Abstract
Multiple myeloma (MM) is an incurable plasma cell malignancy in which p53 is rarely mutated. Thus, activation of the p53 pathway by a small molecule inhibitor of the p53-MDM2 interaction, nutlin, in MM cells retaining wild type p53 is an attractive therapeutic strategy. Recently we reported that nutlin plus velcade (a proteasome inhibitor) displayed a synergistic response in MM. However, the mechanism of the p53-mediated apoptosis in MM has not been fully understood. Our data show that nutlin-induced apoptosis correlated with reduction in cell viability, upregulation of p53, p21, and MDM2 protein levels with a simultaneous increase in pro-apoptotic targets PUMA, Bax and Bak, and down-regulation of anti-apoptotic targets Bcl2 and survivin, and activation of caspase in MM cells harboring wild type p53. Nutlin-induced apoptosis was inhibited when activation of caspase was blocked by the caspase inhibitor. Nutlin caused mitochondrial translocation of p53 where it binds with Bcl2, leading to cytochrome C release. Moreover, blocking the transcriptional arm of p53 by the p53-specific transcriptional inhibitor, pifithrin-α, not only inhibited nutlin-induced upregulation of p53-transcriptional targets but also augmented apoptosis in MM cells, suggesting an association of transcription-independent pathway of apoptosis. However, inhibitor of mitochondrial translocation of p53, PFT-µ, did not prevent nutlin-induced apoptosis, suggesting that the p53 transcription-dependent pathway was also operational in nutlin-induced apoptosis in MM. Our study provides the evidence that nutlin-induced apoptosis in MM cells is mediated by transcription-dependent and -independent pathways and supports further clinical evaluation of nutlin as a novel therapeutic agent in MM.
Introduction
Multiple myeloma (MM), the second most common blood cancer, is a clonal plasma cells neoplasm in the bone marrow.Citation1,Citation2 MM is incurable with current therapies and potential therapeutic targets for new drug development is clinically important.Citation3 We and others have shown that mutations/deletions of p53, a tumor suppressor protein, are relatively rare in MM.Citation4–Citation6 Since about 90% of MM cases retain wild type (wt) p53, therapeutic induction of p53 might be particularly suitable for the treatment of MM.
Human p53 controls cellular responses to stress by inducing transcription of the genes involved in cell cycle regulation, DNA repair and apoptosis.Citation7–Citation10 It is well known that p53 is a short-lived protein, and its cellular level is controlled by the rate at which it is degraded. In response to cellular stress, p53 levels are elevated by a post-translational mechanism.Citation8–Citation11 However, in the absence of stress, p53 is tightly controlled by its negative regulator, MDM2. Because the MDM2 gene is itself a transcriptional target of p53, both proteins form an autoregulatory feedback loop that keeps their level low in unstressed cells.Citation11 MDM2, a p53-specific E3 ubiquitin ligase, not only facilitates proteasomal degradation of p53, but also binds p53 and exports p53 out of the nucleus, inhibiting its transcriptional activity. Therefore, inhibitors of p53-MDM2 binding are expected to stabilize and activate p53.Citation11–Citation13
Because the activity of MDM2 inhibitors depends upon p53 activation in cells expressing wt p53, hematological malignancies that mostly retain wt p53 are potentially attractive targets for MDM2 inhibitor-based therapy.Citation13–Citation16 The first potent and selective small-molecule antagonists of MDM2, the nutlins, activate the p53 pathway in cancer cells harboring wt p53 in vitro and in vivo.Citation14 These non-genotoxic compounds bind MDM2 in the p53 binding pocket with high selectivity and can release p53 from negative control resulting in effective stabilization of p53 and activation of the p53 pathway leading to apoptosis.Citation14–Citation16
Nutlin-induced apoptosis in hematological malignancies is mostly p53-dependent.Citation17–Citation22 We and others have demonstrated that p53-mediated apoptosis in MM cells induced by nutlin is dependent on wt p53 status.Citation23–Citation25 Conventionally p53 mediated apoptosis is followed by p53-transcription-dependent manner.Citation17–Citation24 However, recent studies have shown that p53-mediated apoptosis can be mediated by combination of mechanisms that are both dependent and independent of the transcription of the p53 targets.Citation26–Citation32 In p53-transcription-dependent pathway, activated p53 induces apoptosis through transcription of the p53 target genes which occurs in the nucleus.Citation17–Citation24 On the other hand, p53 may also cause extra-nuclear effects that can induce apoptosis through transcription-independent mechanism.Citation33–Citation35 In transcription-independent pathway, mitochondrial translocation of p53 from nucleus upon activation of the p53 pathway causes direct binding of p53 to Bcl2 family proteins, resulting in the release of cytochrome C and activation of caspase leading to apoptosis.Citation28–Citation35 Depending on the cell types, nutlin-induced apoptosis has been reported to be mediated by either transcription-dependent or -independent or both pathways in hematological malignancies such as acute myeloid leukemia (AML) and chronic lymphocytic leukemia (CLL).Citation33–Citation35 However, the mechanisms of nutlin-induced apoptosis in MM have not been fully elucidated. We therefore examined the p53 signaling pathways associated with nutlininduced apoptosis and provide strong evidence that apoptosis induced by nutlin in MM cells is mediated by p53-transcription-dependent and -independent mechanisms.
Results
Nutlin inhibited cell proliferation, stabilized p53 and induced p53 transcriptional targets in MM cell lines harboring wt p53.
In the first set of experiments, we sought to investigate whether nutlin was able to affect the proliferation of MM cells. The cells were treated with 2.5–10 µM nutlin for 48 hrs and the viability was measured by MTT assay. As shown in , at 10 µM nutlin, the viability of MM1.S and H929 cell lines declined to 30 and 35%, respectively compared with DMSO treated control. No significant reduction was observed in the viability of cells harboring mutant (mt) p53, LP1 or U266 or cells in which p53 expression is blocked, 8226R5 (presence of a stop codon in its DNA binding domain) suggesting a p53-dependent growth inhibition of MM cells by nutlin.
Next we examined whether cellular stress mediated by nutlin lead to elevation of p53 levels and transactivation of p53-target genes. Time course studies showed that, p53 levels started to increase as early as 4 hrs after treatment of MM1.S or H929 cells with 5 µM nutlin and continued to increase till 24 hrs. Since p53 stabilization is tightly linked to p53-dependent MDM2 induction,Citation36,Citation37 MDM2 protein levels were closely correlated with p53 and p21 levels. p21 and MDM2 protein levels continued to increase in a time dependent manner, showing maximum level of p21 expression at 12 hrs in nutlin treated MM1.S cells ().
In our next attempt we studied the dose-dependent activation of the p53 pathway following incubation of different cells with 2.5–10 µM nutlin for 24 hrs. Cells exposed to 10 µM nutlin significantly increased p53 and p21 protein levels in MM1.S and H929 cells (). In contrast, exposure to nutlin did not modulate p53 expression in LP1 or 8226R5 cells. Expression of p53 was initially higher in LP1 cells but did not change with the increasing dose of nutlin (), whereas expression of p53 was not detected in p53 null 8226R5 cells.
Nutlin induced caspase-dependent extrinsic and intrinsic pathways of apoptosis through regulation of apoptotic targets.
Observing the activation of the p53 pathway by nutlin, we next examined its apoptotic effect on MM cells. Treatment of MM1.S cells with nutlin caused a dose- and time-dependent increase in the percentage of annexin-V-positive cells (). For example, at 24 hrs, MM1.S cells treated with 10 µM nutlin showed 30% annexin-V positive cells; however, at 48–72 hrs a wide range of annexin-V positive cells was observed from as high as 50–80%, respectively (). The quantitative apoptosis analysis was done by treating different cells with 5 µM nutlin for 48 hrs and analysed for annexin-V binding by FCM. The FCM data are depicted in . Results showed 40–50% apoptosis in MM1.S or H929 cells, whereas similar treatment did not detect significant apoptosis in U266, LP1 or 8226R5 cells ().
We then examined the role of apoptosis regulated by activation of p53 and its transcriptional targets; these include several pro-apoptotic targets PUMA, Bax and Bak. PUMA and Bax have been shown to be mediators of apoptosis in several malignancies.Citation38,Citation39 We tested the effect of nutlin on the induction of these genes in different MM cells. As shown in , nutlin caused induction of p21 and p27, and a pro-apoptotic gene, PUMA together with repression of an anti-apoptotic gene, Bcl2 in MM1.S and H929 cells but not in mt p53 expressing U266 or p53 null 8226R5 cells. Similar to these results activation of caspase-3 was also observed only in wt p53 expressing cells ().
To systematically explore the apoptosis pathway in MM cells, we further investigated the activation of the caspase cascades mediated by nutlin. Monitoring caspase cleavage by WB analysis revealed activation of caspase-8 and caspase-9 followed by activation of caspase-3 as indicated by increased levels of their active fragments in H929 cells. The activation of other pro-apoptotic markers, Bax and Bak and another anti-apoptotic marker, survivin was also confirmed in these cells (). Following p53 stabilization, activation of caspase-8 and caspase-9 suggests that both extrinsic and intrinsic apoptosis pathways are associated with nutlin-induced caspase-mediated apoptosis in MM cells.
We then examined whether nutlin-induced apoptosis in MM cells is caspase-dependent. Inhibition of caspase activation by using a pan-caspase inhibitor, ZVAD-FMK blocked the activation of caspase-3 in both MM1.S and H929 cells. Moreover, blocking of caspase activation resulted in inhibition of apoptosis as evidenced by decreased activation of PARP which was further confirmed by quantification of % specific apoptosis (). The percent of annexin-V positive cells (48–42%) was substantially decreased in H929 or MM1.S cells treated with nutlin in the presence of ZVAD-FMK (22–20%), suggesting that apoptosis induction by nutlin in MM is caspase dependent.
Nutlin inhibited cell viability, stabilized p53 and induced p53 transcriptional targets in primary MM samples.
We further validated nutlin-induced declining in the cell survival in primary MM samples. As shown in , treatment of primary MM samples with escalating dose of nutlin resulted in dose-dependent inhibition of cell proliferation in 3 of the 5 primary samples which showed 50–70% inhibition of cell proliferation at 10 µM nutlin. The other two samples had minimum cytotoxic effects indicating the presence of nonfunctional p53 in these samples. The functional activity of the p53 was confirmed by studying nutlin-induced activation of the p53 pathway. Representative primary samples from the survival experiments (sample #1 and #2) showed dose-dependent induction of p53 along with upregulation of its targets p21 and MDM2 and downregulation of Bcl2. Dose-dependent increase in the level of cleaved PARP in these samples indicated nutlin-induced apoptosis. In contrast, the level of these proteins was not affected by treatment with nutlin in primary sample #3 which showed consistency with its proliferative responses ().
PFT-α inhibited upregulation of p53 transcriptional targets and potentiated nutlin-induced apoptosis in MM cells harboring wt p53.
Having shown that p53 activated by nutlin was transcriptionally active, we further examined whether nutlin can induce apoptosis in the absence of p53-transcription. MM1.S cells were pre-incubated with PFT-α (15 µM), a pharmacological inhibitor, that has been shown to block p53-mediated transcription. As shown in , PFT-α-treatment did not affect nutlin-induced cellular stabilization of p53 but resulted in inhibition of upregulation of p21, MDM2 and PUMA suggesting that PFT-α is functional in the inhibition of p53-transcription in MM cells. In contrast, pretreatment of LP1 cells harboring mt p53 with PFT-α did not modulate the expression of p53 and its transcriptional targets (). Importantly, however, PFT-α enhanced nutlin-induced apoptosis in MM1.S cells, as indicated by an increase in annexin-V binding (). MM1.S cells treated with nutlin alone showed 25% annexin-V positive cells whereas 13% PFT-α pretreated cells after nutlin treatment were 40% annexin-V positive. PFT-α did not induce significant apoptosis in LP1 cells suggesting that transcriptional blockade induced p53-dependent apoptosis in MM cells ().
Differential expression of p53 target genes in nutlin-induced, PFT-α treated MM cells.
Consistent with the p53 cellular functions, we have previously reported that nutlin induced approximately 50 genes involved in apoptosis, cell cycle regulation, cell growth and differentiation, DNA repair and transcriptional regulation. The most notable potential p53 transcriptional target genes which showed upregulation by gene expression profiling were p21, MDM2, TNFRSF10B/TRAILS-R2, FAS, Bax and GADD45A, some of which, e.g., FAS and GADD45A are implicated in the induction of apoptosis, and CDKN1A/p21 or CDKN2A/p27 are implicated in cell cycle arrest.Citation25 In this study, we confirmed differential expression of some of the potential apoptosis related genes. In addition, microarray analysis was performed to further investigate the specific modulation in gene expression profiling in nutlin-induced PFT-α treated cells. A significant number of differentially expressed p53 target genes were identified and categorized depending on the functions of the genes (). Regarding the pro-apoptotic and anti-proliferative effects of the drugs, a considerable fraction of modulated genes were associated with apoptosis signaling pathways, cell proliferation (noteworthy downregulation of proto-oncogene, MYC) or growth arrest and DNA repair (upregulation of GADD45A). Importantly, genes that are thought to play an important role in the pathogenesis in MM, such as transcription factors MYC and MAF are negatively modulated by PFT-α. The PFT-α induced upregulation of NOXA, GADD45A, ATF3, DDIT3, PPP1R15A and downregulation of MYC, TOB1 and TXNIP in nutlin-treated MM1.S were further verified by qRT-PCR. However, the expression of these p53 target genes were minimally affected in nutlininduced PFT-α treated LP1 cells carrying mt p53, indicating that nutlin-induced apoptosis in MM cells is p53 function dependent ().
