
Abstract
Commentary to:
Prioritization of driver mutations in pancreatic cancer using cancer-specific high-throughput annotation of somatic mutations (CHASM)
Hannah Carter, Josue Samayoa, Ralph H. Hruban and Rachel Karchin
Acknowledgements
Supported, in part, by US Public Health Service Grant CA-75059, awarded by the National Cancer Institute to M.K.
Figures and Tables
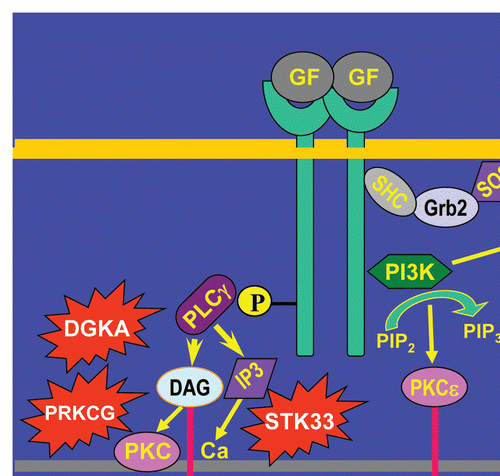
Figure 1 Pathways activated by novel driver mutations intersect with Kras driven aberrant signaling pathways in pancreatic cancer cells. In addition to harboring mutated Kras, pancreatic cancer cells overexpress tyrosine kinase receptors, an example of which is shown binding excessive levels of growth factors (GF). This leads to excessive Ras activation even when Kras is not mutated. Ras becomes active downstream of the adapter proteins SHC and GRB2 and the SOS guanine nucleotide exchange factor. There is activation of additional pathways downstream of ras, as described in the text, but only the RAF, MEK, MAPK cascade is shown. PI3K activation leads to PIP3 generation and subsequent AKT and protein kinase C (PKC) epsilon (PKCε) activation, serving to promote cancer cell survival and chemoresistance. Mutated PI3KCG, which encodes a mutated PI3Kγ, may activate aberrant pathways downstream of gastrointestinal hormones such as cholecystokin (CCK) or gastrin, leading to excessive ras activation and PIP3 generation, further promoting cancer cell survival and proliferation. Concomitantly, there is activation of phospholipase C-gamma (PLCγ), which leads to the generation of inositol-1,4,5-trisphosphate (IP3) which mobilizes intracellular calcium and diacylglycerol (DAG), which activates PKC. Mutated DAG kinase (DGKA), STK33 (which exhibits homology with calcium-calmodulin dependent kinase) and PRKCG, which encodes the gamma isoform of PKC, can in theory “join forces” to lead to enhanced cancer cell proliferation and invasion.
Commentary to: