
Abstract
Epigenetic modifiers such as histone deacetylases (HDACs) have come into focus as novel drug targets for cancer therapy due to their functional role in tumor progression. Since common pan-HDAC inhibitors have adverse side effects and minor anti-cancer activity against solid tumors, enzyme-specific inhibitors were developed. HDAC6 is especially well-suited for specific inhibition due to its unique domain structure and mode of action and has been suggested to provide an exceptionally suitable target for cancer therapy. However, expression and function of HDACs have been insufficiently studied in urothelial cancers (UC), a disease urgently requiring new therapeutic approaches. The present study sought to evaluate HDAC6 as a target for treatment of urothelial cancers with enzyme-specific inhibitors. We observed moderate HDAC6 overexpression in urothelial cancer tissues and a broad range of expression in urothelial cancer cell lines. In the cell lines Tubacin was the most potent inhibitor, compared with Tubastatin and ST-80, but still active only at high micromolar concentrations. HDAC6 expression levels correlated poorly with sensitivity to enzyme inhibition. Combined treatments with heat shock, HSP90 inhibition by 17-AAG, proteasome inhibition by bortezomib, or DNA-damaging agents did not result in significant synergistic effects. Experiments with siRNA-mediated knockdown further underlined that urothelial cancer cells do not critically depend on HDAC6 expression for survival.
Introduction
With about 100 000 new cases per year bladder cancer is the fifth most common cancer type in Europe. While patients with non-muscle invasive papillary urothelial carcinoma expect a rather good prognosis, one third of patients are diagnosed with a muscle-invasive carcinoma and face a poor prognosis. Despite radical cystectomy, the long-term disease-free survival of these patients is below 65% and the 5-y survival rate is below 50%. Although initial responses to standard chemotherapy treatment with cisplatin-based combination regimens are observed in two-third of patients with metastatic disease,Citation1,Citation2 the majority of patients expect a median survival of only 15 mo and long-term remissions are observed in only about 10% of the patients. Thus, there is a growing demand for the development of alternative therapy approaches.
During the last decade, epigenetic regulator proteins have come into focus as novel drug targets for cancer therapy. Mutations and alterations in the expression of enzymes modifying histones and DNA, remodeling chromatin or interpreting histone modifications have been discovered in a wide range of cancers, including urothelial carcinoma.Citation3-Citation5 Histone deacetylases (HDAC) have been highlighted due to their functional role in tumor progressionCitation6 and a large arsenal of inhibitors has been developed to target one or several of these enzymes in cancer therapy. In humans, 11 classical HDACs have been identified which are classified into four classes (class I: HDAC1, HDAC2, HDAC3, and HDAC8; class IIa: HDAC4, HDAC5, HDAC7, and HDAC9; class IIb: HDAC6 and HDAC10; class IV: HDAC11). Although many HDACs deacetylate histones,Citation7 they also act on many other proteins. These include cytoplasmic proteins such as tubulin, cortactin, or heat shock proteins that are especially substrates of HDAC6 which is predominantly localized in the cytoplasm.Citation8,Citation9
Several functions of HDAC6 are well established. The enzyme regulates the cytoskeleton and cell motility via its influence on tubulin and other cytoskeletal proteins (reviewed in ref. Citation10). Moreover, HDAC6 is unique due to its two catalytic domains and a C-terminal zinc finger domain that binds both polyubiquitinated misfolded proteins and the dynein motor. Accordingly, it is involved in cellular stress management induced by misfolded proteins, enabling their degradation by mediating their transport toward aggresomes. HDAC6-deficient cells are therefore hypersensitive to the accumulation of misfolded proteinsCitation8 that causes increased ubiquitin-proteasome-system (UPS) stress. This effect can be exploited for cancer therapy by combining inhibitors of HDAC6 and of the proteasome system such as bortezomib.Citation11 Further, HDAC6 has been shown to activate the chaperone function of heat shock protein 90 (HSP90). Inhibition of HDAC6 disrupts this chaperone function, resulting in polyubiquitination and depletion of pro-growth and pro-survival HSP90 client proteins including BCR-ABL, p53, HER2/ERBB2, AKT, RAF1, and HIF-1α.Citation12,Citation13 The chaperone function of HSP90 is also inhibited by the geldanamycin derivative 17-AAG (17-N-allylamino-17-demethoxygeldanamycin). These observations built the rationale for a combined treatment with HDAC6-specific inhibitors and 17-AAG.
Since common pan-HDAC inhibitors were shown to have adverse side effects and poor anti-cancer activity against lymphoma and solid tumors,Citation14,Citation15 efforts have been undertaken to generate enzyme-specific inhibitors. HDAC6 is well-suited for specific inhibition due to its unique domain structure and mode of action and has been suggested to provide an exceptionally suitable target for cancer therapy.Citation16 Specific HDAC6 inhibitors include Tubastatin A, Tubacin, and ST-80.Citation17-Citation19
Although data on expression and function of HDACs is accumulating, they have been rarely studied in urothelial carcinoma (UC). We have recently published a comprehensive screening analysis of HDAC expression patterns in urothelial carcinoma and tissues.Citation20 Generally, we found that expression levels of HDAC2 and HDAC8 were increased, while levels of HDACs 4, 5, and 7 were diminished. HDAC6 mRNA expression was quite variable in our tissue set as well as in publicly available gene expression data sets with overexpression in some cases. These preliminary data, combined with the special cellular functions of HDAC6 and the availability of HDAC6-specific inhibitors prompted us to investigate HDAC6 in urothelial carcinoma in more detail in order to determine whether it might serve as an appropriate target for therapy of urothelial carcinoma by either specific inhibition alone or in combination with other treatments.
In this study, by comparing cellular toxicity and specificity of three HDAC6-specific inhibitors (Tubacin, Tubastatin A, and ST-80), we identified Tubacin as the most potent inhibitor, albeit still showing efficacy only at high micromolar concentrations. Interestingly, HDAC6 expression levels correlated only rather weakly with sensitivity to enzyme inhibition. Combined treatments with heat shock, 17-AAG, bortezomib, doxorubicin, or etoposide did not result in synergistic effects that would allow applying reduced doses of HDAC6 inhibitors. In parallel experiments we analyzed whether urothelial cancer cells are dependent on HDAC6 by means of siRNA-mediated knockdown. However, growth and survival of urothelial carcinoma cells did not prove critically dependent on HDAC6 expression. We therefore deduce that HDAC6 does not seem to represent a promising target for treatment of urothelial carcinoma.
Results
Urothelial carcinomas show a range of HDAC6 expression levels
In order to investigate whether HDAC6 is aberrantly expressed in urothelial carcinoma, we determined HDAC6 mRNA expression by quantitative real-time PCR in tissues and cell lines (). Expression analysis in urothelial cancer tissues (n = 19) demonstrated moderate, but statistically significant overexpression of HDAC6 compared with normal (n = 10) controls (, P = 0.001). Variations in HDAC6 expression among cancerous tissues were independent from clinicopathological parameters like grade, stage or presence of lymph node metastases (grade 2 vs. grade 3 P = 0.437; ≤pT2 vs. >pT2 P = 0.665; lymph node positive vs. negative P = 0.583, Mann-Whitney U test). Most urothelial cancer cell lines displayed equal or reduced HDAC6 expression compared with normal proliferating uroepithelial cell cultures (UEC). The cell lines VM-CUB1, BFTC-905, HT-1376, and UM-UC-3 showed the lowest expression levels (). Expression exceeded the mean level of normal controls only in two carcinoma cell lines (253J and 639-V). HDAC6 expression in a normal immortalized urothelial cell line (hTERT) was within the range of normal UEC controls originating from different patients.
Figure 1. HDAC6 expression in urothelial cancer cell lines and tissues. (A) Relative HDAC6 expression in cancerous (T) and normal (N) tissues was determined by quantitative real-time PCR analysis and displayed as box-plots. P value was calculated by Mann–Whitney U test. HDAC6 expression values were normalized to TBP as reference gene. (B) Relative mRNA expression of HDAC6 in urothelial cancer cell lines (T) and normal proliferating uroepithelial cell cultures (N, UEC) was measured by quantitative real-time PCR analysis. The dotted line displays the average expression level of the UEC samples. hTERT is an immortalized normal urothelial cell line. HDAC6 protein expression was analyzed in cell lines by western blotting (C; HDAC6 at 131 kDa, α-Tubulin at 50 kDa). Expression of HSP90 and HIF1α was determined in the same manner (C). Immunofluorescence stainings (D) were performed for cell lines with, respectively, high (RT-112, 639-V, 253J), moderate (5637), and low (BFTC-905, VM-CUB1) HDAC6 protein expression. HDAC6 is stained green (FITC); nuclei are stained blue (DAPI). White arrows indicate positively stained filopodia; accumulation of perinuclear speckles in cell lines with a more epithelial phenotype (5637 and RT-112) are highlighted by white arrowheads.
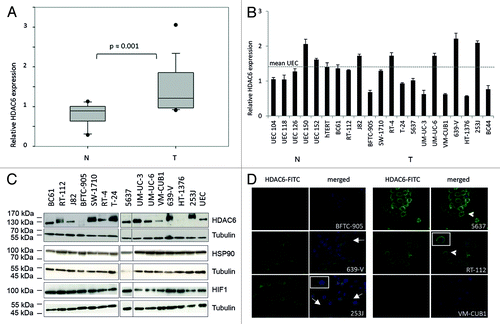
Western blot analysis of HDAC6 protein expression confirmed the variability among the urothelial cancer cell lines (). At the protein level, beside 639-V and 253J cells, further cell lines appeared to express HDAC6 more strongly than normal UEC controls, namely BC61, RT-112, J-82, and UM-UC-3. In addition to BFTC-905, VM-CUB1, and HT-1376, also SW-1710 and RT-4 contained less HDAC6 protein than normal cells. Based on the protein data, we assorted the cell lines into groups () with either high (639-V, 253J, BC61, RT-112, J-82, and UM-UC-3), moderate (T-24, 5637, and UM-UC-6), or diminished expression (BFTC-905, VM-CUB1, HT-1376, SW-1710, and RT-4) and chose according cell lines for further analysis to investigate whether HDAC6 expression level is correlated with sensitivity toward inhibition of enzyme activity. The limited correlation between RNA and protein expression levels in cell lines appeared not to be related to expression of HSP90 or HIF1α as both proteins were equally strong expressed across all cell lines ().
Table 1. Classification of urothelial cancer cell lines regarding HDAC6 protein expression levels
As HDAC6 protein is generally located in the cytoplasm, but can also be found in the nucleus in some cell types, we investigated the localization of HDAC6 protein in urothelial carcinoma cell lines by immunofluorescence staining. For this purpose we stained cell lines with high HDAC6 protein expression (RT-112, 639-V, and 253J), moderate (5637) and low expression (BFTC-905 and VM-CUB1). HDAC6 was generally detected in the cytoplasm (), only the BFTC-905 cell line exhibited also a nuclear signal. Especially cell lines with a more mesenchymal phenotype, like 639-V and 253J, presented prominent positively stained filopodia (white arrows). In cell lines with a more epithelial phenotype (5637 and RT-112) HDAC6 appeared to accumulate preferentially in perinuclear speckles (white arrowheads). In accord with the western blot data, staining in VM-CUB1 cells was very weak.
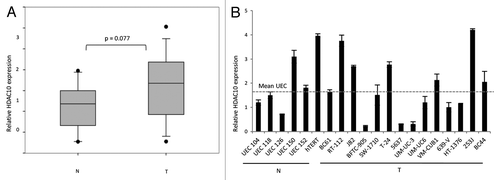
As the second member of HDAC class IIb—HDAC10—is not very well investigated, we also determined the expression of HDAC10 mRNA in urothelial carcinoma tissues () and cell lines (). We observed an expression pattern comparable to HDAC6 with varying expression levels and a tendency toward moderate overexpression in tumor tissues (P = 0.077). HDAC6 and HDAC10 expression did not correlate with each other in urothelial carcinoma cell lines and tissues (Pearson r = 0.38 and 0.25, respectively).
Figure 2. Relative mRNA expression of HDAC10 in urothelial cancer cell lines and tissues. (A) Relative HDAC10 expression in cancerous (T) and normal (N) tissues was determined by quantitative real-time PCR analysis and displayed as box-plot graphs. P value was calculated by Mann–Whitney U test. (B) mRNA expression of HDAC10 in urothelial cancer cell lines (T) and normal proliferating uroepithelial cell cultures (N, UEC) was measured by quantitative real-time PCR analysis and normalized to TBP as reference gene. The dotted line displays the average expression level of the UECs.
Tubacin is the most potent HDAC6-specific inhibitor in urothelial carcinoma cells
Next, we evaluated the sensitivity of urothelial carcinoma cell lines with graduated levels of HDAC6 protein to three different inhibitors of HDAC6 (i.e., Tubacin, Tubastatin A, and ST-80).
At first, we compared the three compounds for their effects on the viability of urothelial cancer cell lines with high HDAC6 expression (RT-112, 639-V, 253J, and UM-UC-3), moderate (5637 and T-24) and low expression (VM-CUB1) using an assay measuring total cellular ATP (). Treatment with SAHA, a pan-HDAC inhibitor, was employed as a positive control for decreased cell viability. Based on our previous experience, we used 10 µM of the compound to induce a significant reduction of cell viability across all analyzed UC lines. Tubacin and Tubastatin A were similarly efficacious with IC50 values between 6 and 12 µM across the cell line panel (). However, 10 µM Tubacin diminished cell viability to below 20% in a larger number of cell lines. ST-80 proved the least potent inhibitor in this assay (). While 639-V and 5637 cells showed an IC50 value of ~80 µM, viability of other cell lines did not decrease below 50% with even 100 µM ST-80 ().
Figure 3. Viability of urothelial cancer cell lines after treatment with three different HDAC6-specific inhibitors. (A) Cell lines were treated with various doses of the HDAC6 inhibitors Tubacin, ST-80, and Tubastatin A for 48 h. Cells administered with SAHA were used as positive control for decreased cell viability and cells treated with DMSO as negative control. The percentage of viable cells is shown on the y-axis. Cell lines are ordered by HDAC6 expression levels (high expression ◇, RT-112-, UM-UC-3; moderate □, 5637-, T-24; low ○, VM-CUB1). (B) Induction of apoptosis was analyzed by combined Annexin V- and propidium iodide-staining and subsequent flow cytometry after treatment with 10 µM Tubacin for 48 h. The percentages of early apoptotic cells in the lower right quadrant Q4 and of late apoptotic/necrotic cells in the upper right quadrant Q2 are indicated. (C) Effects of pharmaceutical HDAC6 inhibition on cell cycle distribution were determined by FACS analysis following PI staining. Relative abundance of cells in the respective cell cycle phases is given for either untreated cells or cells treated with 10 µM Tubacin for 48 h.
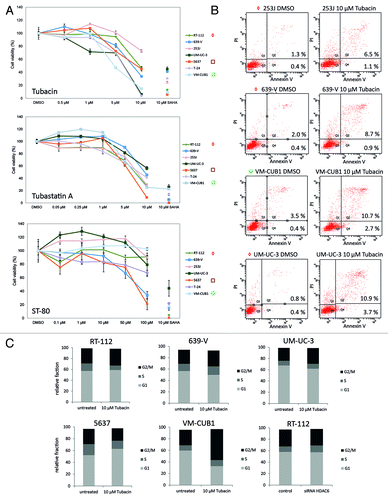
Table 2. IC50 values as determined for HDAC6-specific inhibitors Tubacin, Tubastatin A, and ST-80, the HSP90 inhibitor 17-AAG and the proteasome inhibitor bortezomib in various urothelial cancer cell lines
Toward Tubacin treatment, cell lines with moderate or high HDAC6 expression (5637, 253J, RT-112, and 639-V) appeared to be less sensitive than cell lines with low HDAC6 expression (VM-CUB1 and T-24) (). However, UM-UC-3 was the most sensitive cell line toward Tubacin (IC50 6.6 µM) despite its high HDAC6 protein expression.
