Abstract
Autophagy is a catabolic process that functions in recycling and degrading cellular proteins, and is also induced as an adaptive response to the increased metabolic demand upon nutrient starvation. However, the prosurvival role of autophagy in response to metabolic stress due to deprivation of glutamine, the most abundant nutrient for mammalian cells, is not well understood. Here, we demonstrated that when extracellular glutamine was withdrawn, autophagy provided cells with sub-mM concentrations of glutamine, which played a critical role in fostering cell metabolism. Moreover, we uncovered a previously unknown connection between metabolic responses to ATG5 deficiency and glutamine deprivation, and revealed that WT and atg5−/− MEFs utilized both common and distinct metabolic pathways over time during glutamine deprivation. Although the early response of WT MEFs to glutamine deficiency was similar in many respects to the baseline metabolism of atg5−/− MEFs, there was a concomitant decrease in the levels of essential amino acids and branched chain amino acid catabolites in WT MEFs after 6 h of glutamine withdrawal that distinguished them from the atg5−/− MEFs. Metabolomic profiling, oxygen consumption and pathway focused quantitative RT-PCR analyses revealed that autophagy and glutamine utilization were reciprocally regulated to couple metabolic and transcriptional reprogramming. These findings provide key insights into the critical prosurvival role of autophagy in maintaining mitochondrial oxidative phosphorylation and cell growth during metabolic stress caused by glutamine deprivation.
Introduction
Macroautophagy (referred to hereafter as autophagy) is an evolutionarily conserved cellular process that is activated in response to low nutrient availability to provide building blocks for macromolecule biosynthesis.Citation1,Citation2 Emerging data suggest that basal and induced autophagy play two distinct roles in metabolism.Citation3-Citation6 In nutrient-rich conditions, autophagy occurs constitutively at low levels. However, when cells are starved of nutrients, high levels of induced autophagy allow them to adapt to the starvation by degrading cellular constituents. Autophagy-deficient yeast and mice display lower intracellular levels of amino acids than their wild-type (WT) counterparts during starvation, supporting the idea that autophagy plays a “dual role”: (1) it results in consumption of cellular components for homeostasis; and (2) it promotes cell survival under metabolic stress.Citation7,Citation8
Most mammalian cells in culture require supraphysiologic levels of glutamine for high rates of glycolysis and optimal growth.Citation9,Citation10 Glutamine is a nonessential amino acid (NEAA) that can be converted to α-ketoglutarate, a TCA cycle intermediate, through glutaminolysis.Citation9 When both glucose and glutamine are available, a significant fraction of cellular energy demands, from 40% for normal diploid fibroblasts up to 70% for HeLa cells, is provided by glutamine. In tumor cells, most of the glucose is metabolized to lactate even in the presence of oxygen, and ATP is generated during glycolysis to meet the intracellular energy demand, termed the “Warburg effect.”Citation11-Citation13 The combined utilization of glucose and glutamine provides biochemical precursors to support the biosynthesis of nucleotides and certain NEAAs, such as alanine and aspartate, which are catalyzed by alanine aminotransferase and aspartate aminotransferase, for cell growth and proliferation.Citation14 In addition, glutamine is involved in the formation of glutathione, which is the major thiol-containing, endogenous antioxidant required for maintenance of the cellular redox balance. Glutamine generates glutamate, which together with cysteine and glycine, is required for glutathione synthesis and it also metabolically increases the intracellular level of NADPH, which is used by glutathione reductase to reduce oxidized glutathione to glutathione.Citation15 Many cancer cells display high rates of glutamine use to support mitochondrial oxidative phosphorylation and to provide metabolic intermediates, thus exhibiting “glutamine addiction.”Citation16,Citation17 For example, glutaminolysis initiates an anaplerotic reaction to replenish TCA cycle intermediates, simultaneously producing NADPH and ammonia.Citation6,Citation18 Recently, Eng et al., demonstrated that ammonia from glutaminolysis induces autophagy.Citation19 Furthermore, glutamine efflux is required for MTOR activation and subsequent protein synthesis.Citation20 Although autophagy is viewed as a mechanism that contributes basic components to sustain metabolic function during starvation or stress, how autophagy participates in or regulates metabolic responses to glutamine depletion is currently unclear. Regardless, the contribution from autophagy to various metabolic pathways also remains largely unclear.
Among more than 20 autophagy-related (ATG) proteins, ATG5 is an integral part of the autophagy machinery, as is the ATG5-containing complex (ATG12–ATG5-ATG16L1). This complex catalyzes the conjugation of the C-terminal Gly of ATG8 to phosphatidylethanolamine (PE) via ATG3 to promote incorporation of ATG8–PE (MAP1LC3B) into autophagosomes, thereby allowing autophagosome closure and cargo recruitment.Citation21 Knockouts of the Atg5 gene lead to ablation of ATG5-dependent autophagy.Citation22 Given the prominent role of both glutamine and autophagy in maintaining energy and biosynthetic intermediate levels, we hypothesized that one of the critical biological functions for autophagy is to supply glutamine to fuel cell metabolism. To test this hypothesis, we examined the metabolomic profiles and oxygen consumption in responses to glutamine deprivation using WT and atg5−/− MEFs. Our data suggested that the baseline metabolism of atg5−/− MEFs was in many respects to the early response of WT MEFs to glutamine deprivation. Moreover, real-time RT-PCR analyses uncovered the coordinated regulation of the mRNA levels of key enzymes involved in energy generation in WT MEFs subjected to acute glutamine deprivation and atg5−/− MEFs in full medium. Taken together, these studies provide novel insights into the role of autophagy in regulating oxidative phosphorylation through coupled metabolic and transcriptional reprogramming.
Results
WT and atg5−/− MEFs have distinct basal metabolite profiles
Both autophagy and extracellular nutrients play a critical role in providing cells with glutamine to foster proliferation. To understand the metabolism affected by glutamine deprivation via either autophagy inactivation or depletion of the extracellular source, we subjected WT and Atg5-null MEF cells to an unbiased metabolomics analysis at 0, 6 and 24 h after glutamine removal. Using the Metabolon mass spectrometry-based global biochemical profiling platform, we identified 204 biochemical compounds for further statistical analyses. After normalizing with cell numbers, we found that the basal levels (0 h) of 82 biochemical compounds in atg5−/− MEFs were significantly different to those in WT MEFs; 69 were increased and 13 decreased compared with WT MEFs (p < 0.05, Table S1). Almost all 82 compounds were key metabolic intermediates, suggesting that autophagy inactivation by Atg5 knockout (data shown later) elicits a global change in various biosynthetic and catabolic pathways in MEFs.
To provide a general framework for understanding the contribution of ATG5-dependent autophagy to metabolism during the course of glutamine deprivation, a relative abundance heatmap of 204 metabolites was generated (; Fig. S1). The double dendrogram cluster analysis suggested that the basal metabolic profiles of atg5−/− MEFs (0 h) at full medium resembled those of WT MEFs at 6 h after glutamine withdrawal. Bioinformatic analyses confirmed that 8 out of 11 key metabolic parameters were similar between atg5−/− MEFs in full medium and WT MEFs at 6 h post glutamine withdrawal (). Specifically, the decrease of most nonessential amino acids (NEAAs), altered lipid metabolism, enhanced nucleotide metabolism, and elevated polyamine synthesis were common changes shared by the profiles of atg5−/− MEFs at base line (0 h) and WT MEFs subjected to glutamine depletion (6 h). In contrast, 6 out of the 11 metabolic parameters examined in WT and atg5−/− cells, compared with their respective metabolic status prior to glutamine deprivation, were changed in opposite direction at 6 h after glutamine deprivation. The heatmap of selected metabolites in atg5−/− and WT MEFs () confirmed the trend shown in that at 6 h post glutamine withdrawal atg5−/− cells reversed many of the metabolic changes induced by the ATG5 ablation. Taken together, our data suggest that the early response of the metabolism of WT MEFs to glutamine deprivation is similar to the effect of autophagy impairment on the metabolism of MEFs. The effects of glutamine deprivation are distinct, or opposite, in WT MEFs compared with Atg5-null MEFs (), suggesting that the response to glutamine deprivation is determined by autophagy.
Figure 1. Metabolomics profiling of atg5−/− MEFs in full medium is similar to that of WT MEFs after 6 h of glutamine deprivation. (A) Double dendrogram cluster analyses defining the effect of glutamine deprivation on metabolic intermediates by comparing biomolecule levels in sample sets shown in Figure S1. (B) Compendium of global metabolic profiling. Red ↑, increasing level; Green ↓, decreasing level; -, no difference. (C, upper panel) Hierarchical cluster analysis examining selected biomolecules selected from Figure S1. (Lower panel) Heatmap profile of putrescine, glutamine, reduced and oxidized glutathione, phosphoethanolamine, PRPP, spermine and spermidine in WT and atg5−/− MEFs after glutamine withdrawal (6 and 24 h). (B and C) Signal fold change was normalized to 0 h WT (0 h) as 1. All data points were log2 transformed and used to calculate the Self-Organizing Map. Transformed fold changes are shown in green (decreasing) and red (increasing).