MM cells overexpress the MYC and MAF oncogene, resulting in increased cell proliferation and survival via upregulation of cell cycle and adhesion to bone marrow derived stromal cells.Citation40,Citation41 These genes play an important role in the generation of functionally competent plasma cells and have been shown to control plasma cell differentiation.Citation40 Therefore, inhibition of the function of these genes by PFT-α might potentiate nutlin-induced apoptosis in MM cells ().
MDM2 inhibition by nutlin led to both nuclear and cytoplasmic accumulation of p53 and translocation of p53 to mitochondria.
It has been shown that transcriptional activation of p53-target genes occurs in the nucleus, while cytoplasmic p53 mediates transcription-independent apoptosis if p53 levels reach a certain threshold.Citation28–Citation31,Citation33–Citation35 Therefore, we determined the intracellular localization of p53 in nutlin-treated MM cells. As shown in , after 24 hrs incubation of MM1.S cells with nutlin, individual cells showed both nuclear and cytoplasmic accumulation of p53. Cells exhibited increased p53 staining after nutlin treatment, which is consistent with the results obtained by Western blotting of whole cell lysates prepared from nutlin-stimulated cells. The accumulated cytoplasmic p53 partially co-localized with the mitochondrial marker protein, COXIV ().
We further analyzed the expression of p53 in subcellular fractions of nutlin-treated cells together with non-treated cells (). The upregulation of p53 in nutlin-treated cells was evident in mitochondrial and cytosolic fractions which suggest that treatment of MM cells with nutlin caused effective translocation of nuclear p53 into the mitochondria. Co-incident with mitochondrial p53 translocation is the mitochondrial release of cytochrome C which was evident by reduction of cytochrome C in the mitochondrial fraction and increase of cytochrome C in the cytosolic fraction of nutlin-induced cells (). These data suggest that nutlin-induced apoptosis in MM cells was mediated by transcription-independent mitochondrial permeabilization resulting from translocation of p53 to mitochondria.
p53-Bcl2 interaction was associated with transcription-independent mitochondrial apoptosis.
The Bcl2 protein is largely localized to mitochondria.Citation30 To investigate the direct interaction between p53 and Bcl2, we performed co-imunoprecipitation experiment. Treatment of MM1.S and H929 cells with nutlin resulted in the induction of p53 (). Immunoprecipitation with an anti-Bcl2 antibody resulted in co-precipitation of Bcl2 and p53 from lysates of nutlin-treated cells, but not from cells precipitated without antibody (). The selectivity of the Bcl2-p53 co-immunoprecipitation is emphasized by the observation that the abundant mitochondrial protein COXIV was present in the supernatants remaining after immunoprecipitation () but was entirely undetectable in the immunoprecipitates. These results suggest the interaction between mitochondrial p53 and Bcl2 which is considered a prerequisite for transcription-independent pathway of apoptosis.
Blocking of mitochondrial translocation of p53 did not inhibit nutlin-induced apoptosis in MM cells.
Having shown that mitochondrial translocation of p53 contributed to nutlininduced apoptosis we sought to examine whether blocking of mitochondrial translocation can inhibit apoptosis induction. Since mitochondrial translocation is an early event in the mitochondrial death program, blocking of p53 translocation was performed at 6 and 12 hrs after incubation MM1.S cells with nutlin. As shown in , mitochondrial translocation of p53 was inhibited at 6 hrs after nutlin treatment which was pretreated with PFT-µ for 2 hrs. Notably, the apoptosis induction by nutlin was not further inhibited by PFT-µ (). The inhibition of apoptosis induction was performed at various time points, however, since only a few apoptotic cells were observed at early time points of nutlin treatment, such as 6 and 12 hrs, the data for apoptosis induction at 24 and 48 hrs are shown.
Discussion
Nutlin-induced p53 appears to be fully capable as a transcription factor and an apoptotic inducer and provides a unique molecular tool for studying p53 signaling pathway in human cancer. Depending on the cellular context, transcriptional targets of p53 can either induce or inhibit apoptosis. Despite the prominence of its transcriptional activities, the contribution of nontranscriptional activities to the pro-apoptotic effects of p53 is receiving increasing attention. In this study we attempt to define the molecular mechanisms of both transcriptional and non-transcriptional pro-apoptotic p53 activities in MM.
We examined cell survival and apoptosis in response to activation of the p53 pathway in different MM cell lines and primary MM samples induced by nutlin. We found that nutlin induced p53 activation and apoptosis in MM cells harboring only wt p53 but not mt p53 confirming the data of previous observations in MM.Citation23–Citation25 Going beyond the previous observations, we report here upregulation of further downstream targets of p53 including pro-apoptotic targets; PUMA, Bax and Bak and downregulation of anti-apoptotic targets; Bcl2 and survivin, consistent with the notion that p53 promotes apoptosis by inducing pro-apoptotic targets and repressing transcription of the anti-apoptotic genes.Citation42,Citation43 In addition, we observed that nutlin-induced apoptosis in MM cells was caspase-dependent and followed by both extrinsic and intrinsic pathways.
In this study we demonstrated the requirement of both transcription-dependent and transcription-independent p53 activities for sufficient apoptosis induction by nutlin in MM cells. We found that PFT-α, a p53-transcriptional inhibitor, not only inhibited nutlin-induced upregulation of p53-transcriptional targets such as p21, MDM2 and PUMA but also enhanced the apoptotic activity of nutlin. The increase of the apoptotic rate after PFT-α treatment might be the result of anti-apoptotic action of the p53 transcriptional targets or the result of inhibition of engagement of p53 in transcriptional activity and therefore higher availability for translocation to mitochondria. Our findings led us to examine whether transcription-independent pathway is linked to mitochondrial translocation of p53.Citation44,Citation45 Of note, PFT-α niether modulated the expression of caspase-3 and PARP nor further increased annexin-V binding in MM1.S cells induced by a genotoxic drug, etoposide which activated p53 (data not shown). These results suggest that mechanism of nutlin-induced apoptosis in MM cells differs from the mechanism of apoptosis induced by genotoxic drugs.
In our study, mitochondrial translocation of p53 was supported by two independent experiments: immunofluorescence-based confocal microscopy and WB analysis of subcellular fractions. Microscopic study revealed p53 signals co-localized with the mitochondrial marker, COXIV. Co-localization of p53 with COXIV was also observed in earlier studies with AMLCitation17 and CLL,Citation33 where significant fractions of the cells co-recruited the mitochondrial death pathway to various degrees in response to nutlin. However, in our study, p53's mitochondrial translocation was further demonstrated by detection of elevated p53 levels in mitochondrial fractions of the nutlin-induced cells by WB analysis.