The results on viability paralleled those for induction of cell death as measured by flow cytometry for Annexin V-staining (). Following treatment with 10 µM Tubacin for 48 h, the number of early and late apoptotic cells was more strongly increased in VM-CUB1 cells with low HDAC6 expression as compared with 253J and 639-V cells with high HDAC6 expression. UM-UC-3 showed the strongest induction of apoptosis according with its highest sensitivity in the viability assay. Accordingly, western blot analysis revealed a significant amount of cleaved PARP as a marker for apoptosis only in T-24 cells with moderate HDAC6 expression subsequent to treatment with 10 µM Tubacin, but not in cell lines with high HDAC6 expression like RT-112 and 639-V (data not shown). Cell cycle distribution did not change markedly subsequent to Tubacin treatment. Most cell lines showed a tendency toward G2 arrest, which was only significant in VM-CUB1 cells with low HDAC6 expression. Only 5637 cells responded reproducibly with a G1 arrest (). Inhibition of HDAC6 by siRNA-mediated knock-down did not result in changes of cell cycle distribution (, representative data shown for RT-112 cells). Thus, decreased cell numbers after specific HDAC6 inhibition appear to result rather from increased cell death than from cell cycle arrest.
Subsequently, we investigated the impact of the inhibitors on the acetylation state of α-tubulin, a specific substrate of HDAC6. T-24 cells with low HDAC6 activity displayed relatively weak increases in acetylation only at high doses of either inhibitor (). In contrast, cell lines with high HDAC6 protein expression (639-V and RT-112) responded with significantly increased tubulin acetylation already at lower doses. In all three cell lines, Tubastatin A and especially Tubacin increased tubulin acetylation more strongly than ST-80. In parallel, we confirmed that HDAC6 protein expression did not change after pharmacological inhibition of HDAC6 (data not shown). Therefore all observed effects can be ascribed to inhibition of enzyme activity rather than protein loss.
Figure 4. Immunoblot analysis of α-Tubulin and histone H3 acetylation in urothelial cancer cell lines after treatment with the HDAC6 inhibitors Tubacin, ST-80, and Tubastatin A. Levels of acetylated and total α-Tubulin (50 kDa, top) and Histone H3 (17 kDa, bottom), respectively, were determined in the urothelial cancer cell lines T-24, 639-V, and RT-112 after 24 h treatment with the indicated doses of Tubastatin A, Tubacin, and ST-80. DMSO served as solvent control. Data for histone H3 in 639-V was rearranged from the same membrane for the sake of keeping the same order of samples.
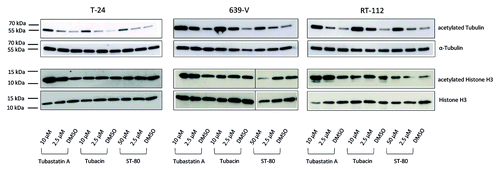
HDAC6 activity is thought to not significantly contribute to global deacetylation of histones. Thus, selective inhibition of HDAC ought not affect global histone acetylation. Accordingly, to determine the specificity of the three compounds for HDAC6 we monitored changes in histone H3 acetylation after treatment of T-24, 639-V, and RT-112 cells with HDCA6-inhibitors. In this experiment Tubacin was the only inhibitor which did generally not alter the acetylation state of histone H3 irrespective of dosage (). Taken together, these data indicate that Tubacin is the most specific and potent HDAC6 inhibitor in urothelial cancer cell lines.
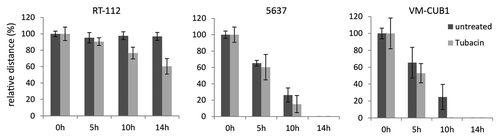
For that reason, we used Tubacin to study the function of HDAC6 activity in regulation of migration of urothelial cancer cells, a common function of HDAC6 in many cell types. However, in several cell lines treatment with Tubacin did not significantly affect migration rates in a wound closure assay ().
Figure 5. Analysis of cell motility of urothelial cancer cell lines following treatment with Tubacin. Effects of pharmaceutical inhibition of HDAC6 by 48 h treatment with Tubacin (IC50 doses) were determined by wound healing assays and compared with untreated cells. Distance measurements were performed at 0, 5, 10, and 14 h after removement of Culture-inserts. Relative distance at the respective time point is displayed on the y-axis.
Combination of pharmacologic HDAC6 inhibition with further induction of cellular stress did not result in broad significant synergistic effects
In order to seek for treatments that might synergize with the limited efficacy of HDAC6 inhibition, we investigated its interaction with five different additional stress stimuli—heat shock, inhibition of the chaperone function of heat shock protein 90 (HSP90) by 17-AAG, proteasome inhibition by bortezomib, or DNA damage induced by doxorubicin or etoposide. Each treatment was investigated combined with the HDAC6-specific inhibitor Tubacin and compared with the pan-HDAC inhibitor SAHA.
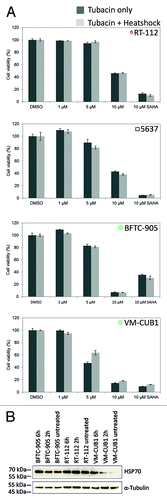
To investigate the interaction of HDAC6 inhibition with heat shock treatment the cell lines RT-112, 5637, BFTC-905, and VM-CUB1 were treated with Tubacin at concentrations ranging from 1 to 10 µM and incubated for 30 min at 42 °C. However, the combined treatment did not result in any significant reduction of cell viability compared with cells treated with Tubacin only (). The cellular reaction to heat-shock was further tested by western blot analysis of HSP70 induction (). Notably, endogenous expression of HSP70 was already significant under basal conditions in accord with previous reportsCitation21,Citation22 and further induction was therefore limited.
Figure 6. Viability of urothelial cancer cell lines after incubation with Tubacin and heat shock treatment. (A) Cell lines were pretreated for 24 h with various doses of Tubacin and then heat-shocked at 42 °C for 30 min. Viability was measured after a 24 h. SAHA was used as positive control for decreased cell viability. The percentage of viable cells is shown on the y-axis. Light gray bar, Tubacin plus heat shock treatment; dark gray bar, Tubacin, but no heat shock treatment. (B) Induction of HSP70 as response to heat shock treatment was shown by western blot analysis.
Prior to investigating combined treatment with the HSP90-inhibitor 17-AAG, we generated a dose response curve to identify appropriate concentrations for the combination experiments () and confirmed the efficacy of HSP90 inhibition by monitoring 17-AAG effects on previously reported downstream factorsCitation13 () in the cell lines RT-112 and VM-CUB1. We used similar concentrations as administered by Karkoulis et al.Citation13 In the dose response curve most urothelial carcinoma cell lines displayed an IC50 value around 0.6 µM for 17-AAG with the exception of the 5637 cell line that was the least sensitive and VM-CUB1 as the most sensitive (; ).
Figure 7. Effects of the HSP90 inhibitor 17-AAG and Tubacin on cell viability of urothelial cancer cell lines. (A) IC50 values for 17-AAG were determined by a dose response analysis with different urothelial cancer cell lines. SAHA was used as positive control for decreased cell viability. The percentage of viable cells is displayed on the y-axis. Cell lines are ordered by HDAC6 expression levels (high expression ◇, RT-112-, UM-UC-3; moderate □, 5637-, T-24; low ○, VM-CUB1). (B) Efficacy of 17-AAG was determined at the molecular level by western blot analysis of known downstream targets (HSP70 70 kDa, E2F1 60 kDa, ERK 40/42kDa, α-Tubulin 50 kDa) after treatment of RT-112 and VM-CUB1 cells with the indicated doses of 17-AAG for 24 h. Effects of single and combined treatment with Tubacin and 17-AAG were determined in a number of urothelial cancer cell lines (C), an extended doses response analysis was performed for 639-V and VM-CUB1 (D).
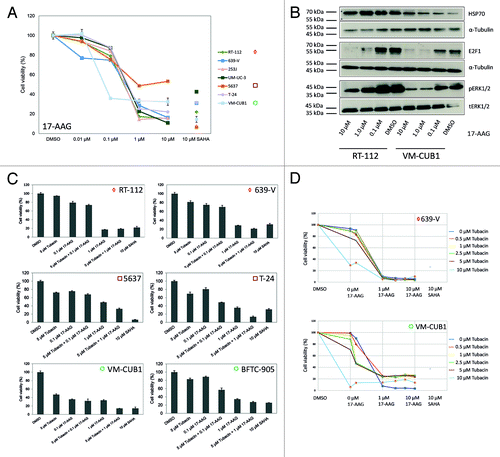
On the molecular level, inhibition of HSP90 by 17-AAG was reported to induce apoptosis and reduce proliferation due to decreased ERK phosphorylation and decreased expression of the cell cycle regulator E2F1, with increased expression of HSP70. Indeed, we observed a dose dependent increase of HSP70 expression and dose-dependent decreases of E2F1 expression and ERK phosphorylation ().