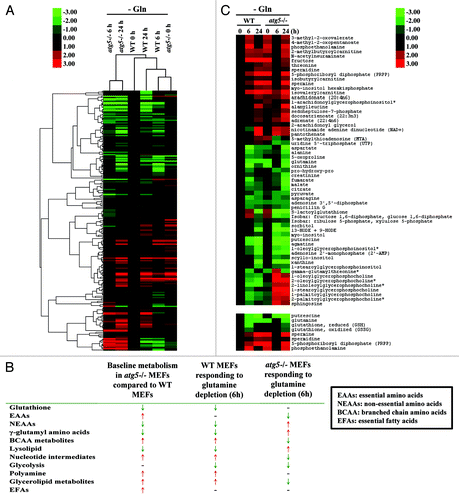
Key metabolic signatures of WT and atg5−/− MEFs during glutamine starvation
To extend our observations, we compared the levels of a number of metabolites in atg5−/− and WT MEFs at different time points of glutamine starvation (, and ). We found that the basal levels (0 h) of the nine essential amino acids (EAAs), except lysine, were higher in the atg5−/− MEFs than WT MEFs (). However, at 6 h after glutamine withdrawal, the levels of all 9 analyzed EAAs were significantly lower in the atg5−/− MEFs than their respective 0 h levels, whereas except for lysine, the levels of the EAAs, in WT MEFs did not exhibit a significant decrease (). The different responses of the EAA levels to 6 h of glutamine starvation in WT MEFs and atg5−/− MEFs raised the possibility that the WT and mutant cells could be using different pathways to maintain EAA levels. To investigate this possibility, we examined the steady-state levels for catabolites of the branched chain amino acids (BCAAs: leucine, isoleucine and valine), isovalerylcarnitine, isobutyrylcarnitine and 2-methylbutyroylcarnitine. Clearly, these catabolites also exhibited the same, opposite trends in WT and atg5−/− MEFs at 6 h post glutamine withdrawal as those of the EAA (). The observed transient decrease in EAAs and BCAA catabolites appears to represent a characteristic response to glutamine deprivation in atg5−/− MEFs.
Figure 2. Autophagy deficiency leads to higher intake of EAA and reduced EAA levels under glutamine depletion. WT and atg5−/− MEFs were grown in serum-supplemented DMEM without glutamine for 0, 6 and 24 h in five replicates. Relative quantification of (A) histidine, (B) isoleucine, (C) leucine, (D) lysine, (E) methionine, (F) phenylalanine, (G) threonine, (H) tryptophan and (I) valine was performed, and the relative levels were determined by setting the levels in non-treated WT MEFs as 1. *p < 0.05; **p < 0.01; ***p < 0.001; Gln, glutamine; n = 15.
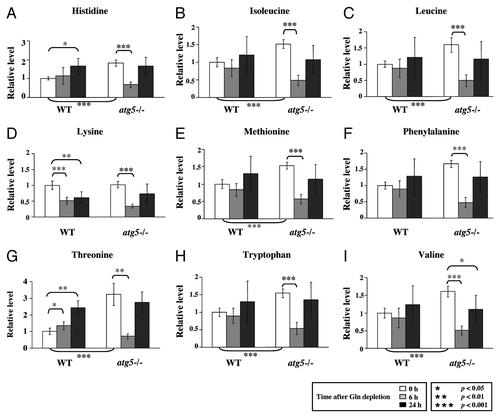
Figure 3. Glutamine withdrawal and autophagy deficiency increase BCAA catabolism. Cell preparation, relative quantification and data analyses were performed as described in . Profiles of leucine metabolites: (A) 4-methyl-2-oxopentanoate and (B) isovalerylcarnitine; isoleucine metabolites: (C) 3-methyl-2-oxovalerate; valine metabolites: (D) isobutyrylcarnitine; (E) 3-methyl-2-oxobutyrate and (F) 2-methylbutyroylcarnitine. *p < 0.05; **p < 0.01; ***p < 0.001; Gln, glutamine; n = 15.
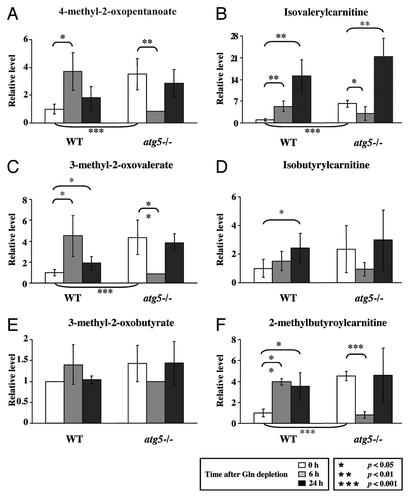
Figure 4. Glutamine depletion affects TCA cycle intermediates. Cell preparation, relative quantification and data analyses were performed as described in . Profiles of (A) glutamine, (B) glutamate, (C) fumarate, (D) malate, (E) citrate, (F) pyruvate and (G) NAD are shown. *p < 0.05; **p < 0.01; ***p < 0.001; Gln, glutamine; n = 15.
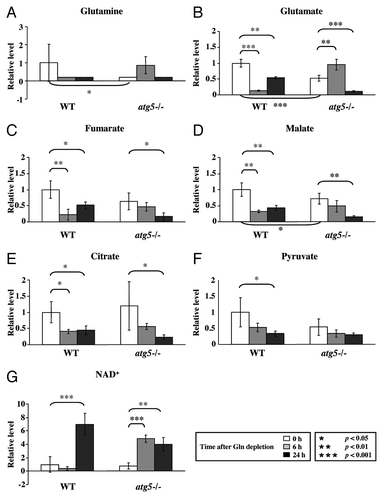
Consistent with the notion that autophagy provides a supply of free amino acids via proteolysis, the basal intracellular glutamine level was lower in atg5−/− MEFs than in WT MEFs in full medium ().Citation7,Citation8 As expected, when we compared their respective responses to glutamine starvation, the glutamine levels became almost undetectable in both cell types by 24 h. To ensure that the intracellular glutamine level was affected by ATG5 level, we tested the response of m5-7 cells, an isogenic atg5−/− MEF line that stably expresses an inducible, tetracycline-off Atg5 construct,Citation23 to glutamine withdrawal in the presence and absence of doxycycline (Dox, 20 ng/ml). A significant decrease of the intracellular glutamine level was observed in m5-7 cells (Fig. S2A, left panel, column 4 vs. column 1) when Dox was added to turn off ATG5 expression (Fig. S2A, right panel). In addition, Dox-treated “ATG5-off” m5-7 cells exhibited lower glucose consumption and reduced ability to produce lactate, whereas the levels of sodium and potassium ion in the medium remained unaffected in response to glutamine withdrawal (Fig. S2B–E).
Because glutamine provides the carbon and nitrogen source for synthesis of NEAAs, including glutamate, aspartate, asparagine, alanine, serine and proline, we compared the level of individual NEAAs in WT and atg5−/− MEFs to determine if they were co-regulated with glutamine in glutamine-deprived atg5−/− MEFs. Five out of six NEAAs examined, including glutamine, were lower in atg5−/− MEFs than in WT MEFs (; Fig. S3A). Compared with the respective levels in WT MEFs in rich medium, a similar decreasing trend in the levels of NEAAs was observed in atg5−/− MEFs maintained in full medium and WT MEFs subjected to glutamine restriction for 6 h (Fig. S3A).
With respect to glutamine levels, the response of atg5−/− MEFs to 6 h glutamine starvation was opposite that of WT MEFs. This paradoxical accumulation of glutamine in atg5−/− MEFs in response to glutamine starvation was unexpected. Our explanation is that glutamine restriction (to decrease ATP levels, see below) combined with an Atg5 deficiency (to decrease release of free amino acids) eventually impaired protein synthesis, thereby increasing glutamine levels in glutamine-deprived atg5−/− MEFs. However, the suggestion that compromised autophagy impedes protein synthesis to increase glutamine levels cannot fully explain the accumulation of EAAs in atg5−/− MEFs, as the levels of NEAAs in the same cells were lower than those in WT cells (Fig. S3A).
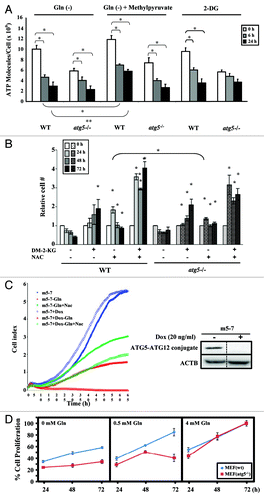
Figure 5. Glutamine depletion inhibits cell proliferation and decreases intracellular ATP levels. (A) ATP levels were reduced by glutamine depletion. WT and atg5−/− MEFs were seeded in complete medium to reach 50% confluence on the day of treatment, and then grown in serum-supplemented DMEM (without glutamine) in the presence or absence of glutamine (4 mM), 2-DG (10 mM) or methylpyruvate (10 mM) for the indicated time periods prior to harvesting. ATP assays were performed as described in Materials and Methods. Cell proliferation was inhibited by glutamine depletion and 2-DG treatment. WT and atg5−/− MEFs were seeded in complete medium to reach 10% confluence on the day of treatment, and then grown in serum-supplemented DMEM in the presence and absence of glutamine (4 mM) or in the full medium with 2-DG (10 mM) for indicated time periods. Relative cell proliferation (determined by the cell proliferation assay described in methods) for each treatment was calculated by comparing to that at 0 h, set as 1. Results from three independent experiments are shown as mean ± SD. The Student’s t-test was performed to determine the difference between (a) control (24/48 h) and -Gln (24/48 h); (b) control (24/48 h) and 2-DG (24/48 h). (B) Both DM-2-KG and NAC rescued cell proliferation. WT and atg5−/− MEFs were seeded in complete medium to reach 10% confluence on the day of treatment, and then grown in glutamine-depleted DMEM with DM-2-KG (7 mM), NAC (2 mM), or both. Cell proliferation was assessed by cell proliferation assay. Significance was calculated using the Student’s t-test between: WT MEFs in Gln(-) (24/48/72 h) and WT MEFs in DM-2-KG/NAC/combination of DM-2-KG and NAC (24/48/72 h); atg5−/− MEFs in Gln(-) (24/48/72 h) and atg5−/− MEFs in DM-2-KG/NAC/combination of DM-2-KG and NAC (24/48/72 h); WT MEFs in NAC (24 h) and atg5−/− MEFs in NAC (24 h). (C) Supplementing with NAC rescues the proliferation of glutamine-deprived cells. Real-time cell proliferation was monitored using an RTCA DP Analyzer. The m5-7 cells were maintained in medium without or with Dox to turn off Atg5 expression (right panel). Glutamine was withdrawn in the presence and absence of NAC supplements and cell growth determined and recorded every 30 min. (D) Minimal glutamine concentration required to support the proliferation of WT and atg5−/− MEFs. Cell proliferation was determined as described in Material and Methods. Briefly, 5000 cells were seeded into 96-well plate over night and incubated overnight prior to switching to the medium with indicated glutamine concentration (0, 0.5 and 4 mM), while 4 mM representing the complete medium. Cell viability was determined at 24, 48 and 72 h afterwards and the % relative cell proliferation was calculated as that of 72 h as 100%. Gln(-), grown in DMEM without glutamine; Gln(+), grown in DMEM with glutamine; DM-2-KG, dimethyl-2-ketoglutarate; 2-DG, 2-deoxyglucose; NAC, N-acetyl-cysteine; Dox, doxycycline. (A and B) Results are shown as mean ± SD from three independent experiments. *p < 0.05.
