
Abstract
Cleavage or shedding of the surface antigen, MHC class I chain-related (MIC) protein (A/B) has been known to be one of the mechanisms by which tumor cells escape host immune surveillance. Thus, any strategy to augment the surface expression of MICA/B could facilitate anticancer immune response. Here, we demonstrate that metabolic perturbation by the glycolytic inhibitor, 3-bromopyruvate (3-BrPA) augments the surface expression of MICA/B in human breast cancer cell lines, MDA-MB-231 and T47D. Data from in vitro studies show that a non-toxic, low-dose of 3-BrPA is sufficient to perturb energy metabolism, as evident by the activation of p-AMPK, p-AKT and p-PI3K. Further, 3-BrPA-treatment also elevated the levels of MICA/B in human breast cancer cell lines. Significantly, 3-BrPA-dependent increase in MICA/B levels also enhanced the sensitivity of cancer cells to natural killer (NK-92MI)-mediated cytotoxicity. In vivo, 3-BrPA-pretreated cells demonstrated greater sensitivity to NK-92MI therapy than their respective controls. The antitumor effect was confirmed by a reduction in tumor size and decreased tumor viability as observed by bioluminescence imaging. Histological examination and TUNEL staining demonstrated that NK-92MI administration promoted apoptosis in 3-BrPA-pretreated cells. Taken together, our data show that targeting energy metabolism could be a novel strategy to enhance the effectiveness of anticancer immunotherapeutics.
Introduction
Breast cancer is one of the most commonly diagnosed cancers and the second leading cause of cancer-related deaths in women in the United States.Citation1,2 Surgery combined with chemotherapy and/ or radiation therapy has become a standard therapeutic option for the management of early-stage breast cancer.Citation3 However, tumor recurrence and/or therapy resistance along with metastatic spread often contribute to poor prognosis and high mortality rates.Citation4-7 Hence there is a need for the development of new therapeutic strategies to improve the outcome of patients with advanced breast cancer.
Several studies have shown that natural killer (NK) cells induce cytotoxicity in cancer cells, at least under experimental conditions, indicating the therapeutic potential of eliciting a host immune response.Citation8,9 The natural ligands for the killer cell lectin-like receptor K1 (KLRK1, better known as NKG2D) on NK cells are the cell surface antigens MHC class I chain-related (MIC)A and MICB. Paradoxically, cancer cells downregulate the expression of MICA/MICB, primarily through the process of shedding or proteolytic cleavage.Citation10 Such a reduction in surface expression of MICA/B facilitates cancer cells to evade NK-cell mediated cytotoxicity. Recent reports have indicated that MICA/B expression can be induced by cellular stress. Thermal stressCitation11 and H2O2Citation12 mediated oxidative stress have been shown to stimulate the surface expression of MICA/B. Since cancer cells have been known to undergo metabolic reprogramming, here we investigated whether the perturbation of energy metabolism deregulates MICA/B shedding. As cancer cells preferentially rely on aerobic glycolysis,Citation13 we hypothesized that disruption of energy metabolism through an antiglycolytic agent such as 3-bromopyruvate (3-BrPA) could affect the proteolytic cleavage or shedding of MICA/B. In this report we demonstrate that human breast cancer cell lines, MDA-MB-231 and T47D pre-treated with a nonlethal dose of the glycolytic inhibitor, 3-BrPA show elevated levels of MICA/B expression and an improved-response to NK cell mediated cytotoxicity. These findings were validated in vivo using mouse models of MDA-MB-231 and T47D. Thus, the outcome of our study suggests that antiglycolytic pre-treatment has the potential to enhance the efficacy of NK-cell mediated therapy against human breast cancer.
Results
MDA-MB-231 and T47D cells tolerate 3-BrPA at low concentrations
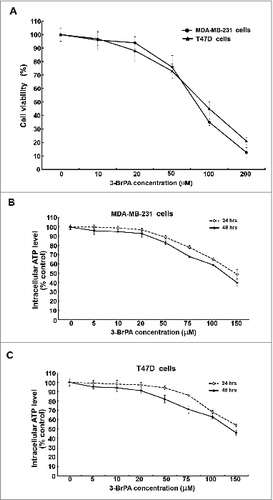
To identify the maximum tolerated dose of 3-BrPA, breast cancer cells were treated with various concentrations of 3-BrPA and cell viability was assessed. As evident from , in both MDA-MB-231 and T47D, the cell viability remained unaffected until 20 μM 3-BrPA, even after 48 h of treatment. The cell viability showed a significant decline beyond 20 μM 3-BrPA. Data from the analysis of intracellular ATP also substantiated that the level of ATP declined from 50 μM 3-BrPA at 48 hours of treatment (). Together the data show that neither the viability nor the cytosolic ATP level is significantly affected at 20 μM 3-BrPA, indicating that the maximum tolerated dose of 3-BrPA is ∼20 μM.
Figure 1. Maximum tolerated dose of 3-BrPA in MDA-MB-231 and T47D cells. Breast cancer cells were evaluated for toxicity in response to 3-bromopyruvate (3-Br-PA). (A) Trypan blue exclusion assay, and (B, C) bioluminescence assay to quantify intracellular ATP levels.
