Abstract
Over 20,000 genes were recently sequenced in a series of 24 pancreatic cancers. We applied CHASM (Cancer-specific High-throughput Annotation of Somatic Mutations) to 963 of the missense somatic missense mutations discovered in these 24 cancers. CHASM identified putative driver mutations (false discovery rate ≤0.3) in three known pancreatic cancer driver genes (P53, SMAD4, CDKN2A). An additional 15 genes with putative driver mutations include genes coding for kinases (PIK3CG, DGKA, STK33, TTK, and PRKCG), for cell cycle related proteins (NEK8), and for proteins involved in cell adhesion (CMAS, PCDHB2). These and other mutations identified by CHASM point to potential "driver genes" in pancreatic cancer that should be prioritized for additional follow-up.
Introduction
Pancreatic cancer is the fourth leading cause of cancer death in the United States. It is estimated that in 2009 close to 42,470 Americans were diagnosed with pancreatic cancer, and that 35,240 died from the disease.Citation1 While the existing therapies for pancreatic cancer are mostly ineffective, the success of gene-specific targeted therapies for lung and colon cancers suggests that a better understanding of the genetic alterations in pancreatic cancer may improve the dismal survival associated with this disease. Indeed, the sequencing of >20,000 coding genes in a series of 24 pancreatic cancers has led to the discovery of a familial pancreatic cancer gene, PalB2, to a better understanding of the DNA alterations associated with cigarette smoking, and to the identification of SMAD4 gene alterations as prognostic markers following surgical resection.Citation2
Genetic alterations that produce bi-allelic inactivation of gene function in a cancer are easily prioritized as likely significant in the development of that neoplasm. Most of the mutations in a cancer, however, are present in only one of the two alleles of the affected gene and/or are not obviously inactivating, and these mutations are much more difficult to classify. The challenge is to determine which of these alterations are “driver” mutations, contributing significantly to tumorigenesis, and which are “passenger” mutations, neutral to cancer fitness.Citation3
We recently developed a high-throughput computational method, called CHASM (Cancer-specific High-throughput Annotation of Somatic Mutations), to prioritize somatic missense mutations found in large-scale tumor sequencing studies. CHASM has been shown to have high sensitivity and specificity for identifying driver mutations, and outperformed other predictors of mutation function.Citation4 When applied to a set of 607 somatic missense mutations found in a sequencing study of glioblastoma multiforme tumors, CHASM was able to correctly identify the isocitrate dehydrogenase (IDH1) R132 mutation recently discovered to act as a driver in this cancer type.Citation5
Here we applied CHASM to 963 of the missense mutations identified in the sequencing of >20,000 coding genes in 24 pancreatic cancers and found that CHASM prioritized missense mutations in most of the known driver genes in pancreatic cancer (TP53, p16/CDKN2A and SMAD4). (Although CHASM is able to correctly identify known KRAS driver mutations, we have omitted them from this list, since they were included in our training data). In addition, many of the mutations prioritized by CHASM (false discovery rate ≤0.3) are in genes that code for proteins with potentially interesting functions, including tyrosine and lipid kinases, cell cycle regulation and cell-adhesion molecules.
Results
Fifty-six of the 963 somatic missense mutations analyzed had an FDR of ≤0.30 (, Suppl. Table 3). Of these 56, 41 mutations occur in known pancreatic driver genes (KRAS, TP53, CDKN2A, SMAD4). Thirty-four of these mutations have previously been identified as drivers and were included in the classifier's training set. Thus, it is likely that our scores inflate the statistical significance of these mutations. Only two of the remaining 15 mutations assigned FDR ≤0.30 were in genes that were mutated more than once in the Discovery set of 24 pancreatic cancers from the original sequencing study. Since recurrent mutation was a criteria for classifying a gene as a significant candidate cancer gene in the original study, 13 of these mutations would not have been prioritized. These 13 mutations occur in 13 genes that were not known driver genes nor were prioritized as likely driver genes in the original sequencing study. That is to say that we wouldn't have picked these genes out as significant without CHASM.