Because γ-glutamyl amino acids are an intermediate of amino acid transport into cells and are converted into their respective amino acids by γ-glutamyl cyclotransferase,Citation24 we analyzed the levels of four γ-glutamyl amino acids: γ-glutamylleucine, γ-glutamylisoleucine, γ-glutamylthreonine and γ-glutamylvaline. All four γ-glutamyl amino acids accumulated in atg5−/− MEFs at 6 h after glutamine depletion (Fig. S3B). The levels of several metabolites also changed at 6 h after glutamine deprivation, but some of these changes were not statistically significant until 24 h of starvation. For example, both WT and atg5−/− MEFs exhibited reduced levels of pyruvate and intermediates of the TCA cycle, including fumarate, malate and citrate, after glutamine depletion (). These reductions were accompanied by an increase of nicotinamide adenine dinucleotide (NAD+) (), the hydrogen acceptor of the TCA cycle. Intriguingly, the NAD+ level was higher in atg5−/− MEFs at 6 h than that in WT MEFs at 6 h ().
Glutamine is essential for cell proliferation and the maintenance of cellular ATP and nucleotide levels
Among these changes, we further focused on investigating if the presence or absence of ATG5 affected cell proliferation and ATP production under conditions of glutamine starvation in order to understand the nature of ATG5-dependent metabolism and to explore its underlying mechanisms. We first assessed ATP levels in WT and atg5−/− MEFs. As we predicted from the lower glutamine level found in atg5−/− MEFs compared with WT MEFs, the ATP level detected in atg5−/− MEF cells is also lower than that detected in WT cells, and the ATP levels in both cell lines fell upon depletion of extracellular glutamine (, left panel). Treatment with the glycolysis inhibitor 2-deoxyglucose (2-DG), resulted in a significant decrease of cellular ATP in WT MEFs (, right panel), which was expected because both glucose and glutamine are major nutrients required for cell growth and proliferation. However, the effect of 2-DG was not significant in the atg5−/− MEFs. Addition of methylpyruvate, a membrane-permeant metabolic intermediate, partially retarded the ATP decrease in glutamine-starved WT MEFs, but not in the starved atg5−/− MEFs (, left panel vs. middle panel). Compared with the cells grown in full medium, glutamine deprivation inhibited the proliferation of both WT and atg5−/− MEFs, as did treatment with 2-DG (Fig. S4A), suggesting that ATG5-dependent autophagy fails to support cell proliferation against extended restriction on glutamine availability or glucose utilization.
We suspected that reduced levels of the antioxidant glutathione (both oxidized and reduced) might account for the suppression of cell proliferation by glutamine starvation in atg5−/− and WT cells (, lower panel). To test this possibility, we supplemented both the WT and atg5−/− glutamine-deprived MEFs with N-acetyl-cysteine (NAC), a precursor for glutathione. In addition, we assessed the role of glutaminolysis-generated α-ketoglutarate, a key intermediate of the TCA cycle, in supporting cell proliferation by supplementing with dimethyl-2-ketoglutarate (DM-2-KG), a membrane-permeable form of α-ketoglutarate. Although the addition of either NAC or DM-2-KG reduced cell death under glutamine deprivation conditions compared with untreated WT cells, the combination of NAC and DM-2-KG could functionally replace glutamine to effectively rescue cell proliferation (). These observations were further confirmed in m5-7 cells using a real-time growth monitor system. Although cell growth was not affected by the addition of Dox to turn off ATG5 expression in full medium (, right panel), the Dox-untreated “ATG5-on” m5-7 cells were able to proliferate better than the Dox-treated “ATG5-off” m5-7 cells, albeit much slower when glutamine was depleted (, left panel). Consistently, NAC treatment partially rescued the proliferation of the glutamine-restricted cells, independent of Dox treatment/ATG5 expression. Taken together, these data suggest that even though glutamine is needed to generate ATP and other macromolecules, maintaining a sufficiently reduced glutathione level, presumably through supplementing with NAC, is beneficial.
To further assess the individual roles of ATP and nucleotide in maintaining cell proliferation during glutamine deprivation, we examined the number of WT and atg5−/− MEF cells subjected to glutamine starvation in the presence and absence of a combination of supplements. Cell proliferation assays showed that supplementing with glutamine at 6 h and 24 h, but not at 48 h, after withdrawal rescued the proliferation of both the WT and atg5−/− MEFs (Fig. S3B). However, the addition of methylpyruvate or/and deoxynucleoside monophosphates or deoxynucleoside triphosphates failed to elicit similar responses. Since reconstitution with glutamine after 24 h of glutamine starvation could rescue both WT and atg5−/− cells from growth inhibition, we conclude that glutamine starvation-induced suppression of cell proliferation is reversible within 24 h irrespective of the ATG5 context (Fig. S4B).
Lastly, we tested if the initial glutamine concentration required by atg5−/− cells for proliferation is different from the concentration required by WT cells. Asynchronous WT and atg5−/− cells were cultured in normal growth medium (4 mM glutamine), media with intermediate concentrations of glutamine (3, 2 and 1 mM glutamine) and glutamine-free medium. All media were supplemented with 25 mM glucose and all cell cultures were followed for up to 72 h. Reduced proliferation of atg5−/− cells was observed when they were cultured in 1 mM glutamine and by WT cells when cultured in 0 mM glutamine (data not shown), suggesting that atg5−/− cells require a higher concentration of glutamine for proliferation than WT cells. We further titrated the glutamine concentration by 0.1 mM increments from 0 to 1 mM in the cell-seeding medium. Our results showed that 0.5 mM glutamine was sufficient to support WT, but not atg5−/− MEF cell proliferation ().
Regulation of mitochondrial oxidative phosphorylation by ATG5 deficiency and glutamine deprivation
In accordance with the reduced TCA cycle intermediates and ATP levels in the atg5−/− cells compared with WT cells ( and ), we postulated that a possible cause for the arrested proliferation of glutamine-starved cells is the interference of mitochondrial function. To test if autophagy deficiency also resulted in impaired mitochondrial respiration under full and glutamine-depleted medium, oxygen consumption (OCR) was examined with and without an uncoupler [p-trifluoromethoxy carbonyl cyanide phenyl hydrazone (FCCP)] to assess the spare respiratory capacity (SRC),Citation25 which is an indicator of the potential reserve capacity for bioenergetic function in cells, using the above-referred experimental setup ().Citation26 At the 0.5 mM glutamine condition, autophagy-competent WT cells showed appreciable OCR, which was elevated by FCCP (). Thus, the autophagy-competent cells effectively tailored mitochondrial respiration to ATP needs and substrate availability. In contrast, atg5−/− cells showed reduced OCR levels and a blunted FCCP response at 0.5 mM glutamine. Therefore, autophagy-defective atg5−/− cells are fundamentally impaired in mitochondrial respiration when glutamine availability is reduced to 0.5 mM ().
Figure 6. Comparable effect of autophagy deficiency and glutamine restriction on mitochondrial function. (A) Real-time oxygen consumption rate in WT and atg5−/− MEFs maintained at different glutamine concentration (0.5 and 4 mM) was determined in the presence or absence of NAC (10 mM) using Seahorse Extracellular Flux (XF-24) analyzer. Oligomycin (5 μg/ml), FCCP (1 μM) and rotenone (1 μM) were added sequentially to determine mitochondrial function. The tracing exhibited the distinct basal OCR between WT MEFs and autophagy-compromised MEFs maintained at different glutamine concentrations (upper panel) and differential response to NAC addition (lower panel) after normalizing with cell numbers. (B) NAC stimulates OCR in atg5−/− MEFs maintained at 0.5 mM glutamine. The bar graph represents mean ± SD from three independent experiments. A two-tailed Student’s t-test was used to calculate statistical significance. *p < 0.01
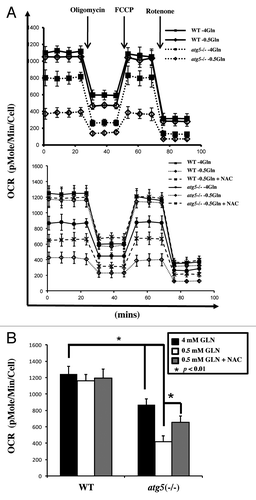
Our data suggested that supplementing with the antioxidant reagent NAC could overcome glutamine deprivation-induced cell death (). We hypothesized that the addition of NAC may improve the oxygen consumption by atg5−/− cells undergoing glutamine deprivation. A comparison of the basal oxygen consumption between WT and atg5−/− cells revealed that the addition of NAC improved the mitochondrial oxidative phosphorylation in glutamine-deprived atg5−/− cells (, lower panel, summarized in ). Taken together, our results suggest that the reactive oxygen species (ROS) level in autophagy-deficient cells was higher than that in autophagy-competent cells, and may interfere with normal mitochondrial function upon glutamine deprivation. In addition, although a decline in intracellular ATP () could be caused by increased ATP utilization in atg5−/− MEF cells, the results from our oxygen consumption rate studies suggested this was not the case.