Nonlethal dose of 3-BrPA induces metabolic perturbation
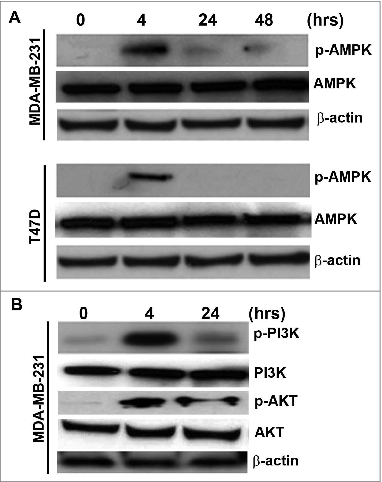
Although cell viability remained unaffected by 20 μM 3-BrPA, we investigated whether it had caused any metabolic stress. Analysis of principal proteins of energy sensing pathway showed that 3-BrPA treatment transiently elevated the level of phosphorylated p-AMPK but not total AMPK in both MDA-MB-231 and T47D cells (). Further analysis also demonstrated a similar elevation in the levels of the phosphorylated forms of phosphatidylinositol 3-kinase (p-PI3K) and p-AKT but not total protein (PI3K and AKT) in MDA-MB-231 cells (). Thus, the data indicate that although 20 μM 3-BrPA is nontoxic and did not affect cell viability, it is sufficient to cause metabolic perturbation as evinced by the activation of this energy-sensing pathway.
Figure 2. Induction of metabolic perturbation by 3-BrPA in MDA-MB-231 and T47D cells. Breast cancer cells were evaluated for metabolic perturbations in response to 3-bromopyruvate (3-BrPA). (A) Western blot showing a transient increase in the p-AMPK upon treatment with 3-BrPA indicating the metabolic stress. (B) Western blot showing the activation of p-PI3K and p-AKT indicating 3-BrPA-dependent perturbation of energy metabolism in MDA-MB-231 cells.
3-BrPA treatment increases the level of MICA/B in MDA-MB-231 cells
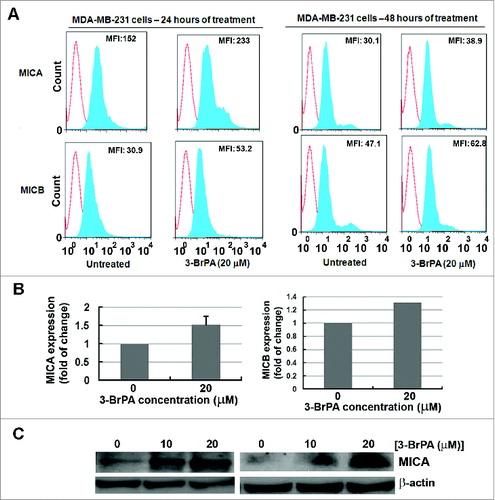
To investigate whether the minimal sublethal perturbation of cellular metabolism has any impact on MICA/B level, breast cancer cells treated with 3-BrPA were subject to cytofluorometry, ELISA and Western blot analysis. Data from flow cytometry demonstrated that 3-BrPA treatment elevated the surface expression of MICA and MICB at 24 and 48 h, as evident by increased mean fluorescence intensity (MFI) (). At 48 h of 3-BrPA treatment, MDA-MB-231 cells showed an increase in the MFI of 8.8 and 15.7 for MICA and MICB, respectively. The increase in the expression of MICA and MICB was also evident by ELISA in which a 0.5-fold increase in the level of cellular MICA was observed (). Further, western blot analysis also confirmed the increase in the expression of MICA in 3-BrPA treated cells (). Thus, 3-BrPA treatment elevated the expression level of MICA/B in MDA-MB-231 cells.
Figure 3. 3-BrPA pre-treatment augments the expression level of MICA/B in MDA-MB-231 cells. MICA and MICB levels in MDA-MB-231 breast cancer cells responding to 20 μM 3-bromopyruvate (3-BrPA). (A) Cytofluorimetric analysis of immunostained cells and (B) ELISA showing 3-BrPA treatment dependent increase in the level of MICA and MICB levels in MDA-MB-231 cells. (C) Western blot showing an increase in the level of MICA in MDA-MB-231 cells.
3-BrPA treatment increases the level of MICA/B in T47D cells
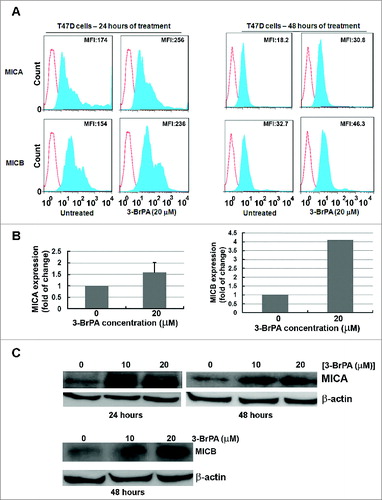
As with MDA-MB-231 cells, data from cytofluorometric analysis of T47D cells also demonstrated that 3-BrPA treatment elevated the surface expression of MICA and MICB (). At 48 h of 3-BrPA treatment T47D cells showed an increase in the MFI of MICA and MICB, 12.6 and 13.6, respectively. The increase in expression of MICA and MICB was also evident by ELISA in which MICB showed a 4-fold increase in 3-BrPA treated cells (). Further, western blot analysis confirmed 3-BrPA treatment-dependent increase in the expression of MICA/B (). Thus, 3-BrPA treatment elevated the expression level of MICA/B in T47D cells.