Five of the 15 mutations assigned FDR ≤0.30, which weren't in known driver genes, were in genes coding for kinases (PIK3CG, DGKA, STK33, TTK and PRKCG), one was in NEK8, a gene coding for a cell cycle related protein,Citation12 and two were in genes coding for cell adhesion proteins (CMAS and PCDHB2).
Next, we summarize the relevance of these mutations.
Kinases.
PIK3CG (R839C). This lipid kinase contains the second highest scoring predicted driver mutation among the set of genes not previously identified as drivers in pancreatic cancer. PIK3CG is a member of the phosphatidylinositol 3-kinases (PI3Ks) family and plays a role in mast cell degranulation,Citation13 lymphocyte activationCitation14 and leucocyte chemotaxis.Citation15,Citation16 Unlike the closely related human oncogene PIK3CA (p110alpha), there is evidence suggesting that PIK3CG functions as a tumor suppressor. Loss of PIK3CG in mice leads to spontaneous, malignant epithelial tumours in the colorectum and PIK3CG can block the growth of human colon cancer cells.Citation17PIK3CG is located in a region on chromosome band 7q22 that is frequently deleted in myeloid malignancies.Citation18 Furthermore, PIK3CG has been found to be downregulated by CpG hypermethylation in Human Colorectal Carcinoma.Citation19
Several X-ray crystal structures exist for PIK3CG, making it attractive for structural analysis. An investigation of the X-ray crystal structure for PIK3CG bound to ATP (PDB ID: 1E8X),Citation20 may explain how the R839C mutant found in the series of pancreatic cancers could lead to a loss of function. Arg 839 is located within the C-terminal catalytic domain. Specifically, residue 839 resides within a highly conserved stretch of sequence that is part of the ATP-binding pocket. This pocket is highly conserved among all PI3Ks and resembles the canonical protein kinase catalytic site. In the crystal structure, Arg 839 is within hydrogen bonding distance of Glu 702 and Gln 705, and appears to anchor in place several residues in direct contact with ATP, including Asp 836 and Lys 833. Inhibitor binding studies have shown that relative to other PI3Ks, PIK3CG has a higher level of conformational plasticity within the ATP binding pocket.Citation21,Citation22 The increased plasticity of the PIK3CG active site combined with the lower electrostatic interaction potential of a cysteine compared to arginine (Cys pKa = 8.33; Arg pKa = 12.48) increases the chance that the R839C mutation causes a destabilization of the ATP binding pocket, resulting in decreased affinity for ATP ().
DGKA (V379I) is a diacylglycerol (DAG) kinase that is important for maintaining membrane phospholipid homeostasis. Its various functions include the conversion of DAG to phosphatidic acid (PA), resynthesis of phosphatidylinositols (PIs) and competition for the second messenger DAG with protein kinase C (PKC).Citation23 DGKA has been implicated in VEGF mediated angiogenesisCitation24 and suppression of TNF-alpha induced apoptosis of human melanoma cells via NF-KB.Citation25 In addition, this kinase is found to be constitutively activated in nucleophosmin/anaplastic lymphoma kinase (NPM/ALK) fusion in malignant lymphomas, where inhibition of DGKA significantly reduced tumor growth.Citation26
Serine/threonine kinase 33 (STK33) (F323L) remains largely uncharacterized but has sequence similarity to calcium/calmodulin dependent kinase (CAMK).Citation27 In addition, STK33 has been identified as a synthetic lethal in KRAS mutant lung cancers.Citation28,Citation29
TTK (D697Y) (also known as MPS1) is a dual specificity kinase (Tyr and Ser/Thr) associated with proliferation through regulation of chromosome alignment during mitosis and with centromere duplication. It also controls nuclear targeting of c-Abl in response to oxidative stress.Citation30 If the protein fails to be degraded, the subsequent production of multiple centromeres could contribute to tumorigenesis. BRAF signaling was shown to interfere with the mitotic spindle checkpoint during cell division by stabilizing TTK levels in melanoma cells.Citation31 This gene has also been implicated in bladder cancer.Citation32
PRKCG (P524R) is the gamma isoform of protein kinase C (PKC). PKC isoforms play diverse physiological roles through their many downstream targets, including transcriptional regulation and cell growth. The gamma isoform is normally only expressed in the nervous system. PRKCG serves as the receptor for phorbol esters, which are a class of tumor promoter.Citation33 This gene is also mutated in spinocerebellar ataxia, leading to aberrant MAPK signaling.Citation34
FLJ25006 (S196L) is an uncharacterized serine/threonine kinase.Citation35 Aside from the fact that is has a C-terminal kinase domain, very little is known about this protein.