Regulation of gene expression by ATG5 deficiency and glutamine restriction
To identify key molecules affected by the Atg5 knockout or glutamine restriction, we analyzed the gene expression profiles of key enzymes involved in the TCA cycle and glutamine metabolism and transport pathways in WT and atg5−/− MEFs subjected to glutamine restriction (0.5 mM) for 6 and 24 h (). Among 20 select transcripts, quantitative RT-PCR analyses revealed that the Atg5 knockout resulted in an increase in Aco1, Cs, Flnb, Ogdh, Sdha, Sucla2, Gpt2, Idh1 and Myc mRNA levels and a decrease in the Ldhb mRNA level (). Parallel changes in the transcript levels of Gpt2, Me1, Ogdh, Sucla2, Slc1a5 and Slc3a2 between WT and atg5−/− MEFs over the time course of glutamine restriction was observed (). In contrast, we noticed an opposite regulation of Aco1, Flnb, Idh1/2 and Ldhb mRNA levels in WT and atg5−/− cells subjected to extended glutamine restriction (0.5 mM, 24 h) (). Taken together with our observations that many metabolic parameters observed in WT MEFs after glutamine deprivation for 6 h were comparable to those observed in atg5−/− MEFs maintained in full medium (–), we hypothesize that ATG5 deficiency results in the activation of some of the same regulatory pathways as those activated in WT MEFs in response to glutamine restriction or deprivation.
Figure 7. Glutamine and autophagy reprogram transcript levels. (A) Glutamine reduction regulates selected gene expression. WT and atg5−/− MEFs were seeded in complete medium to reach 50% confluence on the day of treatment, and then grown in serum-supplemented DMEM with or without glutamine for the indicated time periods prior to RNA isolation. RNA isolation and pathway focused quantitative RT-PCR analyses were performed as described in Materials and Methods. The relative fold change was calculated as each transcript levels in WT MEFs (0 h), after normalizing to that of Gapdh, were set as 1. Results are shown as mean ± SD from three independent experiments. *p < 0.05. Aco1, aconitase 1; Cs, citrate synthase; Flnb, fumarate hydratase 1; Gapdh, glyceraldehyde 3-phosphate dehydrogenase; Gpt2, glutamic pyruvate transaminase; Idh1/2, isocitrate dehydrogenase 1/2; Mdh1, malate dehydrogenase 1; Me1/2, malic enzyme 1/2; Myc, myelocytomatosis oncogene; Ogdh, oxoglutarate dehydrogenase; Sdha, Sdhb, succinate dehydrogenase complex; Slc7a5, cationic amino acid transporter; Slc1a5, neutral amino acid transporter; Slc3a2, activators of dibasic and neutral amino acid transport; Slc7a5, cationic amino acid transporter; Suclg1, Suclg2, Sucla2, succinate-CoA ligase. (B) Our proposed model illustrating the crosstalk between autophagy, glutamine utilization and the TCA cycle. mRNAs encoding enzymes in the TCA cycle and glutamine metabolism, studied by quantitative RT-PCR analyses, are boxed. (C) Glutamine restriction induces Cdkn1a (p21) and Bbc3 (Puma) expression in Atg5−/−, but not WT MEFs. Pairs of mRNAs from (A) were used to determine the transcript level of pro-cell cycle arrest gene (Cdkn1a) and pro-apoptotic regulators (Pmaip1 and Bbc3).
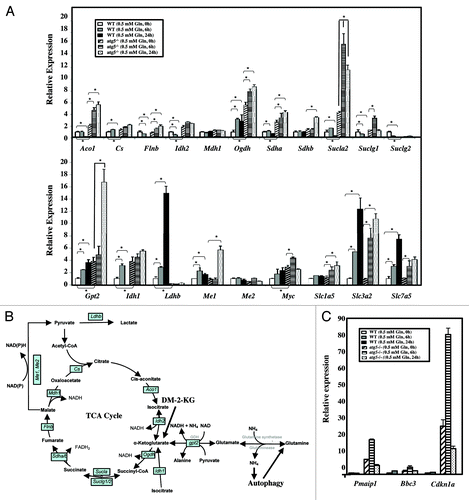
To further address the role of autophagy in coping with metabolic stress, WT and atg5−/− cells were subjected to glutamine restriction (0.5 mM, for 6 and 24 h) and the mRNA levels of genes including proarrest and proapoptotic regulators, such as Pmaip1 (Noxa), Bbc3 (Puma) and Cdkn1a (p21), were analyzed by quantitative RT-PCR. These three genes exhibited higher mRNA levels in atg5−/− cells prior to glutamine restriction () and Cdkn1a and Pmaip1transcript levels were rapidly induced at the early time point (6 h), but extended starvation (24 h) drastically reduced their levels in the atg5−/− MEFs. In contrast, the 0.5 mM glutamine-induced upregulation of proarrest and proapoptotic regulators was not observed in WT MEFs at 0.5 mM glutamine, supporting the hypothesis that autophagy plays an essential role in protecting cells against metabolic stress. Taken together, the data demonstrate that autophagy and glutamine selectively modulate changes in the transcript levels of metabolic, proarrest and proapoptotic regulators.
Glutamine depletion represses autophagy and Atg5 mRNA levels
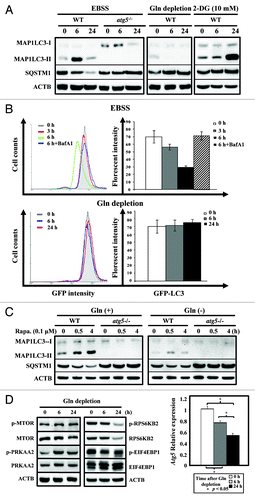
To clarify the role of autophagy in response to glutamine depletion, we examined if depriving cells solely of exogenous glutamine induces autophagy in WT and atg5−/− MEFs. As expected, starvation (EBSS)-treatment, mainly for amino acids and serum, induced a time-dependent change of MAP1LC3 lipidation (LC3-II) and a decrease of the SQSTM1 (p62) level in WT, but not in atg5−/− MEFs, confirming that inactivation of ATG5-dependent autophagy occurred in atg5−/− MEFs and could serve as a control (, left panel). Next we examined the effect of glutamine depletion or glycolysis inhibition on autophagy induction in both WT and atg5−/− MEFs. Although 2-DG-treatment induced a delayed accumulation of MAP1LC3-II (, right panel), the withdrawal of glutamine alone failed to induce MAP1LC3-II lipidation in WT MEFs at 6 and 24 h post treatment (, middle panel). To assess the response of autophagic activity to glutamine depletion in living cells we adapted a quantitative assay that uses GFP-LC3, a well-established autophagosomal marker, and followed its turnover by FACS analysis.Citation27 The reduction in GFP-LC3 fluorescence reflects its delivery into the lysosomes. Consistent with the western analyses of the steady-state levels of endogenous MAP1LC3-I/II (), the FACS assays clearly demonstrated that the GFP fluorescent signal is reduced during EBSS-induced starvation in a time-dependent and lysosomal inhibitor Baf A1-sensitive manner (, upper panel). The GFP-LC3 level remained largely unaffected upon glutamine withdrawal treatment over time (, lower panel). Moreover, glutamine deprivation also decreased the intensity and duration of autophagy induced by rapamycin, a well-known autophagy inducer, because MAP1LC3-II was barely detectable ().
Figure 8. Glutamine depletion fails to induce autophagy. (A) Starvation, but not glutamine depletion, induces autophagy. WT and atg5−/− MEFs were seeded in complete medium to reach 50% confluence on the day of experiment and then grown in EBSS, serum-supplemented DMEM without glutamine, or treated with 2-DG (10 mM), a glycolysis inhibitor, for 0, 6 and 24 h prior to harvesting. Equal amounts of whole cell lysates were analyzed by western blot using anti-MAP1LC3 (LC3), anti-SQSTM1 (p62) and anti-ACTB (actin) antibodies. (B) Glutamine depletion fails to reduce GFP-LC3 fluorescence intensity. (Upper panel) MEF/GFP-LC3 cells were treated with EBSS for 3 and 6 h and then analyzed by FACS. The relative levels of GFP-LC3 fluorescence intensity vs. cell counts are shown in a histogram from a representative experiment (left). MEF/GFP-LC3 cells were cultured in EBSS medium for 3 or 6 h, with or without a 1 h pretreatment of Bafilomycin A1 (Baf A1; 100 nM). For the treated samples, Baf A1 was added to the EBSS media for the additional 6 h incubation. The relative levels of GFP-LC3 intensity from at least three independent experiments are calculated as mean ± SD and shown as a percentage of fluorescence intensity, where control level was designated as 100% (right). (Lower panel) Cells were subjected to glutamine withdrawal and analyzed, without Baf A1-treatment, as shown in (upper panel). (C) Rapamycin induces autophagy in the presence and absence of glutamine. WT and atg5−/− MEFs were seeded in complete medium to reach 50% confluence on the day of experiment and then grown in serum-supplemented DMEM with or without glutamine and analyzed as described in (A). (D) Glutamine depletion induces PRKAA2 (AMPK) and MTOR activation (left panel) and suppresses Atg5 mRNA level (right panel). The phosphorylation status of PRKAA2, MTOR and its downstream RPS6KB2 (p70S6K) and EIF4EBP1 was determined to indicate the activation status of the PRKAA2 and MTOR pathways after glutamine depletion. The expression level of the Atg5 transcript was measured by quantitative RT-PCR analysis. Cells were treated and analyzed as described in (A) with the indicated antibodies and quantitative RT-PCR was performed as described in . Gln, glutamine; 2-DG, 2-deoxyglucose; Rapa, rapamycin.
