Abstract
Autophagy plays an important role in the cellular response to a variety of metabolic stress conditions thus contributing to the maintenance of intracellular homeostasis. Studies in yeast have defined the genetic components involved in the initiation of autophagy as well as the progression through the autophagic cascade. The yeast kinase Atg1 initiates autophagy in response to nutrient limitation in a TOR-dependent manner. The ulk family of genes encodes the mammalian orthologue of yeast Atg1. Our recent work using mouse embryonic fibroblast (MEF) cell lines deficient for both ulk1 and ulk2, has revealed that autophagy induction is more complex in mammals than in yeast. Furthermore, these data confirm the surprising finding that a by-product of amino acid metabolism, ammonia, is a strong inducer of autophagy, as first shown by the Abraham laboratory.
It was initially observed that Ulk1/2 double-deficient (Ulk1/2 DKO) MEFs are unable to mount an autophagic response to amino acid deprivation as shown previously for MEFs deficient for the core autophagy gene Atg5. Surprisingly, the autophagy response to 24 h glucose deprivation is intact in the Ulk1/2 DKO MEFs as opposed to that seen in Atg5-deficient MEFs. Moreover, Ulk1/2 DKO MEFs are able to induce autophagy in response to bioenergetic stress following pharmacological treatment with a glycolytic inhibitor (2-deoxyglucose), an inhibitor of mitochondrial respiration (phenformin), and an AMPK activator (AICAR). Recently, the Guan laboratory suggested that during relatively brief periods (4–5 h) of glucose starvation, the energy-sensor AMPK directly phosphorylates ULK1, leading to autophagy activation. However, we observed that during longer periods of glucose deprivation, Ulk1/2-independent for autophagy induction. These data suggested that Ulk1/2-independent autophagy might be stimulated by an activator(s) distinct from the known energy-sensing signaling pathways. In order to compensate for bioenergetic stress during long-term glucose starvation, cells must catabolize amino acids and fatty acids. As a consequence of elevated amino acid catabolism during glucose deprivation, cellular ammonia levels increase steadily. To test whether ammonia could induce autophagy in Ulk1/2 DKO MEFs, ammonia levels were manipulated either by treatment with extracellular NH4Cl, or by treatment with glucosamine leading to increased ammonia levels generated from the hexosamine biosynthetic pathway. Both treatments induce autophagy in wild-type as well as in Ulk1/2 DKO MEFs but not Atg5-deficient MEFs. Moreover, the autophagy response to ammonia can be abrogated by treatment with cell-permeable pyruvate, which can combine with ammonia to form alanine, which is then secreted from the cells. Surprisingly, the autophagy response to ammonia is independent of mTOR. While many studies have confirmed the critical role of Ulk1/2 in the initiation of autophagy in response to amino acid withdrawal, the data using Ulk1/2 DKO MEFs establishes the existence of an mTOR- and Ulk1/2-independent pathway of autophagy.
The observation originally made by Otto Warburg that cancer cells exhibit distinct metabolism from normal cells has received increasing appreciation in recent years. It is now known that oncogenic signals and/or genetic alterations of metabolic pathways allow cancer cells to reprogram their metabolism to gain proliferative advantage and survive under conditions of nutrient and oxygen limitation. Since autophagy and metabolic reprogramming are both important cellular responses to metabolic demands, it is tempting to hypothesize they are intimately linked. Autophagy occurs constitutively to maintain cellular homeostasis. This process is significantly upregulated by various stress conditions such as nutrient or growth factor limitation, oxidative stress and excess accumulation of damaged organelles. Cancer cells are often exposed to these stress conditions, and therefore autophagy can be considered an important adaptive survival mechanism. Through the catabolic processes of autophagy, cancer cells can acquire necessary fuel to survive under metabolic challenges instead of dying by apoptosis.
It has recently been shown that a variety of amino acids are critical for cell growth. Glutamine can supply the carbon backbone for regenerating TCA cycle intermediates, and can also serve as a nitrogen source. The serine biosynthetic pathway has also been suggested recently to be critical for replenishing the TCA cycle as shown by the Sabatini laboratory. When cancer cells are exposed to nutrient starvation, amino acids can be provided by autophagy. For example, leucine is indispensable for cell growth in cancer models and autophagy can prevent cell death caused by leucine deprivation. It now appears that the ammonia generated from amino acid catabolism following glucose deprivation can also stimulate autophagy. This ammonia-induced autophagy also promotes cell survival and thus represents a promising therapeutic target in cancer cell treatments.
Oncogenic signaling may play a critical role in autophagy induction. The overexpression of Myc in cancer cells leads to increased glutamine catabolism by increasing the level of glutamine transporters and the enzymes involved in glutaminolysis. Since Myc-induced glutamine metabolism increases ammonia levels, Myc amplification in cancer cells may lead to an increase of basal autophagy. Constitutively active Ras expression increases autophagy, which might play an important role in maintaining cell viability as cells alter their metabolism to satisfy their bioenergetic and biosynthetic demand. However, it is not fully understood how metabolic alterations caused by Ras activation directly engage in various cell death/survival pathways in response to oncogenic stress. It could be speculated that certain metabolites like ammonia could play a role.
Despite the identification of a novel autophagy induction pathway by a metabolic byproduct—ammonia—the exact molecular mechanism of how ammonia activates autophagy is not fully understood. Ammonia generated from amino acid metabolism in brain astrocytes has been reported to cause nitrogenous/oxidative stress and osmotic stress, which both can induce autophagy, but further research is needed to elucidate the exact mechanism behind ammonia-induced autophagy.
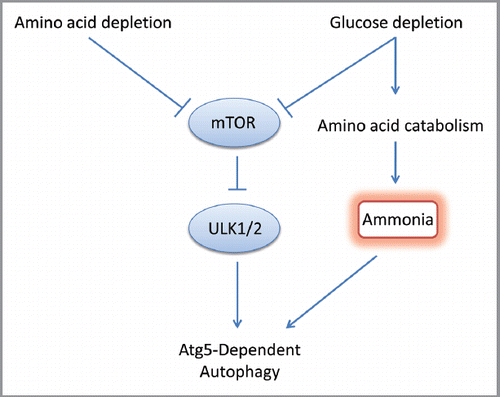
Our study has shown that in mammals at least two independent pathways of autophagy induction exist (). Surprisingly, the initiation of autophagy by ammonia, a by-product of cellular metabolism, is independent of the core genetic initiators (mTOR and Ulk1/2) established by traditional examinations of autophagy. These findings give new insights into how autophagy can provide a survival advantage to cells using alternative bioenergetic substrates such as amino acids to maintain ATP production and mitochondrial integrity.
Figures and Tables
Figure 1 Autophagy can be induced following glucose- and amino acid-starvation via an mTOR- and Ulk1/2-dependent pathway. However, during prolonged glucose starvation, amino acid catabolism generates ammonia, which can directly engage the autophagic machinery, in an mTOR- and Ulk1/2-independent pathway.
Acknowledgments
The authors thank Chao Lu and Pat Ward for critical reading of the manuscript.
Punctum to: Cheong H, Lindsten T, Wu J, Lu C, Thompson CB. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc Natl Acad Sci USA 2011; 108:11121 - 11126; PMID: 21690395; http://dx.doi.org/10.1073/pnas.1107969108