Previous reports have described that mitochondrial p53 localization is specific for p53-dependent apoptosis.Citation45 Our results showing upregulation of pro-apoptotic proteins (transcriptional targets of p53) and, perhaps more importantly, downregulation of anti-apoptotic proteins of the intrinsic apoptotic pathway (Bcl2), suggest that additional molecular events beyond the translocation might be involved. To further understand the mitochondrial pathway of apoptosis, we examined whether direct binding of mitochondrial p53 with Bcl2 can trigger apoptosis. The p53-Bcl2 interaction was observed in nutlin-induced cells harboring wt p53 by co-immunoprecipitation study provides the evidence that p53 can directly bind to mitochondrial Bcl2. In addition, mitochondrial p53 translocation and direct binding to Bcl2 induced mitochondrial outer membrane permeabilization as demonstrated by the release of cytochrome C. It is possible that binding of p53 to Bcl2 neutralizes its inhibitory effect on Bax/Bak restraining the anti-apoptotic effect of Bcl2 thereby leads to mitochondrial apoptosis. Further experiments are required to clarify the role of the p53-Bcl2 interaction in p53-transcription-independent apoptosis.
In contrast to the results in AMLCitation34 and CLLCitation35 where p53 transcriptional independent pathway is the major route for nutlin induced apoptosis, our data in MM showed that transcriptiondependent apoptosis pathway may not be dispensable, i.e., both p53-transcription-dependent and -independent pathways are operational. This may be explained by the differences of the cellular context since p53 mediated apoptosis is cell-type dependent. Based on our data, once p53 is stabilized and accumulated in the nucleus, it either becomes post-translationally modified to recognize specific promoters or it co-operates with transcriptional regulators that deliver p53 to the appropriate site e.g., mitochondria or cytoplasm. It is likely that both of these events are associated with nutlin-induced p53-mediated apoptosis in MM and transcription-dependent and -independent p53 activities converge at the mitochondria. Nuclear p53 via its transcriptional activity induces expression of the PUMA, Bax and Bak, whereas cytosolic or mitochondrial p53 via transcription-independent mechanisms directly activates Bax/Bak and neutralizes the anti-apoptotic effect of Bcl2.
Although gene expression profile by microarray analysis and qRT-PCR identified some of the key p53 gene targets whose blockade by PFT-α results in augmentation of apoptosis in MM cells, further studies are required to provide insights into the molecular mechanism by which PFT-α controls p53-mediated apoptosis. Two of the putative candidate target genes, MYC and MAF, which are known to block apoptosis, may be of importance for further studies since treatment of MM cells with nutlin and PFT-α significantly downregulated the expression of these genes in MM cells. Interestingly, downregulation of these antiapoptotic genes were associated with significant upregulation of a transcription factor, ATF3, which is related to stress response and responsible for unfolded protein response (UPR).Citation46 In addition, it has been reported that ATF3 attenuates cell cycle under stress.Citation47 Therefore, studies on the regulation of these genes by PFT-α in nutlin-induced cells may also provide a clue for identifying the apoptosis signaling pathways in MM, e.g., ERK, JUN N-terminal kinase (JNK) and p38 signaling pathways because each plays a role in ATF upregulation and the JNK/JUN pathway is associated with survival and the stress response.Citation48,Citation49 Elucidating these mechanisms would allow tailoring of specific strategies for therapeutic purposes that could be useful in MM with cytoplasmically sequestered wt p53.
To our knowledge this is the first report to clearly demonstrate that p53-mediated apoptosis in MM cells is followed by both transcription-dependent and -independent mechanisms. Our data support further clinical evaluation of nutlin as a potential novel therapeutic intervention in MM.
Materials and Methods
Myeloma cell lines and primary MM samples.
MM1.S and H929 cell lines harboring wt p53; LP1 and U266 cell lines expressing mt p53; and 8226R5 cell lines not expressing p53 were grown in standard culture medium (IMDM containing 10% fetal bovine serum, 2 mmol/liter L-glutamine, 50 U penicillin and 50 µg/ml streptomycin) at 37°C in a 5% CO2 incubator.
Bone marrow samples from patients with MM were cultured and maintained as described previously.Citation25
Drug treatment.
Nutlin (nutlin-3) was purchased from Cayman Chemical (Ann Arbor, MI, USA) and dissolved in dimethyl sulfoxide (DMSO) to create a 10 mM stock solution and stored at −20°C. Pifithrin-α (PFT-α) was purchased from Biomol (Enzo Life Sciences Inc., PA, USA). Pifithrin-µ (PFT-µ) and etoposide were purchased from Sigma (Sigma Aldrich Inc., MO, USA). In experiments designed to study selective blockade of p53 transcription and p53 translocation, cells were pretreated with PFT-α and PFT-µ, respectively before addition of nutlin. A Caspase family inhibitor (ZVAD-FMK) was purchased from Biovision (Biovision Inc., CA, USA). Cell lines were harvested in log-phase growth and exposed to 2.5–10 µM of nutlin for the time period indicated, with the final DMSO concentration kept constant in each experiment and not exceeding 0.1% (vol/vol).
Antibodies.
Antibodies against the following proteins were used in this study: mouse monoclonal antibodies to p53 (DO-7), p21, histone H1 (H2A), cytochrome C oxidase IV (COXIV, 20E8), β-actin and rabbit polyclonal antibody to p53 (FL-393) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA); mouse monoclonal antibodies to p27, caspase-8 and Bcl2 from BD Biosciences (San Diego, CA, USA); MDM2 and cytochrome C from Calbiochem (San Diego, CA, USA); Rabbit polyclonal antibodies to PUMA, Bax and Bak and mouse monoclonal antibody to caspase-3 and poly (ADP-ribose) polymerase (PARP, Asp214) from Cell Signaling Technology (Cell Signaling, Danvers, MA, USA); caspase-9 from R&D Systems (Minneapolis, MN, USA); survivin from Abcam (Cambridge, MA, USA); anti-tubulin from Sigma. Peroxidase-conjugated goat anti-mouse and anti-rabbit IgG were purchased from Cell Signaling and Santa Cruz Biotechnology, respectively. Alexa Fluor 488 goat anti-rabbit and Alexa Fluor 568 goat anti-mouse secondary antibodies were purchased from Molecular Probe (Eugene, OR, USA).
Cell viability and proliferation assay.
Cell viability was assessed by MTT [3-(4, 5-dimethilthiazol-2yl)-2,5-diphenyl tetrazolium bromide] colorimetric assay. For this, cells were cultured in 96-well micro-titer plates with different concentrations of drugs for 48 hrs period. Then MTT (0.5 mg/ml) was added and the cells were incubated for an additional 4 hrs. This was followed by an addition of acidified isopropanol to the well and overnight incubation at 37°C. Following incubation, the optical density of the cells was read with a microplate reader set at a test wavelength of 570 nm and a reference wavelength of 630 nm. Each experiment was made in triplicate, and the mean value was calculated.
Apoptosis assay.