Cell cycle proteins.
NEK8 (A197P) is a member of the “neverin-mitosis” or NIMA-related kinase (NRK) family of proteins and codes for a protein that plays a role in cell cycle progression from G2 to M-phase. NEK8 has been shown to localize to cilia and regulate both centrosomal and ciliary stability.Citation36,Citation37 Interestingly, pancreatic cancer and precursor pancreatic intraepithelial neoplasia lesions have been found to be devoid of primary cilia.Citation38 Furthermore, mutations in NEK8 have been found to cause polycystic kidney disease.Citation39–Citation41
Cell-adhesion proteins.
CMAS (I275R) catalyzes a reaction that provides substrate for the addition of sialic acid to glycoproteins and glycolipids that are important for the structure and function of tissues.Citation42 This gene has not previously been implicated in cancer however, increase of sialylation of tumor associated carbohydrate antigens (TACA) is a well-known feature of transformed cells.Citation43
PCDBH2 (A323V) is thought to code for a calcium dependent cell-adhesion protein and is thought to be critical for neuronal connections in the brain, though it does appear to be expressed in the pancreas. Epigenetic silencing of protocadherin gene clusters are observed in association with Wilm tumor and gastric cancer.Citation44 These proteins appear to act as tumor suppressors.
Other putative drivers.
PRDM5 (V85I) encodes a transcription factor and is known to act as a tumor suppressor gene.Citation45,Citation46
PRDM5 is a target of epigenetic silencing in colorectal and gastric cancer.Citation47
CALB1 (D199G) is a calcium-binding protein that is part of troponin C superfamily. Expression of CALB1 in a pancreatic islet β-cell line has been shown to protect against cytokine induced apoptosis and necrosis.Citation48
MAPT (G333V) encodes for the microtubule-associated protein tau and is a biomarker for endocrine sensitivity and chemotherapy resistance in estrogen-receptor-positive breast cancer.Citation49
COCH (S400L) is highly conserved among human, mouse and chicken. COCH has been found to be mutated in sensorineural deafness and vestibular disorder DFNA9.Citation50
STN2 (1590S) is part of the endocytic machinery.Citation51 STN2 has not been implicated in any cancer types.
UNC13C (G1126R) plays a role in vesicle maturation during exocytosis.Citation52
Discussion
Pancreatic cancer is fundamentally a genetic disease—a disease caused by inherited and acquired mutations in selected genes.Citation53 Recent advances in sequencing technologies now make it possible to sequence virtually the entire pancreatic cancer genome. The challenge is to determine which of the many genetic alterations identified are biologically significant “driver” mutations, and which are neutral “passenger” mutations.Citation3
We applied the CHASM methodology to a large set of somatic missense mutations previously identified in pancreatic cancer and we prioritize missense mutations in 19 genes (assigned an FDR ≤0.30) as possible drivers in pancreatic cancer. Fifteen of these 19 genes are not known driver genes in pancreatic cancer, and would not have been prioritized had we simply looked at the number of times that gene has been shown to be mutated in pancreatic cancer. Included in the set of predicted driver mutations was a mutation in the PIK3CG gene that is likely to lead to a loss of function. Although members of the PI3K family, specifically PIK3CA, have been found to be among the most mutated genes in various cancer types, mutations within PIK3CG are less common. There are only 4 mutations for PIK3CG listed in the COSMIC database and the R839C mutation is not among them. To our knowledge, this is the first time that the PIK3CG gene has been implicated in pancreatic cancer.