To identify a potential synergism, we combined treatment with a marginally active dose of Tubacin at 5 µM with doses of either 0.1 µM or 1 µM of 17-AAG. More-than additive effects with lower dose concentrations (5 µM Tubacin + 0.1 µM 17-AAG) were observed for the two cell lines T-24 and BFTC-905 (P = 0.01, Student t test) whereas the other cell lines showed only weaker effects with the combination of 5 µM Tubacin and 1 µM 17-AAG (). Less than additive effects were observed in the RT-112 cell line, which was little sensitive to Tubacin. A detailed dose response curve for the combination of Tubacin and 17-AAG obtained for the cell lines 639-V (high HDAC6 expression) and VM-CUB1 (low HDAC6 expression, ) did not indicate synergistic effects in both cell lines.
To investigate the interaction between Tubacin and the proteasome inhibitor bortezomib, experiments were designed in a similar fashion. A dose response curve for bortezomib yielded IC50 values of about 30 nM for most cell lines, with the exception of RT-112 cells which were notably more sensitive (IC50 12.4 nM) and of UM-UC-3 cells which were less sensitive (IC50 53.3 nM, ; ). Based on these results we chose to combine treatments with either 10 nM or 50 nM bortezomib with 5 µM Tubacin to check for synergistic effects (). The lower dose combination with 10 nM bortezomib resulted in more than additive effects (P < 0.001, Student t test) on the BFCT-905 cell line (low HDAC6 protein expression). Less marked synergistic effects were observed for RT-112 cells (P < 0.01, high HDAC6 expression). Similarly, RT-112 cells displayed a synergistic effect with the higher dose combination (P < 0.05), whereas no synergy was notable in VM-CUB1 (low expression), T-24 (moderate expression), and 639-V cells (high expression) with either combination. An extended dose response analysis for these two cell lines did not provide evidence for any synergy with other concentrations (). Notably, across all cell lines, the efficacy of the drug combination did not discernably depend on HDAC6 expression.
Figure 8. Effects of Tubacin, the proteasome inhibitor bortezomib, and agents inducing DNA damage on cell viability of urothelial cancer cell lines. (A) Cell lines were treated with various concentrations of bortezomib to determine IC50 values. The percentage of viable cells is displayed on the y-axis. SAHA was used as a positive control and cells administered with DMSO as negative control. Cell lines are ordered by HDAC6 expression levels (high expression ◇, RT-112-, UM-UC-3; moderate □, 5637-, T-24; low ○, VM-CUB1). To check for synergistic effects cell viability was determined after single and combined treatment in a number of urothelial cancer cell lines (B) and in an extended dose response curve for 639-V and VM-CUB1 (C). Similarly, potential synergistic effects on cell viability were investigated after single and combined treatment of Tubacin with doxorubicin, etoposide, and SAHA over 72 h in 639-V and VM-CUB1 cells (D).
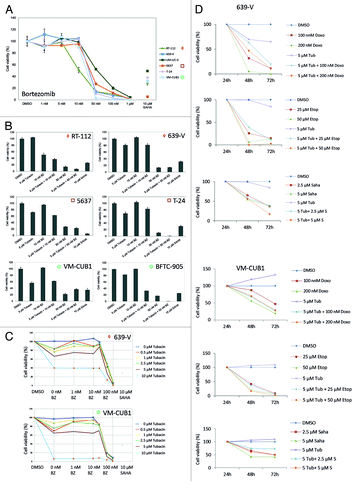
As selective inhibition of HDAC6 has been reported to sensitize cancer cell lines from glioblastoma, breast, prostate, and lung cancer to treatment with DNA damaging compounds,Citation23,Citation24 we investigated combined treatment of Tubacin with doxorubicin or etoposide compared with SAHA for synergistic effects on urothelial cancer cell lines. However, we did not observe any significant synergistic effects (). Notably, even the combination of 5 µM Tubacin with 50 µM etoposide caused only little increases in cellular toxicity compared with treatment with 50 µM etoposide alone. Also note that throughout this experiment treatment with even the relative low concentration of 2.5 µM SAHA was more efficient than treatment with Tubacin.
Inhibition of HDAC6 expression by siRNA knockdown
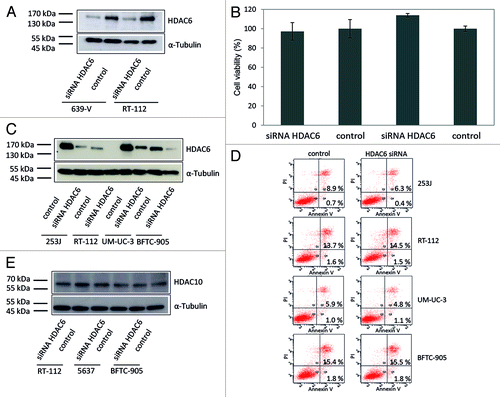
In addition to pharmacological inhibition of HDAC6 enzyme activity, we investigated the effects of siRNA-mediated HDAC6 knockdown on cell viability. Specific siRNA diminished HDAC6 protein expression to low levels as shown by western blotting (). This decrease did not diminish the viability of 639-V and RT-112 cells as measured by total ATP (). Likewise, staining for Annexin V did not reveal significant effects on apoptosis in the cell lines 253J, RT-112, UM-UC-3, and BFTC-905 as compared with controls () despite efficient siRNA knockdown (). Likewise, a combination of siRNA-mediated knock-down and heat shock treatment did not result in decreased cell viability (data not shown). To exclude that the cells compensate HDCA6 knockdown by upregulation of the other HDAC class IIb family member, HDAC10, we also determined HDAC10 protein expression after siRNA knock down of HDAC6. No compensatory expression changes were observed ().
Figure 9. Effect of HDAC6 knockdown by specific siRNA on urothelial cancer cells. (A) The urothelial cancer cell lines 639-V and RT-112 were transfected with a siRNA specific for HDAC6 (siRNA HDAC6) or with a nontargeting control siRNA (control). HDAC6 protein expression was quantified with western blot analysis. α-Tubulin was used as loading control. (B) Effect of HDAC6 knockdown on cell viability was determined after transient transfection by an ATP-dependent assay. (C) Ascertainment of knockdown efficiency in samples used for flow cytometry by western blot analysis. (D) Analysis of apoptosis induction by Annexin V- and propidium iodide-staining and flow cytometry. The percentage of early apoptotic cells in the lower right quadrant Q4 and late apoptotic/necrotic cells in the upper right quadrant Q2 following HDAC6 knock-down are indicated. (E) Exclusion of compensation of HDAC6 knockdown by HDAC10 as assayed by western blot analysis after transfection (HDAC10 67 kDa, α-Tubulin 50 kDa).
Discussion
The recognition that the first generation of relatively unspecific or pan-HDAC inhibitors showed limited clinical efficacy with significant side effectsCitation14,Citation15,Citation25 has pushed the development of compounds inhibiting specifically only a single HDAC enzyme. HDAC6 is particularly well suited for specific inhibition by being notably different from other HDACs with regard to protein structure, cellular localization, and its substrates, while still being involved in the regulation of cellular process like proliferation, migration, invasion and especially cellular stress response.Citation10,Citation26,Citation27 Moreover, HDAC6 has been reported to be overexpressed in a number of different cancer types such as ovarian, gastric, and breast cancer, oral squamous carcinoma and acute myeloid leukemia.Citation11,Citation28-Citation31 In bladder cancer, HDAC6 expression and function has scarcely been investigated. Our present study constitutes the first systematic study of HDAC6 expression and function in this tumor type allowing a first assessment of its potential as a therapeutic target.