To further confirm the results of the LC3 lipidation assays, isogenic m5-7 cellsCitation23 were transiently transfected with an mRFP-GFP tandem monomeric fluorescent-tagged MAP1LC3 (tfLC3)Citation28 and maintained in the presence and absence of Dox regulate ATG5 level (Fig. S5A). At 48 h post-transfection, the cells were subjected to EBSS, 2-DG treatment or glutamine withdrawal. pH-sensitive tfLC3 is a reliable tool for examining autophagosome maturation and autolysosome formation because RFP is much more resistant to lysosomal quenching than GFP in an acidic environment.Citation28 In comparison with the control cells (Fig. S5B and S5C), yellow and red puncta were observed in m5-7 cells in the absence of Dox (20 ng/ml) at 6 h and 24 h, respectively, after EBSS exposure (Fig. S5D). This observation, which is consistent with the time-dependent rise and fall of MAP1LC3-II in , suggested a transient formation of autophagosomes at 6 h, followed by autolysosome maturation at 24 h postglutamine starvation. However, when ATG5 deficiency is induced in m5-7 cells in the presence of Dox, no puncta were detected (Fig. S5E). Likewise, after 24 h of 2-DG treatment, a significant number of yellow puncta were formed in m5-7 cells in the presence of ATG5 (-Dox; Fig. S5F), but not in the absence of ATG5 (+Dox; Fig. S5G), and these results are consistent with the MAP1LC3-II accumulation shown in . Nevertheless, no puncta were observed in glutamine-deprived m5-7 cells with or without Dox (Fig. S5H and S5I) supporting the hypothesis that unlike EBSS or 2-DG treatment, glutamine deprivation failed to induce autophagy in MEFs.
To understand the mechanisms underlying the lack of autophagy induction in glutamine-starved cells, we first assessed whether the PRKAA2 (AMPK)-MTOR pathway is dysregulated in cells upon glutamine deprivation. Consistent with our previous results (), glutamine restriction clearly activated PRKAA2, as reflected by the decrease in ATP content. Because PRKAA2 functions as an upstream activator of MTOR, decreased levels of phosphorylation of the MTOR downstream targets, RPS6KB2 (S6K) and EIF4EBP1, were observed in WT MEFs grown in the full media minus glutamine for 6 and 24 h (, left panel).Citation29,Citation30 These results suggested that both the signaling of PRKAA2 activation and the inhibition of MTOR were not impaired in glutamine-deprived cells. Furthermore, the attenuated phosphorylation of RPS6KB2 and EIF4EBP1, MTOR downstream targets, suggests that prolonged glutamine starvation affects protein synthesis. Finally we measured levels of mRNA that encode the autophagy machinery in WT MEFs that have been subjected to glutamine deprivation. We found that glutamine depletion led to a time-dependent decrease in Atg5 mRNA levels in WT MEFs (, right panel). Because ATG5 is indispensable for autophagy (), we postulate that decreased Atg5 levels could, at least in part, explain why there is a failure to induce autophagy in glutamine-starved cells and why we observed certain metabolic responses to glutamine reduction that were comparable to autophagy inactivation.
Discussion
A critical, unresolved issue of metabolic stress responses is how cells alter metabolism to maintain homeostasis. Although in recent years, increased attention has been given to the connection between autophagy and metabolic alterations, the role of autophagy in the management of cellular responses to extracellular glutamine starvation is not yet well understood.Citation6,Citation31 Herein, we demonstrate that autophagy and glutamine utilization are reciprocally regulated. First, autophagy supplies sub-mM concentrations of glutamine to the cell in order to maintain key metabolite levels. Our unbiased metabolomics revealed that the intracellular level of glutamine in atg5−/− MEFs was lower than the corresponding level in WT MEFs, even in glutamine-rich, full medium (), supporting a role for autophagy in maintaining the glutamine supply. Together with our observations that the basal metabolism of atg5−/− MEFs was, in many respects including reduced intracellular ATP levels, similar to the metabolism of WT MEFs at 6 h postglutamine withdrawal ( and ), the results suggest that autophagy plays a critical role in supporting cell metabolism by providing glutamine, especially under conditions of metabolic stress. Second, our data indicate that glutamine reduction, unlike other nutritional stresses, not only failed to induce autophagy, but also attenuated autophagy induced by the MTOR inhibitor, a potent autophagy inducer. Autophagy is a process in which subcellular constituents are degraded in lysosomes in response to stress.Citation32 Although this catabolic process is believed to provide a survival advantage to stressed cells, ATP is required for at least two steps in the autophagic pathway; sequestration and the operation of the lysosomal proton pump.Citation33 In the present study, both a decreased generation of ATP () and an attenuated expression of endogenous Atg5 mRNA () were detected in glutamine-deprived cells, which presumably overrode the stimulation of autophagy by PRKAA2 activation or MTOR inhibition (; Fig. S5). This observation is consistent with recent reports that ammonia generated from glutaminolysis triggers autophagy and that only the removal of leucine, arginine, lysine or methionine from the medium resulted in a significant induction of autophagy.Citation19,Citation27,Citation34
Based on our results, we speculate that glutamine supplied from autophagy plays critical roles in (1) fueling mitochondrial function and (2) regulating gene expression. It is well established that glutamine-derived α-ketoglutarate is used as an anaplerotic and bioenergetic substrate by mitochondria () and many cell lines use this pathway to supply the bulk of carbon flux through the TCA cycle.Citation35-Citation39 This glutamine dependency is more evident in certain cancer cells. For example, it has recently been shown that elevated levels of phosphoglycerate dehydrogenase (PHGDH) promotes serine pathway flux to boost the anaplerosis of glutamine into the TCA cycle as α-ketoglutarate in ER-negative breast cancer cells.Citation40 Moreover, hypoxia, which is a common feature of solid tumors, promotes reductive glutamine metabolism to support cell growth and viability.Citation41,Citation42 In addition to directly fueling mitochondrial oxidative phosphorylation, glutamine also affects mRNA transcript levels. In this report, we showed that the abundance of mRNAs that encode enzymes involved in glutamine-dependent anaplerosis, including Gpt2, Idh1/2, Ogdh, Sdha and Sucla2, was upregulated in atg5−/− MEFs (). Furthermore, pathway focused quantitative RT-PCR revealed that the levels of 18 out of 21 selected mRNAs that encode proteins involved in glutamine/EAA transport or TCA cycle intermediates that support energy production are affected by glutamine depletion in both cell types (). In addition, the failure to robustly induce Ldhb transcripts by glutamine withdrawal in atg5−/− MEFs () agreed with the observed lower lactate production in glutamine-deprived, Dox-treated “ATG5-off” m5-7 cells (Fig. S2C). We propose that modulation of the selected mRNA levels may be aimed at compensating for decreased α-ketoglutarate in the reduced glutamine environments, although this remains to be proved.
Currently, little is known about the regulation of transcript abundance mediated by metabolic stress. Histone methylation and demethylation, which play important roles in the regulation of gene expression by modulating chromatin structure and the recruitment of transcription factors, is proposed to link metabolic signals to chromatin structure and alter transcription.Citation43 Based on our metabolic and quantitative RT-PCR profiles, it is tempting to speculate that the observed changes in mRNA levels are mediated by the affected levels of metabolites via transcriptional control mechanisms. For example, the activity of histone demethylases could be affected by the decrease or increase of cellular metabolites such as flavin adenine dinucleotide (FAD) and α-ketoglutarate, respectively.Citation44,Citation45 In particular, histone demethylases that contain Jmjc domains are members of the α-ketoglutarate/Fe2+-dependent dioxygenase family and they use α-ketoglutarate as a cofactor to catalyze direct hydroxylation of lysine methylamine groups, to produce succinate and carbon dioxide.Citation46 The hydroxy-methyl group is then spontaneously lost as formaldehyde to liberate methyl groups from modified lysine residues. It has been shown that succinate accumulation in cells deficient for succinate dehydrogenase impairs the activity of histone demethylase KDM6B (JMJD3), leading to the accumulation of the transcriptional-repressive, methylation mark of histone H3 on lysine 27.Citation47 It is plausible that succinate might inhibit α-ketoglutarate-dependent dioxygenases, such as histone demethylases that contain Jmjc domains. Conceivably therefore, a decrease of succinate, presumably from impaired mitochondrial oxidative phosphorylation () due to glutamine reduction, could be responsible for the transcriptional differences observed between cells cultured in glutamine-sufficient vs. glutamine-restricted conditions through the modulation of histone demethylase activity. Despite the identification of a transcript profile regulated by the autophagy product glutamine, the exact molecular mechanism of how glutamine governs transcript abundance is not understood. Further studies are needed to unequivocally define if and how histone methylation and demethylation contribute to the metabolic control of transcription mediated by glutamine levels.