Many of the canonical signaling pathways responsible for mediating essential cellular functions such as growth, proliferation and cell-cell adhesion are known to be deregulated in tumorigenesis, however, the extent to which specific pathway protein constituents contribute to disease progression when mutated is still not well understood. Due to the complex nature of proteinprotein interaction networks (cross talk, redundancy, feedback), each neoplasm can accumulate a different set of alterations that lead to a similar phenotype, and many genes not previously implicated in cancer are likely to play a role. By identifying putative driver genes that are mutated at low frequency, it may be possible to understand which specific components of a pathway are responsible for its deregulation in individual patients. Targeting these genes for functional studies could provide insight into the disease process and may assist in the development of more specific drugs and therapies.Citation54
Materials and Methods
Data collection.
We assembled two sets of missense mutations to train our computational classifier. First, 3,299 missense mutations in 75 genes were selected from the COSMIC databaseCitation6 as likely drivers (Bert Vogelstein, personal communication). Next, we used the CHASM passenger generation algorithmCitation4 to create 4,500 synthetic passenger missense mutations. The passenger mutations were based on di-nucleotide based background mutation rates in pancreatic cancer (Supp. Table 1) and were targeted at genes previously seen to be somatically mutated in either breast, colorectal, glioblastoma multiforme or pancreatic cancers.Citation2,Citation7,Citation8 Finally, we created a third filtered set of 2,700 passenger mutations in genes that have not been previously associated with cancer to use as an empirical null set for estimating p values.
Algorithm.
We used the CHASM protocol to train a random forest (ensemble of decision trees),Citation9,Citation10 using 3,299 likely driver mutations as the positive class and 4,500 synthetic passenger mutations as the negative class (Data Collection). Each missense mutation was quantified by 70 predictive features, calculated from properties of genomic and protein sequence, predicted protein structure and multiple sequence alignments (Suppl. Table 2).Citation4 Each of the 963 somatic pancreatic missense mutations was then scored by the trained forest. Scores range from 0 to 1 and represent the fraction of trees in the forest that classify the mutation as a passenger.
To transform scores into p values, we used the trained forest to score 2,700 filtered passenger mutations from the empirical null (Data Collection). We assume that very few (if any) of these mutations are drivers that were accidentally created by the passenger generation algorithm. Thus, the fraction of filtered passengers with scores below a threshold of interest can be used as an empiritcal p value estimate of the probability that a mutation in that score range is a false positive.
Finally, we obtained q values (FDR) for each of the 963 somatic mutations using the Benjamini-Hochberg algorithm.Citation11
Figures and Tables
Figure 1 Structure of the catalytic domain of PIK3CG (PDB ID: 1E8X). ATP is shown bound within the PIK3CG active site. Residues D836 and K831 are within bonding distance of metal catalyst (green sphere) and ATP alpha/beta phosphates respectively. Residue R839 is within hydrogen bonding distance of both E702 and Q705 and appears to serve as an anchor to correctly position ATP contacts along the N-terminal lobe of the catalytic domain.