Figure 4. 3-BrPA pre-treatment augments the expression level of MICA/B in T47D cells. MICA and MICB levels in T47D breast cancer cells responding to 20 μM 3-bromopyruvate (3-BrPA). (A) Cytofluorimetric analysis of immunostained cells, (B) ELISA and (C) Western blot showing 3-BrPA treatment-dependent increase in MICA and MICB levels in T47D cells.
3-BrPA pre-treatment enhances NK cell-mediated cytotoxicity in vitro
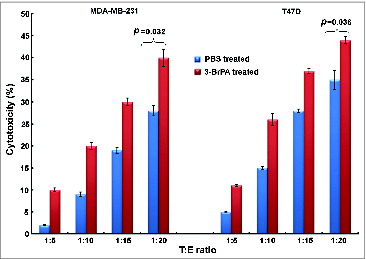
To assess whether 3-BrPA pre-treatment sensitizes breast cancer cells to NK cell-mediated cytotoxicity, MDA-MB-231 and T47D cells were incubated with NK-92MI cells at target (T) and effector (E) ratios of 1:5, 1:10, 1:15 and 1:20. The sensitivity of both MDA-MB-231 and T47D cells to NK cells increased as evident by enhanced cytotoxicity with increasing T:E ratios (). 3-BrPA pre-treated MDA-MB-231 and T47D cells demonstrated a significant increase in NK cell-mediated cytotoxicity as compared to their untreated counterparts (p = 0.032 and p = 0.036 respectively).
Figure 5. 3-BrPA pretreatment enhances NK-cell mediated cytotoxicity in both MDA-MB-231 and T47D breast cancer cells. The indicated breast cancer cells were seeded in 96-well plates (5,000 cells/well) and pretreated with 20 μM 3-bromopyruvate (3-BrPA) or phosphate-buffered saline (PBS) 24 h prior to adding NK-92MI cells at the indicated target (T) to effector (E) cell ratios. After 24 h, the supernatant was removed, and the remaining viable cells were quantified using Cell-Titer Glo Assay kit, which indicated a dose-dependent increase in cytotoxicity with increasing T:E cell ratios.
3-BrPA pre-treatment enhances NK cell-mediated cytotoxicity in vivo
For each luciferase-tagged tumor type (luc-MDA-MB-231 and T47D), mice were divided into 2 groups: vehicle (control) and NK-92MI. Each mouse received 3-BrPA or phosphate buffered saline (PBS) pretreated breast cancer cells, on the left and right flanks, respectively, in order to compare tumor response. In the MDA-MB-231 tumor model, tumor size and bioluminescent imaging (BLI) were used to monitor tumor response before and after NK-92MI treatment for 21days. In T47D tumor-bearing mice, tumor size was measured every 4 days post treatment for 16 days. BLI data show that NK-92MI treatment markedly blocked the progression of 3-BrPA pretreated luc-MDA-MB-231 cells, as compared to the PBS-pretreated cells (). Further, 3-BrPA pretreated luc-MDA-MB-231 tumors were profoundly sensitive to NK-92MI treatment as evidenced by a significant reduction in tumor size ().
Figure 6. 3-BrPA pretreatment enhances NK-cell mediated antitumor effects in MDA-MB-231 breast cancer model. Luciferase tagged MDA-MB-231 cells pretreated with either 20 μM 3-bromopyruvate (3-BrPA) or phosphate-buffered saline (PBS) were implanted subcutaneously in female athymic nude mice and subjected to natural killer (NK) therapy as indicated. (A) Bioluminescent imaging of developing tumors showing that 3-BrPA pretreatment enhances tumor sensitivity to NK-92MI. (B) Tumor images showing reduction in tumor size in 3-BrPA pretreated MDA-MB-231 subjected to NK-92MI therapy. (C) Graph showing significant difference between PBS or 3-BrPA pretreated MDA-MB-231 tumor cells upon NK-92MI treatment.
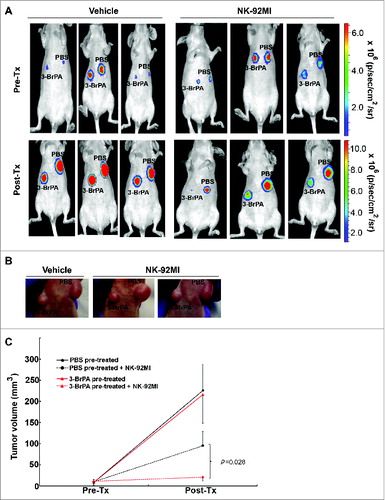
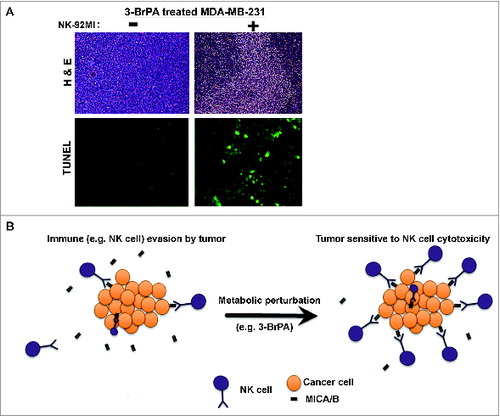
Similarly, NK-92MI treatment blocked the progression of T47D tumors pretreated with 3-BrPA (). Histological examination of 3-BrPA-pretreated luc-MDA-MB-231 tumor sections showed cell death in NK-92MI treated group but not in the untreated group (). Tumor cell death was also confirmed by TUNEL assay that showed an apoptotic signal in NK-92MI treated group but not in the control group ().