Finally, our beneficial effect by NAC-supplemental results suggested that compromised autophagy combined with glutamine reduction increase intracellular ROS production, resulting in decreased oxygen consumption and thereby, ATP production. Although ROS initiates signaling events that facilitate the proliferation of metabolically active cells, cells must also protect themselves against the deleterious effects of pathologically elevated ROS levels.Citation48,Citation49 It is well-established that glutamine buffers cells against oxidative damage by donating both carbon and nitrogen to the major intracellular antioxidant glutathione and also contributes to the maintenance of NADPH levels used to reduce oxidized glutathione to glutathione. We also demonstrated that glutamine deprivation or autophagy impairment results in decreased glutathione levels (), presumably leading to increased ROS. Our metabolomic profiling reveals that γ-glutamyl amino acids accumulated in atg5−/− MEFs after 6 h of glutamine depletion (Fig. S3B). Conceivably therefore, ATG5 deficiency and glutamine deprivation both may suppress the activity of γ-glutamyl cyclotransferase, a key enzyme for the biosynthesis of glutathione, thereby decreasing intracellular glutathione levels.Citation24 In agreement with this hypothesis, supplementation with NAC at 24 h restored the proliferation and oxygen consumption of glutamine-deprived atg5−/− cells ( and ). Taken together, it seems possible that excessive ROS-induced damages, presumably from reduced levels of glutathione, occur in glutamine-restricted cells. However, additional experiments are needed to identify the affected key molecules that are involved in mitochondrial oxidative phosphorylation.
Previous studies have shown that cancer cells can become “addicted” to glutamine. After entering the cell glutamine is converted to glutamate, then enters the TCA cycle and this results in the production of ATP by oxidative phosphorylation.Citation50 In this report, we showed that Atg5 knockout results in decreased glutamine levels, presumably via impaired autophagy. Amino acids provided by autophagy are particularly important when cancer cells are exposed to nutrient starvation. For example, Sheen et al. have shown that leucine is indispensable for the survival of human melanoma.Citation51 It now appears that the glutamine generated from autophagy is crucial for the cellular homeostatic control of MEFs (this report) and cancer cells (data not shown). In addition, our data also suggest that atg5−/− MEFs may utilize extra glutamine to compensate for a lack of basal autophagy, acerbating a “glutamine-deficient” phenotype even when maintained in rich medium. For example, systematic metabolomic profiling indicated that the atg5−/− MEFs accumulated higher levels of EAAs. In the light of a recent report that glutamine efflux is essential for transport of EAAs into cells, it is possible that the glutamine in atg5−/− MEFs was presumably channeled toward maintaining the EAAs, thereby further lowering the intracellular glutamine level.Citation20 Although our current data failed to differentiate the contribution by glutamine from autophagy or extracellular nutrition to maintaining cellular homeostasis, it seems likely that autophagy is the main mechanism for providing glutamine needed to fuel the oxidative mitochondrial metabolism, especially under certain stress conditions.
Although ATG5 has previously been characterized as a protein that is specifically required for autophagy, it has also been shown that mammalian autophagy can occur in an ATG5-independent manner.Citation52 Nonetheless, our findings provide new insights into how autophagy can provide a survival advantage to cells using alternative bio-energetic substrates, such as glutamine, to maintain ATP production and mitochondrial integrity in MEFs. The exclusive decrease of EAAs and BCAA metabolites in glutamine-starved (6 h) atg5−/− MEFs further supports the critical role of glutamine utilization in autophagy-compromised cells. Our findings shed new light on the fine balance and coordination between autophagy and glutamine utilization. Although further investigation will be required to determine the mechanisms regulating glutamine- and ATG5-dependent transcriptional control, our studies should provide a mechanistic perspective for the complex signaling pathways that complement each other in the regulation of autophagy-mediated and glutamine-dependent metabolism. Furthermore, they have provided us with new targets for the prevention and treatment of many diseases that involve dysregulation of metabolism, including cancer.
Materials and Methods
Cell lines and chemicals
WT and atg5−/− MEFs (simian virus 40 T-antigen immortalized) were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% bovine growth serum (Thermo Scientific, SH30541.03) and 1% penicillin/streptomycin (Gibco, 15240), with (Cellgro, 10-013) or without (Cellgro, 15-013) glutamine. The m5-7 cells, that harbored the inducible tet-off Atg5 that was derived from atg5−/− MEFs, were maintained as described previously.Citation23 Dimethyl-2-ketoglutarate (DM-2-KG; 349631), 2-deoxyglucose (2-DG; Sigma, D8375), deoxynucleotides monophosphates (dNMPs; D6375, T7004, D9500, D7750), IMP (I4625), methylpyruvate (371173), N-acetyl-cysteine (NAC; A7250) and transaminase inhibitor aminooxyacetate (C13408) were purchased from Sigma, deoxynucleotides triphosphates (dNTPs) were from Roche (11814362001) and L-glutamine was obtained from Gibco (25030).
Cell proliferation assay
Approximately 5,000 WT and atg5−/− MEFs were seeded into 96-well plates or 16 × E-plates (ACEA, Roche, DC-16-B-NT) and allowed to reach 5% confluence on day 1. On day 2, the medium (100 µl) was switched to a combination of DMEM with the indicated glutamine supplement plus 10% bovine growth serum, dNMPs, dNTPs, methylpyruvate, NAC and DM-2-KG. Cell proliferation was measured by two different methods. First, RTCA DP Analyzer (ACEA, Roche) was used to monitor real-time cell growth using 16 × E-plates. Second, an MTS assay was performed at the end of the indicated period of post-medium switching using CellTiter 96® Aqueous OneSolution Reagent (20 µl, Promega, G3580). After incubation, the absorbance at 490 nm was measured to calculate relative proliferation, which is represented as the mean ± standard deviation (S.D.) from three independent experiments, each performed in duplicate.
Oxygen consumption rate (OCR)
Cellular mitochondrial function was measured using a Seahorse Bioscience XF24 Extracellular Flux Analyzer. The mitochondrial function was assayed by sequential injections of oligomycin (ATP synthase inhibitor; Sigma 75351), FCCP [carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone; mitochondrial oxidative phosphorylation protonophore and uncoupler; Sigma C2920] and rotenone (mitochondrial complex I inhibitor; Sigma R8875) to define basal OCR, ATP-linked OCR, proton leak, maximal respiratory capacity, reserve respiratory capacity and non-mitochondrial oxygen consumption, all according to manufacturer’s instructions. Herein, basal OCR is used to represent the function of mitochondria. Briefly, 2 × 104 cells were seeded into 24-well plates and incubated overnight prior to the sequential addition of a pre-optimized concentration of oligomycin, FCCP and rotenone, respectively. After washing the cells with 1 ml seahorse buffer [DMEM medium without phenol red containing glucose (4.5 g/l), sodium pyruvate (1 mM) and glutamine (0.5 mM)], 600 μl seahorse buffer plus 60 μl each of oligomycin, (50 μg/ml), FCCP (10 μM) and rotenone (10 μM) respectively, was automatically injected. At the end of recording period, cells were lysed and the individual protein concentrations were determined using the Bradford assay. OCR values were calculated after normalizing with the protein concentration and they are plotted as the mean ± SD.
ATP assay
The ENLITEN® ATP Assay System (Promega, FF2000) was used according to manufacturer’s instructions. WT and atg5−/− MEFs were seeded to reach 50% confluence on the day of the experiment and each experimental set was grown in duplicate. At the indicated times, cells in one set were trypsinized and stained with trypan blue to determine cells number and cells in the second set were harvested with PBS (500 μl), and the cell suspension (400 μl) then added to 5% trichloroacetic acid (TCA, 100 μl) according to the manufacturer’s recommendation. TRIS-acetate buffer (900 μl, pH 7.75) was then added to neutralize the TCA solution (100 µl) and to dilute the TCA to a final concentration of 0.1%. The extracts were then further diluted 1:100 and added to an equal volume of rL/L Reagent (Promega, FF2000) that contained D-luciferin and recombinant luciferase prior to measuring the luminescence using a TD-30e luminometer (Turner). The ATP standard (Promega, FF2000) was serially diluted to generate a regression curve for calculation of the exact number of ATP molecules, which was then divided by the cell number to derive the number of ATP molecules per cell. Three independent experiments were performed, and are represented as the mean ± SD.
Whole cell lysate preparation and western blot analyses
Cells were grown in DMEM, with or without glutamine, and supplemented with 10% bovine growth serum or Earle’s balanced salt solution (EBSS; Sigma, E3024) for the indicated time periods. For detection of MAP1LC3-I and MAP1LC3-II, whole cell extracts (WCEs) were prepared with lysis buffer (20 mM Tris-HCL, pH 7.4, 150 mM NaCl, 1% Triton X-100) plus complete protease inhibitor mixture (Roche, 11836145001). WCEs were then mixed with an equal volume of SDS loading buffer and boiled for 3 min. WCEs were then separated by SDS-PAGE followed by immunoblotting with the antibodies: anti-MAP1LC3-I/II (Medical and Biological Laboratories, M115–3), anti-SQSTM1 (American Research Products, 03-GP62-C), anti-phospho-PRKAA2 (Thr172; Cell Signaling, 2535), anti-PRKAA2 (Cell Signaling, 2532), anti-phospho-MTOR (Ser2448; Cell Signaling, 2971), anti-MTOR (Cell Signaling, 2983), anti-phospho-RPS6KB2 (Ser371; Cell Signaling, 9208), anti-RPS6KB2 (Cell Signaling, 9202), anti-phospho-EIF4EBP1 (Thr37/46; Cell Signaling, 2855), anti-EIF4EBP1 (Cell Signaling, 9644) and anti-ACTB (Chemicon, MAB1501R) antibodies. Immunoblots were visualized by staining with the enhanced chemiluminescence detection kit (ECL-Plus, Amersham Biosciences, RPN2132) and detection using a VersaDoc 5000 imaging system (Bio-Rad). Densitometric tracing from images was captured and quantified with Quantity One Software (Bio-Rad). The desired signal in individual samples was normalized to an ACTB (actin) control. Relative protein levels in experimental samples were calculated against the normalized protein level of the untreated sample set as in 1.