For quantitation of apoptotic cells by annexin-V staining, cells with or without drug treatment were washed with PBS, resuspended in annexin-V binding buffer and stained with FITC-annexin-V and PI, according to the manufacturer's instructions (Abcam). Stained cells were analyzed using a FACScan (Becton Dickinson, NJ, USA) flow cytometer and apoptosis quantified as the percent annexin-V positive cells. The drug-specific apoptosis was assessed by the following formula: % specific apoptosis = (test − control) × 100/(100 − control).
Protein extraction, cell fractionation and WB analysis.
Whole cell lysates were prepared by extraction of cell pellets which were lysed for 10 minutes on ice in a buffer composed of 150 mM NaCl, 50 mM Tris-HCl (pH 8.0), 5 mM EDTA, 1% (v/v) Nonidet P-40, 1 mM phenylmethylsulfonyl fluoride (PMSF), 20 µg/ml aprotinin and 25 µg/ml leupeptin. Subcellular fractionation was carried out by using the fractionation kit (Calbiochem) according to the manufacturer's protocol. Protein concentrations were measured by using a Nano Drop 1000 spectrophotometer (Thermo Fisher Scientific Inc., San Diego, CA, USA). Equal amounts of protein extracts were resolved using 12% SDS-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride (PVDF) membrane (Perkin Elmer Inc., Waltham, MA, USA). After blocking for 1 hr at room temperature with PBS containing 5% skim milk or 3% bovine serum albumin (BSA) depending on the antibodies used for probing the blots, the filter was incubated with specific antibodies for at least 2 hrs but not more than 24 hrs. The filter was washed, incubated with a horseradish peroxidase (HRP)-labeled secondary antibody for 1 hr and the blots developed using a chemiluminescent detection system (ECL, Perkin Elmer, Waltham, MA, USA).
Gene expression analysis.
Total RNA was isolated using TRIzol reagent (Invitrogen) and the gene expression profile was evaluated using Illumina RNA analysis Beadchips (Illumina Inc., San Diego, CA, USA) representing ∼48,000 human genes (Human HT12). Raw data was processed using lumi R package.Citation50 Background correction was done in Beadstudio software (Illumina). The quantile normalization methodCitation51 implemented in lumi R package was used to normalize the data. We used LIMMA (linear models for microarray data)Citation52 to identify differentially expressed genes. Expression of key genes in nutlininduced MM1.S cells involved in cell proliferation, apoptosis, cell cycle arrest, transcription and translation, cell signaling, invasion and metastasis was analyzed.
Quantitative real time PCR (qRT-PCR).
To quantify and validate the expression of p53 target genes of interest at their mRNA level, qRT-PCR assays using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a reference gene were performed. Total RNA was isolated and cDNA was synthesized as described previously.Citation25 Samples for the qRT-PCR were prepared using Platinum SYBR Green qPCR SuperMix-UDG with Rox (Invitrogen) and run on the StepOnePlus™ Real-Time PCR System (Applied Biosystems, CA, USA) using a thermal profile of an initial 2 minutes UDG incubation step at 50°C and 2-minutes melting step at 95°C, followed by 40 cycles at 95°C for 20 seconds and 55°C for 40 seconds. The primers used for analysis by qRT-PCR are listed in (Suppl. Table 2). To verify the presence of only one amplicon, a melting curve was processed after each run. After normalization with GAPDH expression, regulation was calculated between treated and untreated cells. All reactions were carried out at least twice in triplicate.
Confocal microscopy.
MM cells were cytospun onto glass slides after treatment with nutlin or DMSO. The slides were washed twice with PBS and fixed with 2% paraformaldehyde. The cells were permeabilized with 0.2% Triton X-100 for 5 minutes, blocked with 2% bovine serum albumin (BSA) for 30 minutes, and incubated overnight at 4°C with rabbit polyclonal anti-p53 (1:200, vol/vol) and mouse monoclonal anti-COXIV (1 µg/ml) antibodies. After washing, the cells were incubated with Alexa Fluor 488 and Alexa Fluor 568 secondary antibody diluted in 2% BSA for 30 minutes at 4°C. Images were obtained using a 60X/1.40 planApo objective lens on an Olympus FV-1000 confocal microscope with Fluoview version 1.7 software (Olympus, NY, USA). Cells not exposed to primary antibodies served as negative controls.
Co-immunoprecipitation of Bcl2 and p53.
Cells were lysed in a buffer containing 0.1% [(3-cholamidopropyl)dimethylamino]-1-propanesulfonate (CHAPS; Cell Signaling Technologies). Lysates were incubated with protein A/G agarose (Santa Cruz Biotechnology) and monoclonal antibody to anti-Bcl2 overnight at 4°C. After centrifugation, the supernatants were removed and stored for future analysis. Beads were washed 3 times with PBS containing 0.2% Triton-X100. 40 µl of gel loading buffer were added to each sample and boiled for 3 minutes prior to SDSPAGE electrophoresis and Western transfer. In control experiments samples were immunoprecipitated without addition of the antibodies.
Figures and Tables
Figure 1 Nutlin induced selective growth inhibition and activated the p53 pathway in MM cells harboring wt p53. (A) Nutlin suppressed the growth of MM cells harboring wt p53. MM cells harboring either wt, mt or null p53 were treated with different dose of nutlin. After 48 hrs, effects of nutlin on the viability of MM cells were assessed using MTT cell viability assay. The survivals of nutlin-treated cells are expressed as percent of the DMSO-treated control. Results are expressed as mean ± SD. (B) Time-dependent activation of the p53 signaling in response to nutlin treatment. Whole cell lysates were prepared at the indicated hours after treatment of MM1.S and H929 cells with 5 µM nutlin and analyzed by WB for the indicated proteins. (C) Dose response of nutlin-induced activation of the p53 pathway in MM cells. At 24 hrs after treatment with 2.5–10 µM nutlin, whole cell lysates from MM cells harboring either wt (MM1.S or H929) or mt (LP1) or null p53 (8226R5) were analysed for the expression of p53 and its two immediate downstream targets, p21 and MDM2 by WB analysis. β-actin was used as loading controls.
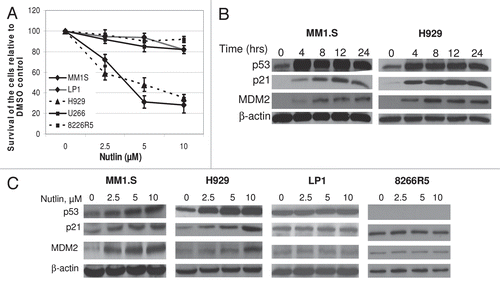
Figure 2 Inhibition of the p53-MDM2 interaction by nutlin induced p53-dependent apoptosis. Annexin-V binding and PI uptake in cells harboring either wt or mt p53 were examined as a marker for induction of apoptosis after treatment of the cells with nutlin. (A) MM1.S cells were incubated with 2.5–10 µM nutlin for different time periods (24–72 hrs) to determine the time and dose responses for nutlin-induced apoptosis in this cell. (B) FCM profiles of MM1.S, H929 and LP1 cells treated for 48 hrs with 5 µM nutlin or DMSO control. Results of a single representative experiment from a set of at least three experiments are shown. (C) Percent specific apoptosis were calculated as described in material and methods and shown in the figure. Results showed significant amount of apoptosis in cells harboring wt p53 but not in cells expressing mt or null p53. Results are expressed as mean ± SD.