Figure 7. 3-BrPA pretreatment enhances NK-cell mediated antitumor effects in T47D breast cancer. T47D cells pretreated with either 20 μM 3-bromopyruvate (3-BrPA) or phosphate-buffered saline (PBS) were implanted subcutaneously in female athymic nude mice and subjected to natural killer (NK) therapy as indicated. (A) 3-BrPA pretreated tumor shows enhanced sensitivity to NK-92MI treatment than the control (PBS pretreated) tumor in T47D model of breast cancer. (B) Graph showing significant difference between PBS or 3-BrPA pretreated T47D tumor cells upon NK-92MI treatment.
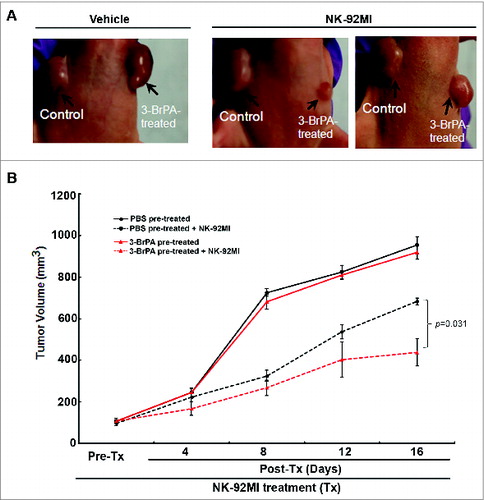
Figure 8. 3-BrPA pretreatment enhances NK-cell mediated cytotoxicity. Histological examination of tumors derived from breast cancer cells pretreated with either 20 μM 3-bromopyruvate (3-BrPA) or phosphate-buffered saline (PBS) and subjected to natural killer (NK) cell therapy as indicated. (A) Hematoxylin and eosin (H&E) and TUNEL staining show that 3-BrPA pretreatment enhanced the sensitivity of MDA-MB-231 tumor cells to NK-92MI treatment resulting in cell death. (B) Schematic showing metabolic perturbation related increase in the surface expression of MICA/B enhances NK-cell mediated cytotoxicity.
Discussion
MICA and MICB are two stress-inducible ligands that bind to the immunoreceptor NKG2D and play an important role in NK cell-mediated cytotoxicity.Citation14 Release or shedding of MIC molecules from the cell surface has been attributed to the evasion of immune surveillance in cancer. Data from the current study unequivocally demonstrate that perturbation of energy metabolism by 3-BrPA augments the expression of MICA/B eventually sensitizing human breast cancer cells to NK cell-mediated cytotoxicity (Fig. 8B).
Early detection and improved chemotherapeutics have considerably extended the survival of breast cancer patients.Citation15 However metastatic and triple negative breast cancer is often refractory to therapies and show poor outcome with higher rates of recurrence.Citation7 Thus, the development of an effective therapy for aggressive tumor phenotypes such as triple negative breast cancer is clearly needed. Immunotherapeutic strategies have been contemplated as alternative approaches to challenge tumor recurrence and chemotherapy-resistance.Citation16 However, major impediments to this approach may include an unfavorable tumor microenvironment, the release of tumor-derived immunosuppressive cytokines, and loss of MHC class surface antigens. The heterogeneous nature of tumor microenvironment has been attributed to hypoxic, high H+, and/or acidic conditions. In this context, a focus on the loss of MICA/B has gained momentum, particularly in light of experimental evidence indicating that HER2/HER3 pathways regulate MICA and MICB expression in breast cancer cells.Citation17 The shedding of MICA and/ or MICB has also been implicated in myeloma progression and immune evasion.Citation18 Further, MICA or MICB conjugated to antitumor antibodies have been shown to sensitize tumor cells to NK-cell mediated cytolysis.Citation8 Until this report, thermal and oxidative stresses were the major stress factors tested for the induction of MICA/B to enhance NK cell-mediated cytotoxicity. Here, we demonstrate that pharmacological targeting of one of the hallmarks of cancer (metabolic reprogramming) augments MICA/B expression thereby potentiating NK cell-mediated anticancer effects. These results were validated in a triple negative breast cancer cell line MDA-MB-231, suggesting its relevance to aggressive forms of human breast cancer.
Cancer cells are known to exhibit an increased rate of glycolysis irrespective of the oxygen availability (aerobic glycolysis) and normal mitochondria.Citation19 This metabolic phenotype also provides selective advantages to cancer cells.Citation20,21 Thus, the altered energy metabolism (e.g., increased glucose utilization and aerobic glycolysis) enables cancer cells to meet their energy demands and has recently been recognized as one of the hallmarks of cancer.Citation22 Although using an antiglycolytic approach has already been considered as a potential therapeutic strategy, it has not been explored in the context of anticancer immunotherapy. In a recent report, Sukumar et al.Citation23 have shown that activation of naïve T cells requires inhibition of glycolysis, a stimulus observed to extend the survival and clonal expansion of antitumor T cells thus facilitating enhanced anticancer effects. Our hypothesis that induction of metabolic stress by 3-BrPA to augment anticancer immune responses in principle emanates from the fact that MICA/B are stress inducible targetsCitation11,12 as well as the notion that cancer cells rely on altered energy metabolism.Citation24 In addition, 3-BrPA is an alkylating agent that is known to cause endoplasmic reticulum stressCitation25 and redox imbalance.Citation26 Thus, any interference in energy metabolic pathway by 3-BrPA could impose physiological stress that, in turn, could promote the induction of MICA/B expression.