We found variable gene expression levels of HDAC6 in urothelial cancer cell lines compared with normal uroepithelial cells, indicating that cell lines might be assorted into three groups according to HDAC6 expression. Analysis of cancer tissues on the RNA level also revealed that HDAC6 expression varied among individual tumors, with an overall significant, albeit moderate increase over normal bladder tissue. Expression was independent of clinical stage, grade or the presence of lymph node metastases. These data confirm the conclusion from our previous global analysis of HDAC isoenzyme expression and in silico analyses of publicly available data setsCitation32-Citation36 that HDAC6 expression is generally variable in urothelial cancers, but that significant overexpression does occur in individual cases, highlighting HDAC6 as a potential target for cancer therapy. Similarly variable results on HDAC6 expression levels have been published for hepatocellular cancers.Citation27,Citation37,Citation38 Furthermore, HDAC6 has been reported to stimulate proliferation in the latter tumor type. In breast cancer, HDAC6 expression was likewise variableCitation29 and particularly strong in small tumors with low histological grade. Accordingly, patients with high HDAC6 expression had a better prognosis. These results on the one hand suggest that HDAC6 may have tissue-dependent functions and on the other hand raise the issue of whether responsiveness to HDAC6-specific inhibitors could be predicted by the level of HDAC6 expression in an individual tumor.
At the protein level, increased HDAC6 expression was observed in a larger number of urothelial carcinoma cell lines. We based the stratification of cell lines for further analysis on the protein data. The obvious differences between mRNA and protein expression in some cell lines indicate that HDAC6 expression is regulated transcriptionally as well as at the protein level. The mechanisms involved in regulation of HDAC6 expression are poorly characterized, but interestingly, regulation by one specific micro-RNA, hsa-miR-433 is documented.Citation39 In order to exclude that factors like HIF1 and HSP90 may be involved we analyzed expression levels of both proteins. Especially HSP90 has been reported to regulate functional integrity and turnover of many proteins e.g., the androgen receptor.Citation40 However, concordant with data from the literature for HSP90Citation13 both proteins were comparably broadly expressed across cell lines. Therefore, HDAC6 and HSP90 may functionally interact in the clearance of misfolded proteins in urothelial carcinoma cells as they do in general, but HSP90 does not appear to regulate HDAC6 protein levels.
Within the cell, HDAC6 is commonly found in the cytoplasmCitation9 concentrating perinuclearly and in filopodia depending on the cell line and its activity state.Citation9,Citation11,Citation29,Citation41 Localization in filopodia may be explained by the interaction between HDAC6 and cortactinCitation42 whereas perinuclear accumulation may be related to the function of HDAC6 as a component of aggresomes.Citation10 In urothelial carcinoma cells, we observed the same tendencies, with preferential staining of filopodia in cells with a more mesenchymal phenotype and preferentially perinuclear staining in epitheloid cell lines. HDAC6 also contains a nuclear localization signal at its N-terminus and can be detected in the nucleus of some cell lines.Citation9,Citation11,Citation43 Among the urothelial carcinoma cell lines, only BFTC-905 cells revealed unambiguous nuclear staining. Lack of detectable nuclear staining in most urothelial carcinoma cell lines is compatible with the current concept that HDAC6 has a limited function in transcriptional regulation, but unlike class I HDACs does not influence global histone acetylation.Citation44
Little is known about the second class IIb isozyme HDAC10 to date. However, since it lacks a ubiquitin-binding domain and tubulin deacetylase activity there may be no functional overlap with HDAC6.Citation10 Only recently, it has been reported to influence neuroblastoma cell survival by promoting autophagy,Citation45 but to suppress cervical cancer metastasis.Citation46 In contrast, increased HDAC10 expression in chronic lymphocytic leukemia patients was associated with poor prognosis.Citation47 In bladder cancer, no investigations on HDAC10 have been published to date. To check whether HDAC10 is interdependent with HDAC6, we measured its expression in some urothelial cancer cell lines and tissues. HDAC10 expression varied among cell lines, too, but did not correlate with HDAC6 expression. In tumor tissues, HDAC10 was overexpressed. During HDAC6 knockdown HDAC10 expression remained unchanged. These data suggest that HDAC10 does not compensate for low HDAC6 expression in a similar fashion as reported for the class I enzymes HDAC1 and HDAC2.Citation48,Citation49
Although a number of HDAC6-specific inhibitors have been developed for research purposes, no such compound has yet become approved for clinical applications. Among the three HDAC6-specific inhibitors Tubacin, Tubastatin A, and ST-80, Tubacin appeared to be most potent exerting effects on tubulin acetylation in urothelial carcinoma cells starting already from a dose of 2.5 µM. The increase in tubulin acetylation appeared dependent on the cellular HDAC6 expression level. Tubacin also appeared to constitute the most specific HDAC6 inhibitor by its lack of effect on overall histone acetylation. Our data on ST-80 in this respect concur with those on leukemia cell lines, where ST-80 also induced histone acetylation from concentrations above 5 µM.Citation50 To our knowledge there is no data available on inhibitory effects of commonly used HDAC6-specific inhibitors on HDAC10. Newly developed compound as such as HPOB or C1A are however reported to be selective for HDAC6 compared with HDAC10.Citation24,Citation51
Subsequently, we compared the three compounds regarding their effects on the viability of urothelial cancer cell lines. In parallel with the data on the molecular effects, ST-80 appeared as the least potent compound with little efficacy even at high micromolar concentrations (100 µM). Again, Tubacin was the most potent compound (IC50 between 6.6 and 12.0 µM). We found that cell lines with higher HDAC6 expression levels were in general less sensitive toward treatment than cell lines with low expression levels. However, since UM-UC-3 cells with high protein expression were the most sensitive (IC50 6.6 µM), HDAC6 protein expression as such does not allow a perfect prediction of therapy response to HDAC6-specific inhibition.
Flow cytometry analyses revealed increased cell death, but no significant effects on cell cycle distribution across UC cell lines subsequent to treatment with 10 µM Tubacin. The observed induction of apoptosis may be explained by the finding that HDAC6 normally inhibits apoptosis by regulating the balance between BCL2 and BAX.Citation52,Citation53 In liver cancer, HDAC6 did not affect proliferation, too, but regulated autophagy.Citation38 Similarly, the newly developed HDAC6-specific inhibitor C1A generated a significant growth inhibitory effect across a number of different cancer cell lines by induction of apoptosis rather than cell cycle arrest, in contrast to the effect of the pan-HDAC inhibitor SAHA.Citation51 Our findings showing only a moderate induction of apoptosis after inhibition of enzyme activity with even higher concentrations of Tubacin, but no induction of apoptosis after diminution of protein expression by siRNA knockdown, indicate that neither HDAC6 protein expression nor enzymatic activity are critical for urothelial carcinoma cells in general. However, some cell lines with low HDAC6 expression levels appeared to be more sensitive to HDAC6 inhibitors than others, suggesting that the cells require a critical level of enzyme activity that was undercut by the treatment. Thus, it could be predicted that patients with moderate or reduced HDAC6 expression would in general have a better chance to benefit from targeted therapy.
Interestingly, UC cell lines do not seem to be more resistant to HDAC6 inhibitors than some other cancer types. For instance, the IC50 for Tubacin in ovarian cancer cells ranged between 10 and 15 µM.Citation11 In combination therapy with a proteasome inhibitor the authors still used 5 µM of the novel inhibitor NK84. A recent study on a single novel bladder cancer cell line (BLX-211) used 100 µM Tubacin to obtain effects on colony formation and migratory capacity.Citation54 When we studied the effect of HDAC6 inhibition on cell motility by treatment with IC50 doses of Tubacin (5–10 µM) we did not observe significant changes in cell migration. The enzyme has been shown to localize to motile, protrusive structures at the leading edge of cells, which we also observed for UC cell lines by immunocytochemical stainings. HDAC6 overexpression promoted chemotactic movement of fibroblasts and lymphocytes, suggesting that HDAC6-mediated tubulin deacetylation regulates microtubule-dependent cell motility.Citation55,Citation56 Nevertheless, there is contradictory data on the importance of HDAC6 in regulation of cellular motility (reviewed in ref. Citation57). It has therefore been proposed that HDAC6 expression per se but not its tubulin deacetylase activity is important for its effect on cell migrationCitation56 and that these effects may be mediated by downstream factors like HSP90.Citation58
As high micromolar drug concentrations are impractical for clinical applications, we sought to find suitable combination regimens that would allow a dose reduction through synergistic effects. HDAC6 is involved in aggresome function, a cellular stress management system induced by misfolded proteins.Citation8 Increased proliferation rates in cancer have been shown to be associated with increased ubiquitin-proteasome-system (UPS) stress and to be targetable in various cancers (ovarian cancer see ref. Citation11, multiple myeloma see ref. Citation59, pancreatic cancer see ref. Citation60). Thus, simultaneous inhibition of the proteasome system by bortezomib and HDAC6 has been shown to act synergistically.Citation59,Citation61,Citation62 A second strategy for combination therapy is based on the fact that HSP90 is an HDAC6 substrate. Inhibition or depletion of HDAC6 has been reported to induce hyperacetylation of HSP90 subsequently inhibiting HSP90 chaperone function. Inhibiting HSP90 function leads to degradation of client proteins, many of which exert oncogenic functions.Citation12,Citation63 Like geldanamycin, its derivate 17-AAG inhibits the chaperone function of HSP90 and its binding to HSP90 is increased by HDAC6 inhibition. Synergistic effects of combined HSP90 and HDAC6 inhibition have been shown for example in leukemia cells at similar doses to those used in our study.Citation63 Our finding concerning single treatment of UC cell lines with 17-AAG were in excellent accord with a previous study.Citation13 While confirming the activity of 17-AAG as such, we observed only minor synergistic effects of Tubacin and 17-AAG on UC cell lines, independent of their HDAC6 expression levels. Similarly, treatment with Tubacin did not enhance the sensitivity of UC cells to the DNA-damaging anticancer drugs doxorubicin and etoposide, unlike glioblastoma, prostate, mammary, and lung cancer cell lines.Citation23,Citation24 The minor effects would not allow to reduce concentrations to a minimum that could be administered in the clinic. Similarly, we did not observe any synergistic effects by combined treatment with Tubacin and heat shock.
Although we had experimentally determined compound concentrations that were in accordance with published data from other cancer types,Citation11,Citation59 a combined treatment of UC cells with Tubacin and the proteasome inhibitor bortezomib did not result in consistent synergistic effects among cell lines, too. Analogous results were obtained for MDA-MB-231 breast cancer cells with similar concentrations.Citation64 Nevertheless, the limited efficacy of combined treatment in urothelial carcinoma compared with other tumor types may be explained by tissue-dependent differences in proliferation rates and associated cellular stress caused by misfolded proteins. Our own observations and those published by Karkoulis et al.Citation13 suggest that UPS stress might play a minor role in urothelial carcinogenesis. However, there is little data on this question available in the literature to date.
In summary, we conclude that urothelial carcinoma patients with normal or low HDAC6 expression might theoretically benefit from treatment with the HDAC6-specific inhibitor Tubacin, but not at practical drug concentrations. With five different potentially interacting treatments, heat shock, HSP90 and proteasomal inhibition as well as induction of DNA-damage with two compounds, no definitive synergistic effects were observed that might overcome this obstacle. Moreover, although cell lines and presumably tumors with low HDAC6 expression may in general be more responsive to the drug, HDAC6 expression as such was not a perfect predictive marker for sensitivity to HDAC6 inhibition. Overall, urothelial cancer may not critically dependent on HDAC6 function. Indeed, various comparisons between Tubacin and the pan-inhibitor SAHA showed lower micromolar concentrations of SAHA to be more efficient in reducing cell viability than Tubacin even at higher concentrations. Thus, our data do not recommend HDAC6 as a good target for therapy of urothelial carcinoma.
Materials and Methods
Patients and tissues
For gene expression the bladder cancer and benign tissue samples were a subset of those described in previous studiesCitation65 comprising 10 benign bladder tissues and 19 bladder cancer tissues from patients with ages ranging from 46 to 86 y (median age: 68 y). The tumor stages and gradings according to the current UICC classification were as follows—pTa G2 in 3 cases, pT2 G3 in 2 cases, pT3 G2 in 2 cases, pT3 G3 in 9 cases, and 3 cases of pT4 G3 tumors. Tissue samples were collected with informed consent of patients. The study was approved by the ethics committee of the medical faculty of the Heinrich Heine University, Study Number 3836.
Cell culture and in vitro exposure of cells to drugs
Cell lines used were provided by Dr MA Knowles (Leeds, UK), Dr J Fogh (New York, USA), Dr Barton Grossman (Houston, USA), or by the DSMZ (Braunschweig, Germany). The well-differentiated urothelial carcinoma cell line BC61 was derived from a papillary bladder cancer in our lab and cultured as previously described.Citation66 Primary urothelial cells (UEC) were prepared from ureters after nephrectomy and were routinely maintained in keratinocyte serum-free medium (Invitrogen) supplemented with 12.5 µg/mL bovine pituitary extract and 0.25 ng/mL epidermal growth factor as described.Citation67 All cell lines were maintained in 75 cm2 cell culture flasks at 37 °C and 5% CO2 in Dulbecco’s modified Eagle medium (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum.
Experiments with drugs were performed 24 h after seeding by exposing the cells to a single dose of the HDAC6 inhibitors Tubacin (Enzo Life Sciences, BML-GR362), ST-80 (see ref. Citation19), or Tubastatin A (Sigma-Aldrich, SML0044), the proteasome inhibitor bortezomib (Selleck Chemicals, S1013), the HSP90 inhibitor 17-AAG (InvivoGen, ant-agl), the pan- HDAC inhibitor SAHA (Cayman Chemicals, 1009929), doxorubicin (Calbiochem, 324389), or etoposide (Calbiochem, 341205) dissolved in DMSO at the indicated concentrations. Control cells were treated with DMSO only.
Transfection with siRNA
For transfection, cells were plated at 2.5 × 105 cells per well in 6-well plates and incubated at 37 °C in an atmosphere of 5% CO2. On the next day, cells were transfected with 10 nM HDAC6 ON-TARGETplus SMARTpool siRNA (J-003499-08, Thermo Scientific- Dharmacon) or an ON_TARGETplus Non-targeting Pool (D-001810-10-05, Thermo Scientific-Dharmacon) using Lipofectamine RNAiMAX (Invitrogen), according to the manufacturer’s recommendations. Cells were then incubated for 72 h before use for further experiments.
Cell viability assay
Cell viability and cell proliferation were measured using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega). This assay measures total cellular ATP as an indicator of viable cells. Human bladder cancer cells were seeded at a density of 3 × 103 (639-V, VM-CUB1, BFTC-905, UM-UC-3, T-24, and 253J) or 4 × 103 (RT-112 and 5637) cells per well into 96-well plates and grown for 24 h. Cells were treated with the indicated drug concentrations or DMSO and further incubated for 48 or 72 h. Cells were incubated with CellTiter-Glo Reagent according to manufacturer’s instructions and luminescence was measured by means of a Wallac Victor 1420 Multilabel Counter (PerkinElmer).
Migration assay
Motility of urothelial cancer cells was determined by wound healing assays using Ibidi Culture-Inserts (Ibidi, 80209). Cells were pretreated with the respective IC50 doses of Tubacin for 24 h or left untreated and then seeded into cell culture inserts with equal cell numbers across all samples. After removing the inserts distance measurements were performed at the indicated time points by means of microscopy and Nikon NIS- Elements Software.
RNA expression analysis
Total cellular mRNA was isolated by using the TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. For RT-PCR, 1 μg total RNA was reversed transcribed using the QuantiTect Reverse Transcription Kit (Qiagen), but with an extended incubation time of 30 min at 42 °C. QRT-PCR was performed with QuantiTect SYBR Green RT-PCR Kit (Qiagen) according to the manufacturer’s instructions. To determine expression levels of HDAC6 (QT00002709) and HDAC10 (QT00007252) we used QuantiTect Primer assays (Qiagen). Expression of the housekeeping gene TATA-box binding protein (TBP) was determined with the following primers (forward: 5′-ACAACAGCCT GCCACCTTA-3′; reverse: 5′-GAATAGGCTG TGGGTCAGT-3′). QRT-PCR was performed using an initial denaturation at 95 °C for 15 min and 45 cycles of amplification including denaturation at 94 °C for 15 s, annealing at 55 °C at 30 s and elongation at 72 °C for 30 s. Reactions were performed with ABI PRISM® 7900 HT (Life Technologies). TBP was used as a reference gene. Technical duplicates had less than 10% difference.