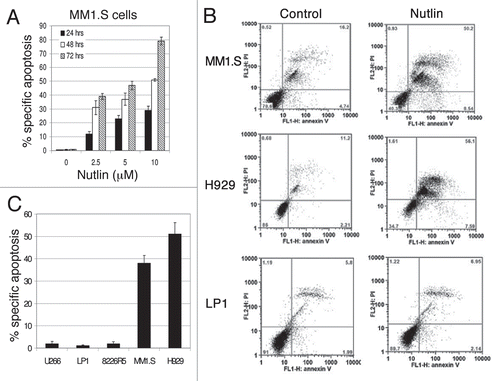
Figure 3 Nutlin-induced apoptosis was mediated through regulation of apoptotic targets. (A) MM cells were treated with 5 µM nutlin for 24 hrs. Whole cell lysates from the indicated cells were analysed for expression of the indicated proteins by WB analysis. Nutlin-induced upregulation of the p53 along with a pro-apoptotic target, PUMA and downregulation of an anti-apoptotic protein, Bcl2 was observed in wt p53 harboring MM1.S and H929 cells. By contrast, no significant changes in the levels of these proteins were detected in mt p53 expressing U266 or p53 null 8226R5 cells. Apoptosis in these cells was further confirmed by cleavage of caspase-3. (B) Nutlin-induced apoptosis is mediated by both extrinsic and intrinsic pathways in MM cells. H929 cells were treated with different doses of nutlin for 24 hrs and activation of caspase together with pro- and anti-apoptotic targets were examined by WB analysis. Nutlin induced activation of both caspase-8 and caspase-9 followed by induction of caspase-3. The activation of caspase in H929 cells was associated with induction of pro-apoptotic proteins, PUMA, Bax and Bak and repression of anti-apoptotic proteins, Bcl2 and survivin. (C) Nutlin-induced apoptosis in MM cells was caspase-dependent. MM1.S and H929 cells were treated with 10 µM pan-caspase inhibitor for 1 hr before treating the cells with 5 µM nutlin for 24 hrs. Activation of caspase and PA RP was examined by WB analysis. Inhibition of capsase-activation by pan-caspase inhibitor, ZVAD-FMK resulted in blocking of caspase activation and thus inhibition of apoptosis.
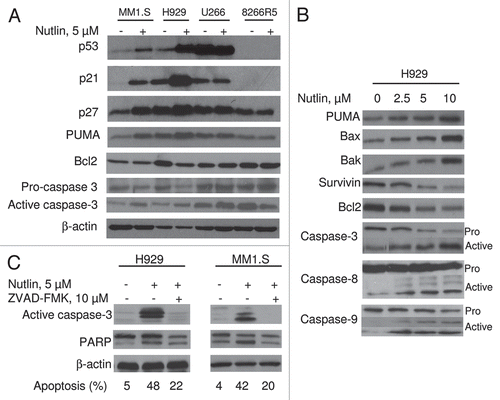
Figure 4 Nutlin inhibited the growth of primary MM samples and stabilized p53. (A) Primary MM samples were treated with different dose of nutlin for 48 hrs and survival of the cells was measured by MTT cell survival assay. Nutlin induced dose-dependent inhibition of the proliferation of cells in 3 of the 5 primary samples. The survivals of nutlin-treated cells are expressed as percent of the DMSO-treated control. Results are expressed as mean ± SD. (B) Primary MM samples were treated with different dose of nutlin for 24 hrs and activation of the p53 pathway was examined by induction of p53 and its downstream targets. Nutlin induced p53 activation in two representative samples in (A), however, the induction of p53 was not observed in the sample #3, in which inhibition of proliferation was not observed by MTT assay.
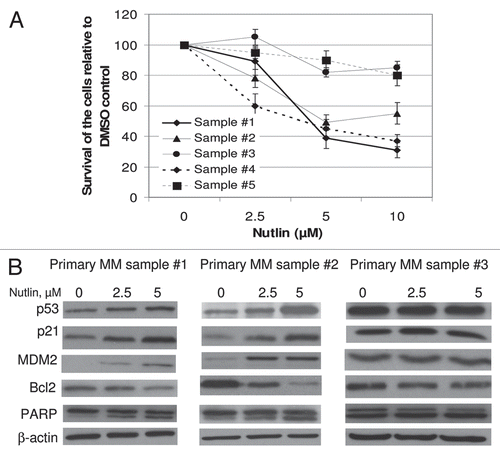
Figure 5 Transcriptional blockade by PFT-α induced apoptosis of MM1.S cells. Cells were pre-treated with 15 µM PFT-α for 4 hrs followed by 5 µM nutlin for additional 6 hrs. (A) Immunoblot indicating that pretreatment of MM1.S cells with PFT-α repressed p53 transcriptional targets p21, MDM2, PUMA and Bax upon nutlin treatment, while not altering p53 levels. (B) Immunoblot showing expression of p53 and its transcriptional targets in nutlin-induced PFT-α treated LP1 cells. (C) Quantitation of apoptosis assay by FCM for nutlin-induced MM1.S and LP1 cells pretreated with PFT-α. Pretreatment of MM1.S cells with PFT-α augmented the apoptotic response to nutlin. PFT-α did not interfere with nutlin-induced apoptosis in LP1 cells. Data are mean ± SD of duplicate measurements. (D) Gene expression analysis by qRT-PC R in nutlin-induced MM1.S and LP1 cells in the presence or absence of p53 transcriptional inhibitor, PFT-α.
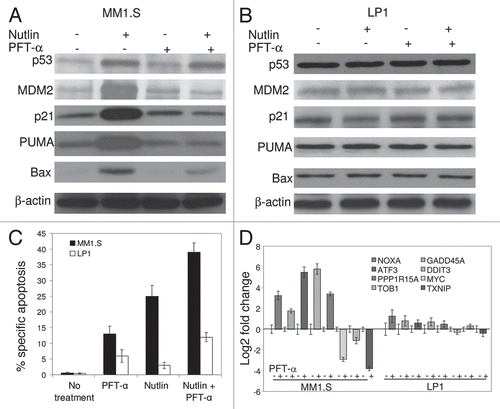
Figure 6 Nutlin-induced apoptosis was associated with translocation of p53 to mitochondria. (A) Nutlin-treated MM1.S cells were stained for p53 (green) and mitochondrial marker protein COXIV (red) and visualized by confocal microscopy. Localization of p53 to mitochondria is indicated by the yellow-orange color in the merged images. A preferential translocation of cytoplasmic p53 to mitochondria suggests that cytoplasmic p53 mediates apoptosis mainly at the level of mitochondria. (B) Nutlin-induced MM cells were fractionated to yield nuclear, mitochondrial and cytosolic fractions. Accumulation of p53 in mitochondrial and cytosolic fractions in addition to nuclear fraction was assessed by western blotting. Nutlin stabilized wt p53 in both nucleus and cytoplasm and promoted translocation of p53 to mitochondria followed by cytochrome (cyt. C) release. The purity of the subcellular fractions was verified by using H2A, COXIV and β-tubulin for detecting nuclear-, mitochondrial- and cytosol-specific proteins, respectively.