The ability to evade immune surveillance is one of the hallmarks of cancer Citation22,27, and it has been well established that tumor cells escape immune detection through immunosuppressive networks. The factors that challenge the functional efficiency of antitumor-immune cells (e.g., T cells and NK cells) have been broadly classified as lymphocyte or T cell-associated factors and tumor-associated factors.Citation28 The former includes immunosuppressive elements, such as regulatory T cells and myeloid derived suppressive dendritic cells, among others. The tumor-associated factors include alteration in the expression of antigens (e.g., MHC) that enables cancer cells to escape immune detection, and the acidic microenvironment which is hostile to invading antitumor immune cells.Citation29 Interestingly, MICA/B are also known to be stress-inducible molecules on the basis of lines of evidence derived from exposure of cells to oxidative and thermal stresses. Thus, induction of cellular stress by metabolic perturbation is likely to elevate the expression levels of MICA/B. The results from the current study establish that a low, non-cytotoxic dose of 3-BrPA is sufficient to induce metabolic perturbation in order to elevate the surface expression level of MICA/B. 3-BrPA has been shown to promote antitumor effects in many solid malignancies including liver,Citation30,31 breast,Citation32 pancreas,Citation33 brainCitation34 and colon cancers.Citation35 However, at the dose tested in this study, no cytotoxicity or antitumor effects “per se” have been reported. Remarkably, the nonlethal dose of 3-BrPA is sufficient to enhance the efficacy of NK cell-mediated immunotherapy. In summary, our findings demonstrate that metabolic perturbation elevates the expression profile of cancer cell surface antigens, such as MICA and MICB. Furthermore, such an alteration in MICA/B levels enhances the antitumor immune response, as indicated by improved efficacy of NK-cell mediated therapy. Thus, our results provide a novel framework to potentiate immunotherapy by employing nontoxic levels of an anti-metabolite such as 3-BrPA. Further characterization of the global effects of nonlethal, low dose 3-BrPA on lactate output and the tumor microenvironment might reveal additional enhancements in the effectiveness of immunotherapy against human breast cancer.
Materials and Methods
Cell culture, chemicals and reagents
Human breast cancer cell lines, MDA-MB-231 and T47D, and human NK-92MI cells were purchased from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured as per supplier's instructions. In brief, MDA-MB-231 cells were cultured in L-15 medium supplemented with 2 mM L-glutamine (Invitrogen, Carlsbad, CA), 10% fetal bovine serum (FBS;Hyclone, Thermoscientific, USA) and 50 U/mL penicillin and streptomycin (Sigma-Aldrich, St. Louis, MO) at 37°C in a humidified atmosphere without CO2. T47D cells were cultured in RPMI 1640 medium supplemented with 2 mM L-glutamine (Invitrogen) supplemented with 10% FBS (Hyclone) and 50 U/mL penicillin and streptomycin (Sigma-Aldrich) at 37°C in a humidified atmosphere with 5% CO2. The highly potent cytotoxic human NK cell line NK-92MI (originally isolated from a patient with non-Hodgkin lymphoma) has the ability to continuously grow in vitro, and unlike its parental cell line NK-92, this cell line does not require IL-2 supplementation. These cells were cultured in α-Minimum Essential Medium, [α-MEM], obtained from ATCC, supplemented with 2 mM L-glutamine, 0.2 mM inositol, 0.02 mM folic acid, 0.1mM β-mercaptoethanol, 12.5% FBS and 12.5% horse serum. Unless otherwise mentioned, all chemicals including 3-BrPA and protease and phosphatase inhibitor cocktails were purchased from Sigma Chemical Co. (St. Louis, MO). Antibodies used in the protein gel blotting included:anti-MICA, anti-MICB, anti-MICA/B, and HRP-linked goat anti-rabbit or anti-mouse IgG (Santa Cruz Biotechnology, CA); anti-AMPK, anti-p-AMPK, anti-PI3K, anti-p-PI3K (p110α), anti-AKT and anti-p-AKT (Cell Signaling Technologies, Danvers, MA, USA); and β-actin (Sigma-Aldrich). The ECL Plus detection reagent and the necessary materials for chemiluminescent detection of immunoblots were procured from GE-Healthcare (Piscataway, NJ). D-Luciferin potassium salt used as the substrate for bioluminescence imaging was obtained from Gold Biotechnology Inc., (St. Louis, MO). For apoptosis analysis, the terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) kit was purchased from Millipore Co. (Bedford, MA).
Determination of maximum tolerated dose and intracellular ATP level
To determine the nonlethal, maximum tolerated dose of 3-BrPA for both the MDA-MB-231 and T47D breast cancer cell lines, we evaluated cell viability using the Trypan-blue exclusion method. Quantification of intracellular ATP level was performed using Cell-Titer Glo Assay kit (Promega Corp., Madison, WI) and FLUOstar Omega plate reader (BMG Labtech, UK). In brief, cells were plated in 96-well flat-bottomed opaque plates the day before 3-BrPA treatment and cellular ATP level was determined as per the kit manufacturer's protocol.