Global metabolic profiling
The nontargeted metabolic profiling platform employed for this analysis combined three independent platforms: ultrahigh performance liquid chromatography/tandem mass spectrometry (UHPLC/MS/MSCitation2) optimized for basic species, UHPLC/MS/MSCitation2 optimized for acidic species, and gas chromatography/mass spectrometry (GC/MS). WT and atg5−/− MEFs (five replicates, three independent experiments) were cultured in glutamine-deficient DMEM supplemented with 10% bovine growth serum for 0, 6 and 24 h to reach 50% confluence prior to harvesting. Briefly, equal numbers (3 × 106) of cells in each replicate of WT and atg5−/− MEFs were processed as described previously.Citation50,Citation53 Cells were homogenized in a fixed, minimum volume of water and 100 μl withdrawn for subsequent analyses. Using an automated liquid handler (Hamilton LabStar), protein was precipitated from the homogenized cells with methanol that contained four standards used to report the extraction efficiency. The resulting supernatants were split into equal aliquots for analysis on the three platforms. Aliquots, dried under nitrogen and vacuum-desiccated, were subsequently either reconstituted in 0.1% formic acid in water (50 μl, acidic conditions) or in 6.5 mM ammonium bicarbonate in water, pH 8 (50 μl, basic conditions) for the two UHPLC/MS/MSCitation2 analyses or derivatized to a final volume of 50 μl for GC/MS analysis using equal parts of bistrimethyl-silyl-trifluoroacetamide and a solvent mixture of acetonitrile:dichloromethane:cyclohexane (5:4:1) with 5% triethylamine at 60°C for one hour. In addition, three types of controls were analyzed in concert with the experimental samples: samples generated from pooled experimental samples served as technical replicates throughout the data set, extracted water samples served as process blanks, and a cocktail of standards spiked into every analyzed sample allowed instrument performance monitoring. Experimental samples and controls were randomized across a one-day platform run.
For UHPLC/MS/MSCitation2 analysis, aliquots were separated using a Waters Acquity UPLC (Waters) and analyzed using an LTQ mass spectrometer (Thermo Fisher Scientific, Inc.) that consisted of an electrospray ionization source and linear ion-trap mass analyzer. The MS instrument scanned 99–1000 m/z and alternated between MS and MSCitation2 scans using dynamic exclusion with approximately 6 scans per second. Derivatized samples for GC/MS were separated on a 5% phenyldimethyl silicone column with helium as the carrier gas and a temperature ramp from 60°C to 340°C, then analyzed on a Thermo-Finnigan Trace DSQ MS (Thermo Fisher Scientific, Inc.) operated at unit mass resolving power with electron impact ionization and a 50–750 atomic mass unit scan range.
Metabolites were identified by automated comparison of the ion features in the experimental samples to a reference library of chemical standard entries that included retention time, molecular weight (m/z), preferred adducts, and in-source fragments as well as associated MS spectra and were curated by visual inspection for quality control using software developed at Metabolon.Citation54
Medium BioProfile and cellular glutamine measurement
m5-7 cells (5 × 105) were cultured in 6-well plates in the presence or absence of doxycycline (Dox, 20 μg/ml; Sigma, D9891) and deprived of glutamine for 0, 6 and 24 h. The level of glucose, lactate, Na+ and K+ in the culture medium was then measured using BioProfile 100 Plus (Nova Biomedical Corporation). Intracellular glutamine levels were measured using the EnzyChromTM Glutamine Assay Kit (BioAssay Systems). Cell number was determined by trypan blue exclusion assay for normalization.
Quantitative RT-PCR analyses
RNA isolation was performed using an RNeasy Mini Kit according to the manufacturer’s instructions. (Qiagen, 74104) and reverse transcribed into cDNA using an iScriptTMcDNA Synthesis Kit (Bio-Rad, 170-8891). Synthesized cDNA and iQTM SYBR® Green Supermix (Bio-Rad, 170-8882) were mixed for subsequent quantitative real-time PCR performed on an iQTM single color real-time PCR detection system (Bio-Rad). The PCR program consisted of one cycle of 95°C for 3 min, followed by up to 40 cycles of 95°C for 15 sec and 55°C for 45 sec. The amplicon specificity was confirmed using dissociation curves after the reactions were complete. The primer pairs for Cs, Aco, Ogdh, Suclg1, Suclg2, Sucla2, Sdha, Sdhb, Flnb, Mdh1, Idh1, Idh2, Gpt2, Ldhb, Me1, Me2, Myc, Slc7a5, Slc1a5, Slc3a2 and Gapdh are shown in Table S2. The transcript levels of genes were normalized to those of Gapdh and calculated using the ∆CT equation as follows: relative expression = 2-∆CT, where ∆CT = CT (gene) − CT (Gapdh).
Fluorescence microscopy analyses
m5-7 cells that were maintained were transiently transfected with mRFP-GFP-LC3 and cultured, in the presence or absence of Dox, either in complete medium or treated with EBSS, 2-DG or glutamine depleted medium for 6 and 24 h. The cells were then fixed in 3.7% paraformaldehyde (Sigma, P6148) in PBS and examined using an Olympus IX81 inverted fluorescence microscope. Fluorescent images were acquired and saved using Zeiss LSM Image Browser software.
Measurement of GFP-LC3 intensity by fluorescence-activated cell sorter (FACS) analysis
WT MEF cells that stably expressed GFP-LC3 were seeded subconfluently into 6-well plates the day before glutamine depletion. Following the indicated treatment periods, cells were harvested with trypsin/EDTA, washed with PBS, and subjected to FACS. Analysis of 1 × 105 cells/sample was performed by a CyAnTM ADP 9 Color Flow cytometer (DAKO; Analytic Cytometry Core Facility in City of Hope Medical Center), and viable cell counts were plotted as GFP fluorescence intensity by FlowJo Software (Tree Star, Inc.). The level of GFP fluorescence intensity in each treated sample was normalized to the level of resting, vehicle treated controls set at 100%. The relative level of GFP-LC3 intensity in each treatment was calculated from at least three independent experiments.
Statistical analysis
Each cell biology experiment was performed in triplicate for representative means and images. For statistical analysis and data display purposes for the metabolomics data, raw values were normalized by sample cell number. Any missing values were assumed to be below the limits of detection and these values were imputed with the compound minimum (minimum value imputation). Statistical analysis of log-transformed data was performed using “R” (http://cran.r-project.org), which is a freely available, open-source software package. Welch’s t-tests were performed to compare data between experimental groups. Multiple comparisons were accounted for by estimating the false discovery rate using q-values. The complete linkage hierarchical cluster and heatmap were generated using Gene Cluster 3.0. A probability value of less than 0.5 (p < 0.05) was considered significant.