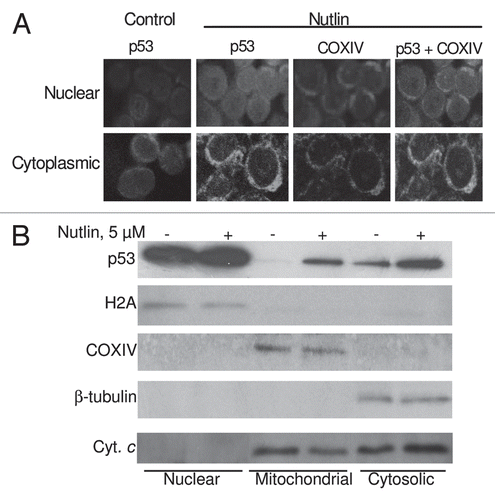
Figure 7 Role of the p53-Bcl2 interaction in nutlin-induced apoptosis. (A) Nutlin-induced p53 binding to mitochondrial Bcl2 protein. MM1.S and H929 cells were treated with 5 µM nutlin or DMSO control for 6 hrs. After which CHAPS lysates together with MM1.S whole cell lysates (w.c.l.) were prepared and analyzed directly or (B) following immunoprecipitation with anti-Bcl2 antibody. Nutlin-induced upregulation of p53 was shown in MM1.S whole cell lysates as well as CHAPS lysates of both MM1.S and H929 cells. Similar to this observation, p53 protein expression precipitated by Bcl2 in the cytosolic extracts of MM1.S and H929 cells was also increased after treatment with nutlin. (C) Immunoprecipitation supernatants were analyzed for COXIV to document equal inputs.
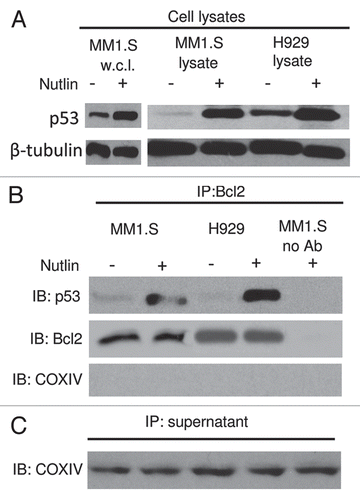
Figure 8 Blocking of mitochondrial translocation of p53 did not inhibit apoptosis induction by nutlin. (A) MM1.S cells pretreated with PFT-µ, a specific inhibitor of mitochondrial translocation of p53, were treated with 5 µM nutlin for 6 hrs. Accumulation of p53 was studied by mitochondrial fractionation of the cells followed by WB analysis. Treatment of cells with PFT-µ resulted in inhibition of mitochondrial translocation of p53. β-tubulin and COXIV served as loading controls as well as cytosolic and mitochondrial markers, respectively. However, PFT-µ did not prevent either mitochondrial cytochrome (cyt. c) release or cleavage of PA RP in nutlin-induced cells. (B) PFT-µ did not inhibit apoptosis induction by nutlin in MM cells. MM1.S cells pretreated with PFT-µ were treated with 5 µM nutlin and apoptosis was measured by annexin-V staining at two different time periods. Consistent with the results of cytochorme C release and PARP cleavage shown in (A), pretreatment of MM1.S cells with PFT-µ did not inhibit the apoptotic response to nutlin.
Table 1 Differentially expressed genes in nutlin-induced PFT-α treated MM1.S cells
Additional material
Download Zip (39.2 KB)Acknowledgements
This work was supported in part by research grants from Canadian Institute of Health Research (CIHR) and Leukemia and Lymphoma Society of Canada (LLSC). We thank D. Branch and A. Schimmer for their helpful suggestions and critical review of the manuscript, and P. Hu for his assistance with microarray data analysis.
References
- Fonseca R, Stewart KA. Targeted therapeutics for multiple myeloma. The arrival of a risk-stratified approach. Mol Cancer Ther 2007; 6:802 - 810
- Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics 2002. CA Cancer J Clin 2005; 55:74 - 108
- Kyle RA, Rajkumar SV. Multiple myeloma. N Engl J Med 2004; 351:1860 - 1873
- Chang H, Qi C, Yi QL, Reece D, Stewart AK. p53 gene deletion detected by fluorescence in situ hybridization is an adverse prognostic factor for patients with multiple myeloma following autologous stem cell transplantation. Blood 2005; 105:358 - 360
- Chng WJ, Price-Troska T, Gonzalea-Paz N, Van Wier S, Jacobus S, Blood E, et al. Clinical significance of TP53 mutation in myeloma. Leukemia 2007; 13:582 - 584
- Chang H, Yeung J, Qi C, Xu W. Aberrant nuclear p53 protein expression detected by immunohistochemistry is associated with hemizygous p53 deletion and poor survival for multiple myeloma. Br J Haematol 2007; 138:324 - 329
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; 408:307 - 310
- Bargonetti J, Manfredi JJ. Multiple roles of the tumor suppressor p53. Curr Opin Oncol 2002; 14:86 - 91
- Rich T, Allen RL, Wyllie AH. Defying death after DNA damage. Nature 2000; 407:777 - 783
- Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature 2000; 408:433 - 439
- Shangary S, Wang S. Targeting the MDM2-p53 interaction for cancer therapy. Clin Cancer Res 2008; 14:5318 - 5324
- Tovar C, Rosinski J, Filipovic Z, Higgins B, Kolinsky K, Hilton H, et al. Small-molecule MDM2 antagonist reveals aberrant p53 signaling in cancer: implications for therapy. Proc Natl Acad Sci USA 2006; 103:18888 - 18893
- Shangary S, Wang S. Small molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu Rev Pharmacol Toxicol 2009; 49:223 - 241
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303:844 - 848
- Shangary S, Qin D, McEachem D, Liu M, Miller RS, Qiu S, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci USA 2008; 105:3933 - 3938
- Saha MN, Micallef J, Qiu L, Chang H. Pharmacological activation of the p53 pathway in hematological malignancies. J Clin Pathol 2010; 63:204 - 209
- kojima K, konopleva M, Samudio IJ, Shikami M, Cabreira-Hansen M, McQueen T, et al. MDM2 antagonists induce p53-dependent apoptosis in AML: implications for leukemia therapy. Blood 2005; 106:3150 - 3159
- Secchiero P, Barbarotto E, Tiribelli M, Zerbinati C, di Iasio MG, Gonelli A, et al. Functional integrity of the p53-mediated apoptotic pathway induced by the non-genotoxic agent nutlin-3 in B-cell chronic lymphocytic leukemia (B-CLL). Blood 2006; 107:4122 - 4129
- Coll-Mulet L, Iglesias-Serret D, Santidrian AF, Cosialls AM, de Frias M, Castaño E, et al. MDM2 antagonists activate p53 and synergize with genotoxic drugs in B-cell chronic lymphocytic leukemia cells. Blood 2006; 107:4109 - 4114
- Gu L, Zhu N, Findley HW, Zhou M. MDM2 antagonist nutlin-3 is a potent inducer of apoptosis in pediatric acute lymphoblastic leukemia cells with wild type p53 and overexpression of MDM2. Leukemia 2008; 22:730 - 739