Western blotting and ELISA
The expression level of MICA/B protein in MDA-MB-231 and T47D cells pretreated with low dose 3-BrPA was also determined by western blotting and ELISA techniques. For protein gel blotting, breast cancer cells were washed in PBS, lysed in RIPA lysis buffer (Sigma-Aldrich) with protease and phosphatase inhibitor cocktails (both from Sigma-Aldrich) at 4°C using a dounce homogenizer. The lysates were centrifuged at 12,000 × g for 15 min at 4°C to remove any cell debris. The clear supernatants were collected and the protein concentrations were analyzed using a 2-D Quant protein assay kit (GE Healthcare) as previously described.Citation36 Immunoblotting was performed using specific antibodies, as per supplier's instructions. For the ELISA analysis, breast cancer cells were washed in PBS, lysed by 15 min shaking in RIPA lysis buffer (Sigma-Aldrich) with a protease inhibitor cocktail and a phosphatase inhibitor cocktails (both from Sigma-Aldrich) at 4°C. Cell debris was cleared at 12,000 × g for 15 min. The supernatants were collected and the protein concentrations were analyzed using a 2-D Quant protein assay Kit (GE Healthcare, Pittsburgh, PA, USA) according to the manufacturer's protocol. MICA and MICB concentrations were determined via DuoSet ELISA according to the manufacturer's instructions (ab59569 and ab100593; Abcam Co., MA, USA).
Fluorescence cytometry
Surface expression of MICA/B in MDA-MB-231 and T47D cells cultured in the presence or absence of low dose 3-BrPA was analyzed by cytoflourometric analysis. In brief, breast cancer cells were washed in PBS, collected with ethylenediaminetetraacetic acid (EDTA;Sigma-Aldrich), and analyzed for cell surface expression of proteins, MICA/B, MICA, and MICB, by direct or indirect immunofluorescence using specific monoclonal antibodies (mAbs), including: phycoerythrin (PE)-labeled MICA/B (6D4; BioLegend, San Diego, CA, USA) PE-labeled MICA (159227; R&D Systems, Minneapolis, MN, USA), or PE-labeled MICB (236511; R&D Systems). PE-labeled anti-mouse IgG (Santa Cruz Biotechnology Inc., Dallas, TX, USA) was used as secondary Ab as needed. Fluorescein isothiocyanate (FITC)- or PE-labeled anti-mouse IgG (Santa Cruz Biotechnology Inc.) were used as isotype controls. Stained cells were acquired on a FACSCalibur cytometer (BD Biosciences, San Jose, CA, USA) and analyzed using FlowJo software 6.4.7 (Tree Star, Ashland, OR).
Generation of luc-MDA-MB-231 cells for bioluminescence imaging
For bioluminescence studies, the luciferase reporter plasmid pcDNA 3.1-CMV-fl (firefly luciferase) was generated as previously described.Citation31 MDA-MB-231 cells stably expressing luciferase gene were generated by transfection with pcDNA 3.1-CMV-fl plasmid using Turbofectin 8.0 (Origene Technologies) followed by clonal selection with G418 (Invitrogen) containing growth medium. Clones expressing highest luciferase activity were selected and used for further studies. For convenience, these stable cells will be referred to hereafter as luc-MDA-MB-231.
Histology and TUNEL assay
For histology, tissues were routinely fixed in phosphate-buffered 10% formalin (Polysciences Co., Warrington, PA), dehydrated by graded ethanol, embedded in Paraplast Plus wax (McCormick Scientific), sectioned at 5 μm, mounted on slides,oven dried, and deparaffinized. The tissue sections were subject to hematoxylin and eosin (H&E) staining and viewed under a light microscope. Apoptosis was assessed using TUNEL detection kit (Millipore Corp.) following the manufacturer's protocol. Sections were also counterstained with 4′,6-diamidino-2-phenylindole (DAPI) to visualize nuclei. Finally, tissue sections were covered with cover slips mounted with Prolong Gold Antifade Reagent (Invitrogen) and allowed to dry in the dark. The slides were viewed under a Nikon ECLIPSE TS 100 fluorescent microscope and the images were captured using a Nikon Coolpix digital camera (Nikon Instruments, Inc., Melville, NY).
NK cell-mediated cytotoxicity assay
The NK cell-mediated cytolytic assay was performed as previously described with modifications.Citation17 MDA-MB-231 and T47D cells treated with either 3-BrPA or PBS were subjected to target (T) and effector (E) ratios of 1:5, 1:10, 1:15 and 1:20. In brief, 5000 target cells were seeded in 96-well plates and the next day effector cells (NK-92MI) were added. After 24 h incubation, the supernatant of the culture containing suspended NK-92MI cells and floating dead cells were removed, and the remaining viable cells were quantified using Cell-Titer Glo Assay kit (Promega Inc.). The decrease in viable cells indicated the level of cytotoxicity as compared to the PBS control.