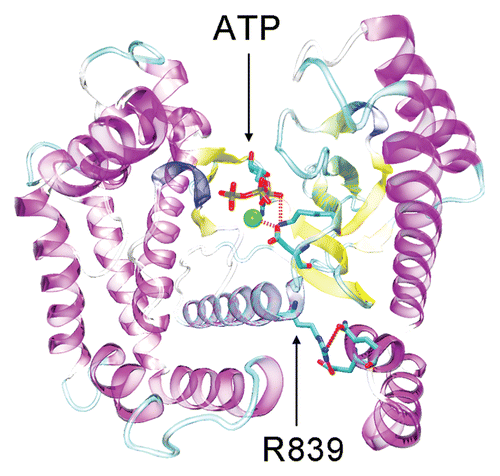
Table 1 Fifty-six somatic missense mutations with CHA SM FDR ≤ 0.30
Additional material
Download Zip (138.9 KB)Acknowledgements
This research was made possible with U.S. Government support under and awarded by: DoD, Air Force Office of Scientific Research, National Defense Science and Engineering Graduate (NDSEG) Fellowship, 32 CFR 168a to H.C, National Science Foundation CAREER award DBI 0845275 and NIH NCI R21 CA135866 to R.K. Thanks to Bert Vogelstein for providing a manually curated list of likely driver missense mutations.
References
- Society AC. Cancer Facts and Figures 2009 2009; Atlanta American Cancer Society, Inc
- Jones S, Zhang Z, Parsons DW, Lin J, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancer revealed by tumor genome analysis. Science 2008; 321:1801 - 1806
- Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al. Patterns of somatic mutation in human cancer genomes. Nature 2007; 446:153 - 158
- Carter H, Chen S, Isik L, Tyekucheva S, Velculescu VE, Kinzler KW, et al. Cancer-specific high-throughput annotation of somatic mutations: computational prediction of driver missense mutations. Cancer Res 2009; 69:6660 - 6667
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009; 360:765 - 773
- Forbes S, Clements J, Dawson E, Bamford S, Webb T, Dogan A, et al. Cosmic 2005. Br J Cancer 2006; 94:318 - 322
- Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006; 314:268 - 274
- Parsons DW, Jones S, Zhang X, Lin J, Leary RJ, Angenendt P, et al. An integrated genomic analysis of glioblastoma multiforme. Science 2008; 321:1807 - 1812
- Amit Y, Geman D. Shape quantization and recognition with randomized trees. Neural Comput 1997; 9:1545 - 1588
- Breiman L. Classification and regression trees Regression trees The Wadsworth statistics/probability series 1984; Wadsworth International Group
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol 1995; 289 - 300
- Bowers AJ, Boylan JF. Nek8, a NIMA family kinase member, is overexpressed in primary human breast tumors. Gene 2004; 328:135 - 142
- Kim MS, Radinger M, Gilfillan AM. The multiple roles of phosphoinositide 3-kinase in mast cell biology. Trends Immunol 2008; 29:493 - 501
- Ji H, Rintelen F, Waltzinger C, Bertschy Meier D, Bilancio A, Pearce W, et al. Inactivation of PI3Kgamma and PI3Kdelta distorts T-cell development and causes multiple organ inflammation. Blood 2007; 110:2940 - 2947
- Ferguson GJ, Milne L, Kulkarni S, Sasaki T, Walker S, Andrews S, et al. PI(3)Kgamma has an important context-dependent role in neutrophil chemokinesis. Nat Cell Biol 2007; 9:86 - 91
- Thomas MS, Mitchell JS, DeNucci CC, Martin AL, Shimizu Y. The p110gamma isoform of phosphatidylinositol 3-kinase regulates migration of effector CD4 T lymphocytes into peripheral inflammatory sites. J Leukoc Biol 2008; 84:814 - 823
- Sasaki T, Irie-Sasaki J, Horie Y, Bachmaier K, Fata JE, Li M, et al. Colorectal carcinomas in mice lacking the catalytic subunit of PI(3)Kgamma. Nature 2000; 406:897 - 902
- Kratz CP, Emerling BM, Bonifas J, Wang W, Green ED, Le Beau MM, et al. Genomic structure of the PIK3CG gene on chromosome band 7q22 and evaluation as a candidate myeloid tumor suppressor. Blood 2002; 99:372 - 374