| Abbreviations: | ||
| PRKAA2 | = | AMP-dependent protein kinase |
| MTOR | = | mechanistic target of rapamycin |
| NEAA | = | nonessential amino acids |
| PE | = | phosphatidylethanolamine |
| EAA | = | essential amino acids |
| PRPP | = | 5-phosphoribosyl-1-pyrophophate |
| IMP | = | inosine 5′-monophosphate |
| 2-DG | = | 2-deoxyglucose |
| dNTP | = | deoxynucleotide triphosphate |
| dNMP | = | deoxynucleotide monophosphate |
| TCA | = | trichloroacetic acid |
| TCA cycle | = | tricarboxylic acid cycle or Kreb cycle |
| GFP-LC3 | = | human MAP1LC3 gene is fused in frame with GFP in the expression vector |
| tfLC3 | = | tandem fluorescent-tagged MAP1LC3 |
| BCAA | = | branched chain amino acid |
| NAD+ | = | nicotinamide adenine dinucleotide |
| AMP | = | adenosine monophosphate |
| ADP | = | adenosine diphosphate |
| DM-2-KG | = | dimethyl-2-ketoglutarate |
| NAC | = | N-acetyl-cysteine |
Additional material
Download Zip (1.5 MB)Acknowledgments
We are sincerely grateful to Drs. Chih-Pin Liu, Rama Natarajan, Mei-Ling Kuo, Paul Lee, Wei Jia and Henry Forman for their helpful suggestions and critical reading of manuscript. This work was supported in part by National Institute of Health Research Grants R01DE10742 and R01DE14183 (to D.K.A.), Nesvig Foundation Grant Award (to D.K.A. and M.K.), City of Hope’s Women’s Cancers Program Award (to Y.-R.C.), and National Science Council (Taiwan) Grant NSC97-2917-I-002–101 (to T.-C.L.). We also thank Dr. Guihua Sun for the conducting heatmap analyses, members of the Ann laboratory for helpful discussions, Ying-Yin Chao and Michael Reid for technical assistance and Drs. Margaret Morgan and Keely Walker for editing.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/21228
References
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27 - 42; http://dx.doi.org/10.1016/j.cell.2007.12.018; PMID: 18191218
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313 - 26; http://dx.doi.org/10.1016/j.cell.2010.01.028; PMID: 20144757
- Wang W, Guan KL. AMP-activated protein kinase and cancer. Acta Physiol (Oxf) 2009; 196:55 - 63; http://dx.doi.org/10.1111/j.1748-1716.2009.01980.x; PMID: 19243571
- Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 2005; 5:726 - 34; http://dx.doi.org/10.1038/nrc1692; PMID: 16148885
- Mizushima N. The pleiotropic role of autophagy: from protein metabolism to bactericide. Cell Death Differ 2005; 12:Suppl 2 1535 - 41; http://dx.doi.org/10.1038/sj.cdd.4401728; PMID: 16247501
- Rabinowitz JD, White E. Autophagy and metabolism. Science 2010; 330:1344 - 8; http://dx.doi.org/10.1126/science.1193497; PMID: 21127245
- Onodera J, Ohsumi Y. Autophagy is required for maintenance of amino acid levels and protein synthesis under nitrogen starvation. J Biol Chem 2005; 280:31582 - 6; http://dx.doi.org/10.1074/jbc.M506736200; PMID: 16027116
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature 2004; 432:1032 - 6; http://dx.doi.org/10.1038/nature03029; PMID: 15525940
- DeBerardinis RJ, Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010; 29:313 - 24; http://dx.doi.org/10.1038/onc.2009.358; PMID: 19881548
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 2008; 7:11 - 20; http://dx.doi.org/10.1016/j.cmet.2007.10.002; PMID: 18177721
- Zielke HR, Ozand PT, Tildon JT, Sevdalian DA, Cornblath M. Reciprocal regulation of glucose and glutamine utilization by cultured human diploid fibroblasts. J Cell Physiol 1978; 95:41 - 8; http://dx.doi.org/10.1002/jcp.1040950106; PMID: 641112
- Donnelly M, Scheffler IE. Energy metabolism in respiration-deficient and wild type Chinese hamster fibroblasts in culture. J Cell Physiol 1976; 89:39 - 51; http://dx.doi.org/10.1002/jcp.1040890105; PMID: 8468
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324:1029 - 33; http://dx.doi.org/10.1126/science.1160809; PMID: 19460998
- Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci 2010; 35:427 - 33; http://dx.doi.org/10.1016/j.tibs.2010.05.003; PMID: 20570523
- Matés JM, Pérez-Gómez C, Núñez de Castro I, Asenjo M, Márquez J. Glutamine and its relationship with intracellular redox status, oxidative stress and cell proliferation/death. Int J Biochem Cell Biol 2002; 34:439 - 58; http://dx.doi.org/10.1016/S1357-2725(01)00143-1; PMID: 11906817
- Bode BP, Fuchs BC, Hurley BP, Conroy JL, Suetterlin JE, Tanabe KK, et al. Molecular and functional analysis of glutamine uptake in human hepatoma and liver-derived cells. Am J Physiol Gastrointest Liver Physiol 2002; 283:G1062 - 73; PMID: 12381519
- Szeliga M, Obara-Michlewska M. Glutamine in neoplastic cells: focus on the expression and roles of glutaminases. Neurochem Int 2009; 55:71 - 5; http://dx.doi.org/10.1016/j.neuint.2009.01.008; PMID: 19428809
- Eng CH, Abraham RT. Glutaminolysis yields a metabolic by-product that stimulates autophagy. Autophagy 2010; 6:968 - 70; http://dx.doi.org/10.4161/auto.6.7.13082; PMID: 20724823
- Eng CH, Yu K, Lucas J, White E, Abraham RT. Ammonia derived from glutaminolysis is a diffusible regulator of autophagy. Sci Signal 2010; 3:ra31; http://dx.doi.org/10.1126/scisignal.2000911; PMID: 20424262
- Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009; 136:521 - 34; http://dx.doi.org/10.1016/j.cell.2008.11.044; PMID: 19203585
- Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem 2007; 282:37298 - 302; http://dx.doi.org/10.1074/jbc.C700195200; PMID: 17986448
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 2001; 152:657 - 68; http://dx.doi.org/10.1083/jcb.152.4.657; PMID: 11266458
- Hosokawa N, Hara Y, Mizushima N. Generation of cell lines with tetracycline-regulated autophagy and a role for autophagy in controlling cell size. FEBS Lett 2006; 580:2623 - 9; http://dx.doi.org/10.1016/j.febslet.2006.04.008; PMID: 16647067
- Meister A. Selective modification of glutathione metabolism. Science 1983; 220:472 - 7; http://dx.doi.org/10.1126/science.6836290; PMID: 6836290
- Choi SW, Gerencser AA, Nicholls DG. Bioenergetic analysis of isolated cerebrocortical nerve terminals on a microgram scale: spare respiratory capacity and stochastic mitochondrial failure. J Neurochem 2009; 109:1179 - 91; http://dx.doi.org/10.1111/j.1471-4159.2009.06055.x; PMID: 19519782
- Dranka BP, Hill BG, Darley-Usmar VM. Mitochondrial reserve capacity in endothelial cells: The impact of nitric oxide and reactive oxygen species. Free Radic Biol Med 2010; 48:905 - 14; http://dx.doi.org/10.1016/j.freeradbiomed.2010.01.015; PMID: 20093177
- Shvets E, Fass E, Elazar Z. Utilizing flow cytometry to monitor autophagy in living mammalian cells. Autophagy 2008; 4:621 - 8; PMID: 18376137
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3:452 - 60; PMID: 17534139
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003; 115:577 - 90; http://dx.doi.org/10.1016/S0092-8674(03)00929-2; PMID: 14651849
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 2008; 30:214 - 26; http://dx.doi.org/10.1016/j.molcel.2008.03.003; PMID: 18439900
- Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010; 330:1340 - 4; http://dx.doi.org/10.1126/science.1193494; PMID: 21127244
- Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ 2005; 12:Suppl 2 1542 - 52; http://dx.doi.org/10.1038/sj.cdd.4401765; PMID: 16247502
- Plomp PJ, Wolvetang EJ, Groen AK, Meijer AJ, Gordon PB, Seglen PO. Energy dependence of autophagic protein degradation in isolated rat hepatocytes. Eur J Biochem 1987; 164:197 - 203; http://dx.doi.org/10.1111/j.1432-1033.1987.tb11011.x; PMID: 3830181
- Cheong H, Lindsten T, Wu J, Lu C, Thompson CB. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc Natl Acad Sci U S A 2011; 108:11121 - 6; http://dx.doi.org/10.1073/pnas.1107969108; PMID: 21690395
- Reitzer LJ, Wice BM, Kennell D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem 1979; 254:2669 - 76; PMID: 429309
- Brand K. Glutamine and glucose metabolism during thymocyte proliferation. Pathways of glutamine and glutamate metabolism. Biochem J 1985; 228:353 - 61; PMID: 2861809
- DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A 2007; 104:19345 - 50; http://dx.doi.org/10.1073/pnas.0709747104; PMID: 18032601
- Lemons JM, Feng XJ, Bennett BD, Legesse-Miller A, Johnson EL, Raitman I, et al. Quiescent fibroblasts exhibit high metabolic activity. PLoS Biol 2010; 8:e1000514; http://dx.doi.org/10.1371/journal.pbio.1000514; PMID: 21049082
- Yang C, Sudderth J, Dang T, Bachoo RM, McDonald JG, DeBerardinis RJ. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res 2009; 69:7986 - 93; http://dx.doi.org/10.1158/0008-5472.CAN-09-2266; PMID: 19826036
- Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011; 476:346 - 50; http://dx.doi.org/10.1038/nature10350; PMID: 21760589
- Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, et al. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A 2011; 108:19611 - 6; http://dx.doi.org/10.1073/pnas.1117773108; PMID: 22106302
- Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2012; 481:380 - 4; PMID: 22101433
- Teperino R, Schoonjans K, Auwerx J. Histone methyl transferases and demethylases; can they link metabolism and transcription?. Cell Metab 2010; 12:321 - 7; http://dx.doi.org/10.1016/j.cmet.2010.09.004; PMID: 20889125
- Anand R, Marmorstein R. Structure and mechanism of lysine-specific demethylase enzymes. J Biol Chem 2007; 282:35425 - 9; http://dx.doi.org/10.1074/jbc.R700027200; PMID: 17897944
- Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006; 439:811 - 6; http://dx.doi.org/10.1038/nature04433; PMID: 16362057
- Klose RJ, Zhang Y. Regulation of histone methylation by demethylimination and demethylation. Nat Rev Mol Cell Biol 2007; 8:307 - 18; http://dx.doi.org/10.1038/nrm2143; PMID: 17342184
- Cervera AM, Bayley JP, Devilee P, McCreath KJ. Inhibition of succinate dehydrogenase dysregulates histone modification in mammalian cells. Mol Cancer 2009; 8:89; http://dx.doi.org/10.1186/1476-4598-8-89; PMID: 19849834
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A 2010; 107:8788 - 93; http://dx.doi.org/10.1073/pnas.1003428107; PMID: 20421486
- Wellen KE, Thompson CB. Cellular metabolic stress: considering how cells respond to nutrient excess. Mol Cell 2010; 40:323 - 32; http://dx.doi.org/10.1016/j.molcel.2010.10.004; PMID: 20965425
- Evans AM, DeHaven CD, Barrett T, Mitchell M, Milgram E. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal Chem 2009; 81:6656 - 67; http://dx.doi.org/10.1021/ac901536h; PMID: 19624122
- Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, et al. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009; 461:654 - 8; http://dx.doi.org/10.1038/nature08455; PMID: 19794493
- Ohta T, Masutomi N, Tsutsui N, Sakairi T, Mitchell M, Milburn MV, et al. Untargeted metabolomic profiling as an evaluative tool of fenofibrate-induced toxicology in Fischer 344 male rats. Toxicol Pathol 2009; 37:521 - 35; http://dx.doi.org/10.1177/0192623309336152; PMID: 19458390
- Dehaven CD, Evans AM, Dai H, Lawton KA. Organization of GC/MS and LC/MS metabolomics data into chemical libraries. J Cheminform 2010; 2:9; http://dx.doi.org/10.1186/1758-2946-2-9; PMID: 20955607
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 2003; 100:9440 - 5; http://dx.doi.org/10.1073/pnas.1530509100; PMID: 12883005