In vivo studies
Animal studies were performed as approved by the Johns Hopkins University Animal Care and Use Committee. For the in vivo experiments female athymic nude mice (Crl:NU-Foxn1/nu strain; Charles River Laboratory, Germantown, MD, USA), aged 4–6-weeks old and weighing 20–25 g, were used in accordance with institutional guidelines. Mice were maintained in laminar flow rooms at constant temperature and humidity, with food and water given ad libitum. Tumor xenografts were initiated with subcutaneous injection of either luc-MDA-MB-231 or T47D cells (4–5 x 106cells) growing in log phase, either pretreated with 3-BrPA or vehicle (saline). Tumors were allowed to grow for 2–3 weeks up to about 2–5 mm in the largest diameter. Tumor viability was confirmed by BLI. BLI was performed as described previously.Citation31 In brief, mice were injected with D-luciferin intraperitoneally (150 mg/kg) and bioluminescent images were acquired using the IVIS 200 small animal imaging system (Xenogen Corp.) at multiple time intervals (1 min, 5 min and 15 min). A total of 20 nude mice with tumor xenografts were allocated randomly into different groups and subjected to intravenous (i.v.) injection with 1x106 NK cells or phosphate buffered saline (PBS). After one week, the mice were injected again with NK cells or PBS. The tumor response was assessed by viability (by bioluminescence imaging) and size measurement (using calipers). The end point was 2 weeks after the final treatment (injection of NK cells). The mice were then sacrificed and tumor tissues were harvested and subject to histopathology.
Statistical analysis
Data correspond to at least 3 independent experiments and are expressed as mean ± SEM. Statistical comparison of data sets were evaluated by the Student's t-test.
Disclosure of Potential Conflicts of Interest
Dr. Geschwind is the founder of Presciencelabs LLC, a biotech firm currently developing 3-BrPA for clinical use in cancer.
Funding
This work was supported by the grant from the United States Department of Defense (award no. W81XWH-11-1-0343), Abdulrahman Abdulmalik Research Fund and Charles Wallace Pratt Research Fund.
References
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin 2009; 59:225-49; PMID:19474385; http://dx.doi.org/10.3322/caac.20006
- DeSantis C, Siegel R, Bandi P, Jemal A. Breast cancer statistics, 2011. CA Cancer J Clin 2011; 61:409-18; PMID:21969133; http://dx.doi.org/10.3322/caac.20134
- Arriagada R, Le MG, Guinebretiere JM, Dunant A, Rochard F, Tursz T. Late local recurrences in a randomised trial comparing conservative treatment with total mastectomy in early breast cancer patients. Ann Oncol 2003; 14:1617-22; PMID:14581268
- Atkinson RL, Zhang M, Diagaradjane P, Peddibhotla S, Contreras A, Hilsenbeck SG, Woodward WA, Krishnan S, Chang JC, Rosen JM. Thermal enhancement with optically activated gold nanoshells sensitizes breast cancer stem cells to radiation therapy. Sci Transl Med 2010; 2:55ra79; PMID:20980696; http://dx.doi.org/10.1126/scitranslmed.3001447
- Braun S, Pantel K, Muller P, Janni W, Hepp F, Kentenich CR, Gastroph S, Wischnik A, Dimpfl T, Kindermann G, et al. Cytokeratin-positive cells in the bone marrow and survival of patients with stage I, II, or III breast cancer. N Engl J Med 2000; 342:525-33; PMID:10684910; http://dx.doi.org/10.1056/NEJM200002243420801
- Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nat Rev Cancer 2004; 4:448-56; PMID:15170447; http://dx.doi.org/10.1038/nrc1370
- Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin Cancer Res 2007; 13:4429-34; PMID:17671126; http://dx.doi.org/10.1158/1078-0432.CCR-06-3045
- Germain C, Larbouret C, Cesson V, Donda A, Held W, Mach JP, Pelegrin A, Robert B. MHC class I-related chain A conjugated to antitumor antibodies can sensitize tumor cells to specific lysis by natural killer cells. Clin Cancer Res 2005; 11:7516-22; PMID:16243826; http://dx.doi.org/10.1158/1078-0432.CCR-05-0872
- Sutlu T, Alici E. Natural killer cell-based immunotherapy in cancer: current insights and future prospects. J Intern Med 2009; 266:154-81; PMID:19614820; http://dx.doi.org/10.1111/j.1365-2796.2009.02121.x
- Salih HR, Rammensee HG, Steinle A. Cutting edge: down-regulation of MICA on human tumors by proteolytic shedding. J Immunol 2002; 169:4098-102; PMID:12370336
- Dayanc BE, Bansal S, Gure AO, Gollnick SO, Repasky EA. Enhanced sensitivity of colon tumour cells to natural killer cell cytotoxicity after mild thermal stress is regulated through HSF1-mediated expression of MICA. Int J Hyperthermia 2013; 29:480-90; PMID:23902341; http://dx.doi.org/10.3109/02656736.2013.821526
- Yamamoto K, Fujiyama Y, Andoh A, Bamba T, Okabe H. Oxidative stress increases MICA and MICB gene expression in the human colon carcinoma cell line (CaCo-2). Biochim Biophys Acta 2001; 1526:10-2; PMID:11287116; http://dx.doi.org/S0304-4165(01)00099-X
- Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: Progress and prospects. Mol Cancer 2013; 12:152, 4598-12-152; PMID:24298908; http://dx.doi.org/10.1186/1476-4598-12-152
- Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, Spies T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999; 285:727-9; PMID:10426993; http://dx.doi.org/10.1126/science.285.5428.727
- Early Breast Cancer Trialists' Collaborative Group (EBCTCG). Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: An overview of the randomised trials. Lancet 2005; 365:1687-717; PMID:15894097; http://dx.doi.org/S0140-6736(05)66544-0
- Smyth MJ, Godfrey DI, Trapani JA. A fresh look at tumor immunosurveillance and immunotherapy. Nat Immunol 2001; 2:293-9; PMID:11276199; http://dx.doi.org/10.1038/86297
- Okita R, Mougiakakos D, Ando T, Mao Y, Sarhan D, Wennerberg E, Seliger B, Lundqvist A, Mimura K, Kiessling R. HER2/HER3 signaling regulates NK cell-mediated cytotoxicity via MHC class I chain-related molecule A and B expression in human breast cancer cell lines. J Immunol 2012; 188:2136-45; PMID:22301547; http://dx.doi.org/10.4049/jimmunol.1102237
- Jinushi M, Vanneman M, Munshi NC, Tai YT, Prabhala RH, Ritz J, Neuberg D, Anderson KC, Carrasco DR, Dranoff G. MHC class I chain-related protein A antibodies and shedding are associated with the progression of multiple myeloma. Proc Natl Acad Sci U S A 2008; 105:1285-90; PMID:18202175; http://dx.doi.org/10.1073/pnas.0711293105
- Ward PS, Thompson CB. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012; 21:297-308; PMID:22439925; http://dx.doi.org/10.1016/j.ccr.2012.02.014
- Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer 2004; 4:891-9; PMID:15516961; http://dx.doi.org/10.1038/nrc1478
- Gatenby RA, Gillies RJ. Glycolysis in cancer: A potential target for therapy. Int J Biochem Cell Biol 2007; 39:1358-66; PMID:17499003; http://dx.doi.org/S1357-2725(07)00115-X
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/10.1016/j.cell.2011.02.013
- Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, Roychoudhuri R, Palmer DC, Muranski P, Karoly ED, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest 2013; 123:4479-88; PMID:24091329; http://dx.doi.org/10.1172/JCI69589
- Dang CV. Links between metabolism and cancer. Genes Dev 2012; 26:877-90; PMID:22549953; http://dx.doi.org/10.1101/gad.189365.112
- Ganapathy-Kanniappan S, Geschwind JF, Kunjithapatham R, Buijs M, Syed LH, Rao PP, Ota S, Kwak BK, Loffroy R, Vali M. 3-bromopyruvate induces endoplasmic reticulum stress, overcomes autophagy and causes apoptosis in human HCC cell lines. Anticancer Res 2010; 30:923-35; PMID:20393016
- Ihrlund LS, Hernlund E, Khan O, Shoshan MC. 3-bromopyruvate as inhibitor of tumour cell energy metabolism and chemopotentiator of platinum drugs. Mol Oncol 2008; 2:94-101; PMID:19383331; http://dx.doi.org/10.1016/j.molonc.2008.01.003
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100:57-70; PMID:10647931
- Rosenberg SA. Progress in human tumour immunology and immunotherapy. Nature 2001; 411:380-4; PMID:11357146; http://dx.doi.org/10.1038/35077246
- Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer 2005; 5:263-74; PMID:15776005; http://dx.doi.org/10.1038/nrc1586
- Geschwind JF, Ko YH, Torbenson MS, Magee C, Pedersen PL. Novel therapy for liver cancer: Direct intraarterial injection of a potent inhibitor of ATP production. Cancer Res 2002; 62:3909-13; PMID:12124317
- Ganapathy-Kanniappan S, Kunjithapatham R, Torbenson MS, Rao PP, Carson KA, Buijs M, Vali M, Geschwind JF. Human hepatocellular carcinoma in a mouse model: Assessment of tumor response to percutaneous ablation by using glyceraldehyde-3-phosphate dehydrogenase antagonists. Radiology 2012; 262:834-45; PMID:22357885; http://dx.doi.org/10.1148/radiol.11111569
- Buijs M, Vossen JA, Geschwind JF, Ishimori T, Engles JM, Acha-Ngwodo O, Wahl RL, Vali M. Specificity of the anti-glycolytic activity of 3-bromopyruvate confirmed by FDG uptake in a rat model of breast cancer. Invest New Drugs 2009; 27:120-3; PMID:18553054; http://dx.doi.org/10.1007/s10637-008-9145-0
- Ota S, Geschwind JF, Buijs M, Wijlemans JW, Kwak BK, Ganapathy-Kanniappan S. Ultrasound-guided direct delivery of 3-bromopyruvate blocks tumor progression in an orthotopic mouse model of human pancreatic cancer. Target Oncol 2013; 8:145-51; PMID:23529644; http://dx.doi.org/10.1007/s11523-013-0273-x
- El Sayed SM, El-Magd RM, Shishido Y, Chung SP, Diem TH, Sakai T, Watanabe H, Kagami S, Fukui K. 3-bromopyruvate antagonizes effects of lactate and pyruvate, synergizes with citrate and exerts novel anti-glioma effects. J Bioenerg Biomembr 2012; 44:61-79; PMID:22318356; http://dx.doi.org/10.1007/s10863-012-9409-4
- Zhou Y, Tozzi F, Chen J, Fan F, Xia L, Wang J, Gao G, Zhang A, Xia X, Brasher H, et al. Intracellular ATP levels are a pivotal determinant of chemoresistance in colon cancer cells. Cancer Res 2012; 72:304-14; PMID:22084398; http://dx.doi.org/10.1158/0008-5472.CAN-11-1674
- Ganapathy-Kanniappan S, Geschwind JF, Kunjithapatham R, Buijs M, Vossen JA, Tchernyshyov I, Cole RN, Syed LH, Rao PP, Ota S, et al. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is pyruvylated during 3-bromopyruvate mediated cancer cell death. Anticancer Res 2009; 29:4909-18; PMID:20044597