- Semba S, Itoh N, Ito M, Youssef EM, Harada M, Moriya T, et al. Downregulation of PIK3CG, a catalytic subunit of phosphatidylinositol 3-OH kinase, by CpG hypermethylation in human colorectal carcinoma. Clin Cancer Res 2002; 8:3824 - 3831
- Walker EH, Pacold ME, Perisic O, Stephens L, Hawkins PT, Wymann MP, et al. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin and staurosporine. Mol Cell 2000; 6:909 - 919
- Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, et al. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell 2006; 125:733 - 747
- Williams R, Berndt A, Miller S, Hon WC, Zhang X. Form and flexibility in phosphoinositide 3-kinases. Biochem Soc Trans 2009; 37:615 - 626
- Raben DM, Wattenberg BW. Signaling at the membrane interface by the DGK/SK enzyme family. J Lipid Res 2009; 50:35 - 39
- Baldanzi G, Mitola S, Cutrupi S, Filigheddu N, van Blitterswijk WJ, Sinigaglia F, et al. Activation of diacylglycerol kinase alpha is required for VEGF-induced angiogenic signaling in vitro. Oncogene 2004; 23:4828 - 4838
- Yanagisawa K, Yasuda S, Kai M, Imai S, Yamada K, Yamashita T, et al. Diacylglycerol kinase alpha suppresses tumor necrosis factor-alpha-induced apoptosis of human melanoma cells through NFkappaB activation. Biochim Biophys Acta 2007; 1771:462 - 474
- Bacchiocchi R, Baldanzi G, Carbonari D, Capomagi C, Colombo E, van Blitterswijk WJ, et al. Activation of alpha-diacylglycerol kinase is critical for the mitogenic properties of anaplastic lymphoma kinase. Blood 2005; 106:2175 - 2182
- Mujica AO, Brauksiepe B, Saaler-Reinhardt S, Reuss S, Schmidt ER. Differential expression pattern of the novel serine/threonine kinase, STK33, in mice and men. Febs J 2005; 272:4884 - 4898
- Downward J. Finding the weakness in cancer. N Engl J Med 2009; 361:922 - 924
- Scholl C, Frohling S, Dunn IF, Schinzel AC, Barbie DA, Kim SY, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell 2009; 137:821 - 834
- Nihira K, Taira N, Miki Y, Yoshida K. TTK/Mps1 controls nuclear targeting of c-Abl by 14-3-3-coupled phosphorylation in response to oxidative stress. Oncogene 2008; 27:7285 - 7295
- Cui Y, Guadagno TM. B-Raf(V600E) signaling deregulates the mitotic spindle checkpoint through stabilizing Mps1 levels in melanoma cells. Oncogene 2008; 27:3122 - 3133
- Olesen SH, Thykjaer T, Orntoft TF. Mitotic checkpoint genes hBUB1, hBUB1B, hBUB3 and TTK in human bladder cancer, screening for mutations and loss of heterozygosity. Carcinogenesis 2001; 22:813 - 815
- Geiges D, Meyer T, Marte B, Vanek M, Weissgerber G, Stabel S, et al. Activation of protein kinase C subtypes alpha, gamma, delta, epsilon, zeta and eta by tumor-promoting and nontumor-promoting agents. Biochem Pharmacol 1997; 53:865 - 875
- Verbeek DS, Goedhart J, Bruinsma L, Sinke RJ, Reits EA. PKCgamma mutations in spinocerebellar ataxia type 14 affect C1 domain accessibility and kinase activity leading to aberrant MAPK signaling. J Cell Sci 2008; 121:2339 - 2349
- Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol 11:9 - 22
- Otto EA, Trapp ML, Schultheiss UT, Helou J, Quarmby LM, Hildebrandt F. NEK8 mutations affect ciliary and centrosomal localization and may cause nephronophthisis. J Am Soc Nephrol 2008; 19:587 - 592
- Plotnikova OV, Pugacheva EN, Golemis EA. Primary cilia and the cell cycle. Methods Cell Biol 2009; 94:137 - 160
- Seeley ES, Carriere C, Goetze T, Longnecker DS, Korc M. Pancreatic cancer and precursor pancreatic intraepithelial neoplasia lesions are devoid of primary cilia. Cancer Res 2009; 69:422 - 430