- Drakos E, Thomaids A, Medeiros LJ, Li J, Leventaki V, Konopleva M, et al. Inhibition of p53-murine double minute-2 interaction by nutlin-3A stabilizes p53 and induces cell cycle arrest and apoptosis in Hodgkin lymphoma. Clin Cancer Res 2007; 13:3380 - 3387
- Drakos E, Atsaves V, Schlette E, Li J, Papanastasi I, Rassidakis GZ, et al. The therapeutic potential of p53 reactivation by nutlin-3a in ALK+ anaplastic large cell lymphoma with wild-type or mutated p53. Leukemia 2009; 23:2290 - 2299
- Stuhmer T, Chatterjee M, Hildebrandt M, Herrmann P, Gollasch H, Gerecke C, et al. Nongenotoxic activation of the p53 pathway as a therapeutic strategy for multiple myeloma. Blood 2005; 106:3609 - 3617
- Ooi MG, Hayden PJ, Kotoula V, McMillin DW, Charalambous E, Daskalaki E, et al. Interactions of the Hdm2/p53 and proteasome pathways may enhance the antitumor activity of bortezomib. Clin Cancer Res 2009; 15:7153 - 7160
- Saha MN, Jiang H, Jayakar J, Reece D, Branch DR, Chang H. MDM2 antagonist nutlin plus proteasome inhibitor velcade combination displays a synergistic anti-myeloma activity. Cancer Biol Ther 2010; 9:937 - 945
- Schuler M, Green DR. Transcription, apoptosis and p53: catch-22. Trends Genet 2005; 21:181 - 187
- Sun Y. p53 and its downstream proteins as molecular targets of cancer. Mol Carcinog 2006; 45:409 - 415
- Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human Bax gene. Cell 1995; 80:293 - 299
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell 2003; 11:577 - 590
- Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 2007; 26:1324 - 1337
- Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004; 303:1010 - 1014
- Leu JI, Dumont P, Hafey M, Murphy ME, George DL. Mitochondrial p53 activates Bax and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol 2004; 6:443 - 450
- Kojima K, Konopleva M, McQueen T, O'Brien S, Plunkett W, Andreeff M. Mdm2 inhibitor Nutlin-3A induces p53-mediated apoptosis by transcription-dependent and transcription-independent mechanisms and may overcome Atm-mediated resistance to fludarabine in chronic lymphocytic leukemia. Blood 2006; 108:993 - 1000
- Vaseva A, Natalia DM, Ute MM. The transcription-independent mitochondrial p53 program is a major contributor to nutlin-induced apoptosis in tumor cells. Cell cycle 2009; 8:1711 - 1719
- Steele AJ, Prentice AG, Hoffbrand AV, Yogashangary BC, Hart SM, Nacheva EP, et al. p53-mediated apoptosis of CLL cells: evidence for a transcription independent mechanism. Blood 2008; 112:3827 - 3834
- Freedman DA, Wu L, Levine AJ. Functions of the MDM2 oncoprotein. Cell Mol Life Sci 1999; 55:96 - 107
- Iwakuma T, Lozano G. MDM2, an introduction. Mol Cancer Res 2003; 1:993 - 1000
- Mackus WJ, Kater AP, Grummels A, Evers LM, Hooijbrink B, Kramer MH, et al. Chronic lymphocytic leukemia cells display p53-dependent drug-induced PUMA upregulation. Leukemia 2005; 19:427 - 434
- Thompson T, Tovar C, Yang H, Carvajal D, Vu BT, Xu Q, et al. Phosphorylation of p53 on key serines is dispensable for transcriptional activation and apoptosis. J Biol Chem 2004; 279:53015 - 53022
- Yaari-Stark S, Shaked M, Nevo-Caspi Y, Jacob-Hircsh J, Shamir R, Rechavi G, et al. Ras inhibits endoplasmic reticulum stress in human cancer cells with amplified Myc. Int J Cancer 2010; 126:2268 - 2281
- Hurt EM, Wiestner A, Rosenwald A, Shaffer AL, Campo E, Grogan T, et al. Overexpression of c-maf is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma. Cancer Cell 2004; 5:191 - 199
- Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J Biol Chem 2002; 277:3247 - 3257
- Mirza A, McGuirk M, Hockenberry TN, Wu Q, Ashar H, Black S, et al. Human survivin is negatively regulated by wild type p53 and participates in p53-dependent apoptotic pathway. Oncogene 2002; 21:2613 - 2622
- Arima Y, Nitta M, Kuninaka S, Zhang D, Fujiwara T, Taya Y, et al. Transcriptional blockade induces p53-dependent apoptosis associated with translocation of p53 to mitochondria. J Biol Chem 2005; 280:19166 - 19176
- Marchenko ND, Zaika A, Moll UM. Death signal-induced localization of p53 protein to mitochondria: a potential role in apoptotic signaling. J Biol Chem 2000; 275:16202 - 16212
- Cullinan SB, Diehl JA. Coordination of ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway. Int J Biochem Cell Biol 2006; 38:317 - 332
- Wek RC, Jiang HY, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans 2006; 34:7 - 11
- Bogoyevitch MA, Gillespie-Brown J, Ketterman AJ, Fuller SJ, Ben-Levy R, Ashworth A, et al. Stimulation of the stress-activated mitogenactivated protein kinase subfamilies in perfused heart. p38/RK mitogen-activated protein kinases and c-Jun N-terminal kinases are activated by ischemia/reperfusion. Circ Res 1996; 79:162 - 173
- Dent P, Yacoub A, Contessa J, Caron R, Amorino G, Valerie K, et al. Stress and radiation induced activation of multiple intracellular signaling pathways. Radiation Res 2003; 159:283 - 300
- Du P, Kibbe WA, Lin SM. Lumi: a pipeline for processing Illumina microarray. Bioinformatics 2008; 24:1547 - 1548
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A Comparison of Normalization Methods for High Density Oligonucleotide Array Data Based on Bias and Variance. Bioinformatics 2003; 19:185 - 193
- Smyth GK. Linear models and empirical Bayes methods for assessing differentially expressed in microarray experiments. Stat Appl Genet Mol Biol 2004; 3:3