16 melanoma cells, IFNγ-mediated G1 cell cycle arrest was associated with signal transduction through IFNγ receptor, phosphorylation of Stat1, downregulation of Skp2 and accumulation of its target p27 Cyclin-dependent kinase inhibitor, but did not appear to involve other pathways known to mediate cell cycle arrest. Similarly, the suppression of cell proliferation and G1 cell cycle arrest through pStat1, Skp2 and p27 were also observed in FBL-3 and to a lesser extent in other tumor cells, (e.g., p815, CT26 and 3LL). However, EL-4 cells were insensitive to IFNγ because they lack signal transduction through the IFNγ receptor. Although the mechanisms involved in inhibiting cell proliferation may differ among various tumor cell lines, insufficient IFNγ, IFNγ insensitivity due to downregulation of IFNγ receptors, or defects in IFNγ signal transduction might commonly be involved in tumor escape from ACT.
It has been reported that combinations of IFNγ and TNFα strongly suppress tumor growth and induce cell senescence, i.e., permanent growth arrest in G0/G1.Citation8 Perhaps because pmel-1 CTLs produce large amounts of IFNγ but not TNFα, suppression of tumor growth in our model was only transient. Therefore, combining TNFα-producing T helper type 1 (Th1) CD4+ T cells or Toll-like receptor (TLR) ligand-stimulated macrophages might contribute to improve CD8+ ACT. Moreover, a strategy to induce poly-functional CD8+ T cells producing IFNγ, TNFα, and IL-2 would likely further enhance the antitumor effects of ACT.
We have demonstrated that IFNγ produced by relatively small numbers of tumor-infiltrating CTLs can promptly arrest proliferation of a large number of tumor cells in G1 phase, rather than kill them. This dominance of G1 cell cycle arrest over direct cytotoxicity may be widespread; however, both the intrinsic nature of the tumor and the quality of the immune response determine the balance between G1 cycle arrest and lytic activity. Our results might help explain recent clinical reports that durable antitumor responses are sometimes observed in patients receiving immune checkpoint inhibitors. In such patients, antitumor immunity contributes to tumor growth arrest and induction of slow cancer regression instead of rapid elimination of tumor cells. Appropriate strategies to maintain tumor cells in a quiescent/dormant state for extended periods as well as to induce cell death are highly desirable.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol 2006; 6:836-48; PMID:17063185; http://dx.doi.org/10.1038/nri1961
- Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer 2008; 8:299-308; PMID:18354418; http://dx.doi.org/10.1038/nrc2355
- Noji S, Hosoi A, Takeda K, Matsushita H, Morishita Y, Seto Y, Kakimi K. Targeting spatiotemporal expression of CD137 on tumor-infiltrating cytotoxic T lymphocytes as a novel strategy for agonistic antibody therapy. J Immunother 2012; 35:460-72 10.1097/CJI.0b013e31826092db; PMID:22735804; http://dx.doi.org/10.1097/CJI.0b013e31826092db
- Hosoi A, Matsushita H, Shimizu K, Fujii S-i, Ueha S, Abe J, Kurachi M, Maekawa R, Matsushima K, Kakimi K. Adoptive cytotoxic T lymphocyte therapy triggers a counter-regulatory immunosuppressive mechanism via recruitment of myeloid-derived suppressor cells. Int J Cancer 2014; 134:1810-22; http://dx.doi.org/10.1002/ijc.28506
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. Visualizing Spatiotemporal Dynamics of Multicellular Cell-Cycle Progression. Cell 132:487-98; PMID:18267078
- Matsushita H, Hosoi A, Ueha S, Abe J, Fujieda N, Tomura M, Maekawa R, Matsushima K, Ohara O, Kakimi K. Cytotoxic T lymphocytes block tumor growth both by lytic activity and IFN-γ-dependent cell cycle arrest. Cancer Immunol Res 2015; 3:26-36; PMID:25127875
- Dighe A, Richards E, Old L, Schreiber R. Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFN gamma receptors. Immunity 1994; 1:447-56; PMID:7895156; http://dx.doi.org/10.1016/1074-7613(94)90087-6
- Braumuller H, Wieder T, Brenner E, Aszmann S, Hahn M, Alkhaled M, Schilbach K, Essmann F, Kneilling M, Griessinger C, et al. T-helper-1-cell cytokines drive cancer into senescence. Nature 2013; 494:361-5; PMID:23376950; http://dx.doi.org/10.1038/nature11824