- Liu S, Lu W, Obara T, Kuida S, Lehoczky J, Dewar K, et al. A defect in a novel Nek-family kinase causes cystic kidney disease in the mouse and in zebrafish. Development 2002; 129:5839 - 5846
- Smith LA, Bukanov NO, Husson H, Russo RJ, Barry TC, Taylor AL, et al. Development of polycystic kidney disease in juvenile cystic kidney mice: insights into pathogenesis, ciliary abnormalities and common features with human disease. J Am Soc Nephrol 2006; 17:2821 - 2831
- Sohara E, Luo Y, Zhang J, Manning DK, Beier DR, Zhou J. Nek8 regulates the expression and localization of polycystin-1 and polycystin-2. J Am Soc Nephrol 2008; 19:469 - 476
- Munster AK, Weinhold B, Gotza B, Muhlenhoff M, Frosch M, Gerardy-Schahn R. Nuclear localization signal of murine CMP-Neu5Ac synthetase includes residues required for both nuclear targeting and enzymatic activity. J Biol Chem 2002; 277:19688 - 19696
- Cazet A, Julien S, Bobowski M, Krzewinski-Recchi MA, Harduin-Lepers A, Groux-Degroote S, et al. Consequences of the expression of sialylated antigens in breast cancer. Carbohydr Res 2010; 345:1377 - 1383
- Dallosso AR, Hancock AL, Szemes M, Moorwood K, Chilukamarri L, Tsai HH, et al. Frequent long-range epigenetic silencing of protocadherin gene clusters on chromosome 5q31 in Wilms' tumor. PLoS Genet 2009; 5:e1000745
- Deng Q, Huang S. PRDM5 is silenced in human cancers and has growth suppressive activities. Oncogene 2004; 23:4903 - 4910
- Duan Z, Person RE, Lee HH, Huang S, Donadieu J, Badolato R, et al. Epigenetic regulation of protein-coding and microRNA genes by the Gfi1-interacting tumor suppressor PRDM5. Mol Cell Biol 2007; 27:6889 - 6902
- Watanabe Y, Toyota M, Kondo Y, Suzuki H, Imai T, Ohe-Toyota M, et al. PRDM5 identified as a target of epigenetic silencing in colorectal and gastric cancer. Clin Cancer Res 2007; 13:4786 - 4794
- Rabinovitch A, Suarez-Pinzon WL, Sooy K, Strynadka K, Christakos S. Expression of calbindin-D(28k) in a pancreatic islet beta-cell line protects against cytokine-induced apoptosis and necrosis. Endocrinology 2001; 142:3649 - 3655
- Andre F, Hatzis C, Anderson K, Sotiriou C, Mazouni C, Mejia J, et al. Microtubule-associated protein-tau is a bifunctional predictor of endocrine sensitivity and chemotherapy resistance in estrogen receptor-positive breast cancer. Clin Cancer Res 2007; 13:2061 - 2067
- Robertson NG, Resendes BL, Lin JS, Lee C, Aster JC, Adams JC, et al. Inner ear localization of mRNA and protein products of COCH, mutated in the sensorineural deafness and vestibular disorder, DFNA9. Hum Mol Genet 2001; 10:2493 - 2500
- Martina JA, Bonangelino CJ, Aguilar RC, Bonifacino JS. Stonin 2: an adaptor-like protein that interacts with components of the endocytic machinery. J Cell Biol 2001; 153:1111 - 1120
- Wu CH, Apweiler R, Bairoch A, Natale DA, Barker WC, Boeckmann B, et al. The Universal Protein Resource (UniProt): an expanding universe of protein information. Nucleic Acids Res 2006; 34:Database issue D187 - D191
- Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol 2008; 3:157 - 188
- Hucl T, Rago C, Gallmeier E, Brody JR, Gorospe M, Kern SE. A syngeneic variance library for functional annotation of human variation: application to BRCA2. Cancer Res 2008; 68:5023 - 5030