Western blotting
Western blot analysis was used for evaluating protein expression. Total protein was extracted by lysing the cells in a buffer containing 150 mM NaCl, 1% Triton X-100, 0.5% desoxycholate, 1% Nonidet P-40, 0.1% SDS, 1 mM EDTA, 50 mM Tris (pH 7.6), and protease inhibitor cocktail (10 µL/mL, P-8340, Sigma-Aldrich) for 30 min on ice.
Protein concentration was determined by BCA protein assay (Pierce) and samples were separated in SDS-page gels and transferred to PVDF membranes (Millipore). The membrane was blocked with 5% non-fat milk in TBS-T (150 mM NaCl, 10 mM Tris, pH 7.4, and 0.1% Tween-20), washed and then probed with primary antibodies. Antibodies included a rabbit polyclonal antibody to HDAC6 (H-300, Santa Cruz, 1:5000), mouse monoclonal antibodies to HDAC10 (D9, Santa Cruz, 1:500) and α-Tubulin (T-5168, Sigma-Aldrich, 1:50 000). Further antibodies were used against acetylated Tubulin (T-7451, Sigma-Aldrich, 1:17 000), HSP70 (06-468, Upstate, 1:2000), E2F1 (sc-251, Santa Cruz, 1:1000), phosphorylated ERK (4370, Cell Signaling, 1:1000), total ERK (9102, Cell Signaling, 1:500), HSP70 (06-468, Upstate, 1:2000), HSP90 (F-8, Santa Cruz, 1:2000), HIF1α (ab2185, Abcam, 1:1000), and cleaved PARP (9541, Cell Signaling, 1:1000). The membrane was incubated with the primary antibody at room temperature for 1 h or at 4 °C overnight. After washing, the membrane was incubated with the suitable horseradish peroxidase-conjugated secondary antibody (Santa Cruz) for 1 h and exposed using an ECL™ Advanced Western Blotting Detection Kit (Amersham) according to the manufacturer’s recommendations.
Immunofluorescence stainings
Cells were seeded onto glass slides and allowed to grow for 24 h. Cells were washed and fixed with 4% formaldehyde in PBS for 30 min at room temperature. Cells were washed twice with PBS before being incubated with 0.5% Triton X-100 for 3 min for cell permeabilization. After washing, slides were blocked with 1% BSA in PBS for 30 min. Primary and FITC-conjugated anti-rabbit secondary antibodies to HDAC6 were diluted 1:100 and 1:50 and incubated for 1 h each. Nuclear staining was achieved by administration of DAPI (1 µg/mL) for 5 min. Images were taken on Nikon Eclipse 400 microscope (Nikon) with the NIS-Elements D2-30 software.
Flow cytometry
For flow cytometry, 5 × 105 cells were seeded in 6 cm-dishes and incubated for 24 h. Cells were treated with 10 µM Tubacin or DMSO. In another experiment, cells were transfected with HDAC6 siRNA or a non-targeting control. After 48 h, attached cells and cells in the supernatant were collected. For assessing apoptotic cell death and necrosis, cells were incubated with Annexin V-FITC (Immunotools), Annexin V binding buffer and propidium iodide (2 µg/ mL in PBS, Sigma-Aldrich). Cell cycle was analyzed by staining the cells with a buffer containing 0.1% sodium citrate, 0.1% Triton X-100 and 50 µg/mL of propidium iodide.Citation68 Cells were analyzed by means of a FACSCalibur (BD Biosciences) and CellQuest II software.
Histone extraction
Histones were extracted for detection of acetylation levels according to a published protocolCitation69 by means of TCA-precipitation. Extract concentration was determined by BCA protein assay (Pierce) and 1 µg of each samples was separated in 15% SDS-Page gels and transferred to PVDF membranes (Millipore) according to the aforementioned western blotting protocol. We detected acetylated histone H3 (1:2000, Active Motif 39139) and total histone H3 (1:1000, Cell Signaling 3638).
Statistical analysis
Statistical analyses were performed using SPSS 18 (SPSS) and Prism 5 (GraphPad Software). Significance was measured by the no-parametric Mann–Whitney U test and Student t test. Differences in viability after treatment with Tubacin and/or 17-AAG/bortezomib were compared with untreated controls by Bonferroni posttest. P values of <0.05 were considered as significant. Dose-response curves and IC50 values were approximated by nonlinear regression analysis using SigmaPlot 12 (Systat Software Inc.).
| Abbreviations: | ||
| DAPI | = | 4′, 6-diamidine-2-phenylindol |
| FITC | = | fluorescein isothiocyanate |
| HDAC | = | histone deacetylase |
| HSP90 | = | heat shock protein 90 |
| TCA | = | trichloroacetic acid |
| UEC | = | normal uroepithelial cells (proliferating in cell culture) |
| UPS | = | ubiquitin-proteasome-system |
| UC | = | urothelial cancer |
| PVDF | = | polyvinylidene fluoride |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The study was in part funded by a grant of the German Research Foundation to G.N. (NIE 1398/1-1). The authors thank Marija Lenzen for experimental support.
References
- von der Maase H, Sengelov L, Roberts JT, Ricci S, Dogliotti L, Oliver T, Moore MJ, Zimmermann A, Arning M. Long-term survival results of a randomized trial comparing gemcitabine plus cisplatin, with methotrexate, vinblastine, doxorubicin, plus cisplatin in patients with bladder cancer. J Clin Oncol 2005; 23:4602 - 8; http://dx.doi.org/10.1200/JCO.2005.07.757; PMID: 16034041
- Sternberg CN, Bellmunt J, Sonpavde G, Siefker-Radtke AO, Stadler WM, Bajorin DF, Dreicer R, George DJ, Milowsky MI, Theodorescu D, et al, International Consultation on Urologic Disease-European Association of Urology Consultation on Bladder Cancer 2012. ICUD-EAU International Consultation on Bladder Cancer 2012: Chemotherapy for urothelial carcinoma-neoadjuvant and adjuvant settings. Eur Urol 2013; 63:58 - 66; http://dx.doi.org/10.1016/j.eururo.2012.08.010; PMID: 22917984
- Butler JS, Koutelou E, Schibler AC, Dent SY. Histone-modifying enzymes: regulators of developmental decisions and drivers of human disease. Epigenomics 2012; 4:163 - 77; http://dx.doi.org/10.2217/epi.12.3; PMID: 22449188
- Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell 2012; 150:12 - 27; http://dx.doi.org/10.1016/j.cell.2012.06.013; PMID: 22770212
- Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S, Wu R, Chen C, Li X, Zhou L, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet 2011; 43:875 - 8; http://dx.doi.org/10.1038/ng.907; PMID: 21822268
- Ahmad M, Hamid A, Hussain A, Majeed R, Qurishi Y, Bhat JA, Najar RA, Qazi AK, Zargar MA, Singh SK, et al. Understanding histone deacetylases in the cancer development and treatment: an epigenetic perspective of cancer chemotherapy. DNA Cell Biol 2012; 31:Suppl 1 S62 - 71; http://dx.doi.org/10.1089/dna.2011.1575; PMID: 22462686
- Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer 2001; 1:194 - 202; http://dx.doi.org/10.1038/35106079; PMID: 11902574
- Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003; 115:727 - 38; http://dx.doi.org/10.1016/S0092-8674(03)00939-5; PMID: 14675537
- Li Y, Shin D, Kwon SH. Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J 2013; 280:775 - 93; PMID: 23181831
- Boyault C, Sadoul K, Pabion M, Khochbin S. HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene 2007; 26:5468 - 76; http://dx.doi.org/10.1038/sj.onc.1210614; PMID: 17694087
- Bazzaro M, Lin Z, Santillan A, Lee MK, Wang MC, Chan KC, Bristow RE, Mazitschek R, Bradner J, Roden RB. Ubiquitin proteasome system stress underlies synergistic killing of ovarian cancer cells by bortezomib and a novel HDAC6 inhibitor. Clin Cancer Res 2008; 14:7340 - 7; http://dx.doi.org/10.1158/1078-0432.CCR-08-0642; PMID: 19010849
- Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, Rocha K, Kumaraswamy S, Boyapalle S, Atadja P, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem 2005; 280:26729 - 34; http://dx.doi.org/10.1074/jbc.C500186200; PMID: 15937340
- Karkoulis PK, Stravopodis DJ, Margaritis LH, Voutsinas GE. 17-Allylamino-17-demethoxygeldanamycin induces downregulation of critical Hsp90 protein clients and results in cell cycle arrest and apoptosis of human urinary bladder cancer cells. BMC Cancer 2010; 10:481; http://dx.doi.org/10.1186/1471-2407-10-481; PMID: 20828379
- Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, Chiao JH, Reilly JF, Ricker JL, Richon VM, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007; 109:31 - 9; http://dx.doi.org/10.1182/blood-2006-06-025999; PMID: 16960145
- Luu TH, Morgan RJ, Leong L, Lim D, McNamara M, Portnow J, Frankel P, Smith DD, Doroshow JH, Wong C, et al. A phase II trial of vorinostat (suberoylanilide hydroxamic acid) in metastatic breast cancer: a California Cancer Consortium study. Clin Cancer Res 2008; 14:7138 - 42; http://dx.doi.org/10.1158/1078-0432.CCR-08-0122; PMID: 18981013
- Kapoor S. Inhibition of HDAC6-dependent carcinogenesis: emerging, new therapeutic options besides belinostat. Int J Cancer 2009; 124:509; http://dx.doi.org/10.1002/ijc.23975; PMID: 18942705
- Butler KV, Kalin J, Brochier C, Vistoli G, Langley B, Kozikowski AP. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J Am Chem Soc 2010; 132:10842 - 6; http://dx.doi.org/10.1021/ja102758v; PMID: 20614936
- Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci U S A 2003; 100:4389 - 94; http://dx.doi.org/10.1073/pnas.0430973100; PMID: 12677000
- Scott GK, Marx C, Berger CE, Saunders LR, Verdin E, Schäfer S, Jung M, Benz CC. Destabilization of ERBB2 transcripts by targeting 3′ untranslated region messenger RNA associated HuR and histone deacetylase-6. Mol Cancer Res 2008; 6:1250 - 8; http://dx.doi.org/10.1158/1541-7786.MCR-07-2110; PMID: 18644987
- Niegisch G, Knievel J, Koch A, Hader C, Fischer U, Albers P, Schulz WA. Changes in histone deacetylase (HDAC) expression patterns and activity of HDAC inhibitors in urothelial cancers. Urol Oncol 2013; 31:1770 - 9; http://dx.doi.org/10.1016/j.urolonc.2012.06.015; PMID: 22944197
- Garg M, Kanojia D, Seth A, Kumar R, Gupta A, Surolia A, Suri A. Heat-shock protein 70-2 (HSP70-2) expression in bladder urothelial carcinoma is associated with tumour progression and promotes migration and invasion. Eur J Cancer 2010; 46:207 - 15; http://dx.doi.org/10.1016/j.ejca.2009.10.020; PMID: 19914824
- Yu HJ, Chang YH, Pan CC. Prognostic significance of heat shock proteins in urothelial carcinoma of the urinary bladder. Histopathology 2013; 62:788 - 98; http://dx.doi.org/10.1111/his.12087; PMID: 23425286
- Namdar M, Perez G, Ngo L, Marks PA. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc Natl Acad Sci U S A 2010; 107:20003 - 8; http://dx.doi.org/10.1073/pnas.1013754107; PMID: 21037108
- Lee J-H, Mahendran A, Yao Y, Ngo L, Venta-Perez G, Choy ML, Kim N, Ham W-S, Breslow R, Marks PA. Development of a histone deacetylase 6 inhibitor and its biological effects. Proc Natl Acad Sci U S A 2013; 110:15704 - 9; http://dx.doi.org/10.1073/pnas.1313893110; PMID: 24023063
- Modesitt SC, Sill M, Hoffman JS, Bender DP, Gynecologic Oncology Group. A phase II study of vorinostat in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol 2008; 109:182 - 6; http://dx.doi.org/10.1016/j.ygyno.2008.01.009; PMID: 18295319
- Arsenault D, Brochu-Gaudreau K, Charbonneau M, Dubois CM. HDAC6 deacetylase activity is required for hypoxia-induced invadopodia formation and cell invasion. PLoS One 2013; 8:e55529; http://dx.doi.org/10.1371/journal.pone.0055529; PMID: 23405166
- Ding G, Liu HD, Huang Q, Liang HX, Ding ZH, Liao ZJ, Huang G. HDAC6 promotes hepatocellular carcinoma progression by inhibiting P53 transcriptional activity. FEBS Lett 2013; 587:880 - 6; http://dx.doi.org/10.1016/j.febslet.2013.02.001; PMID: 23402884
- Park SY, Jun JA, Jeong KJ, Heo HJ, Sohn JS, Lee HY, Park CG, Kang J. Histone deacetylases 1, 6 and 8 are critical for invasion in breast cancer. Oncol Rep 2011; 25:1677 - 81; PMID: 21455583
- Zhang Z, Yamashita H, Toyama T, Sugiura H, Omoto Y, Ando Y, Mita K, Hamaguchi M, Hayashi S, Iwase H. HDAC6 expression is correlated with better survival in breast cancer. Clin Cancer Res 2004; 10:6962 - 8; http://dx.doi.org/10.1158/1078-0432.CCR-04-0455; PMID: 15501975
- Sakuma T, Uzawa K, Onda T, Shiiba M, Yokoe H, Shibahara T, Tanzawa H. Aberrant expression of histone deacetylase 6 in oral squamous cell carcinoma. Int J Oncol 2006; 29:117 - 24; PMID: 16773191
- Bradbury CA, Khanim FL, Hayden R, Bunce CM, White DA, Drayson MT, Craddock C, Turner BM. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia 2005; 19:1751 - 9; http://dx.doi.org/10.1038/sj.leu.2403910; PMID: 16121216
- Lee JS, Leem SH, Lee SY, Kim SC, Park ES, Kim SB, Kim SK, Kim YJ, Kim WJ, Chu IS. Expression signature of E2F1 and its associated genes predict superficial to invasive progression of bladder tumors. J Clin Oncol 2010; 28:2660 - 7; http://dx.doi.org/10.1200/JCO.2009.25.0977; PMID: 20421545
- Dyrskjøt L, Kruhøffer M, Thykjaer T, Marcussen N, Jensen JL, Møller K, Ørntoft TF. Gene expression in the urinary bladder: a common carcinoma in situ gene expression signature exists disregarding histopathological classification. Cancer Res 2004; 64:4040 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-03-3620; PMID: 15173019
- Dyrskjot L, Zieger K, Orntoft TF. Recent advances in high-throughput molecular marker identification for superficial and invasive bladder cancers. Front Biosci 2007; 12:2063 - 73; http://dx.doi.org/10.2741/2211; PMID: 17127444
- Mengual L, Burset M, Ars E, Lozano JJ, Villavicencio H, Ribal MJ, Alcaraz A. DNA microarray expression profiling of bladder cancer allows identification of noninvasive diagnostic markers. J Urol 2009; 182:741 - 8; http://dx.doi.org/10.1016/j.juro.2009.03.084; PMID: 19539325
- Sanchez-Carbayo M, Socci ND, Lozano J, Saint F, Cordon-Cardo C. Defining molecular profiles of poor outcome in patients with invasive bladder cancer using oligonucleotide microarrays. J Clin Oncol 2006; 24:778 - 89; http://dx.doi.org/10.1200/JCO.2005.03.2375; PMID: 16432078
- Kanno K, Kanno S, Nitta H, Uesugi N, Sugai T, Masuda T, Wakabayashi G, Maesawa C. Overexpression of histone deacetylase 6 contributes to accelerated migration and invasion activity of hepatocellular carcinoma cells. Oncol Rep 2012; 28:867 - 73; PMID: 22766642
- Jung KH, Noh JH, Kim JK, Eun JW, Bae HJ, Chang YG, Kim MG, Park WS, Lee JY, Lee SY, et al. Histone deacetylase 6 functions as a tumor suppressor by activating c-Jun NH2-terminal kinase-mediated beclin 1-dependent autophagic cell death in liver cancer. Hepatology 2012; 56:644 - 57; http://dx.doi.org/10.1002/hep.25699; PMID: 22392728
- Simon D, Laloo B, Barillot M, Barnetche T, Blanchard C, Rooryck C, Marche M, Burgelin I, Coupry I, Chassaing N, et al. A mutation in the 3′-UTR of the HDAC6 gene abolishing the post-transcriptional regulation mediated by hsa-miR-433 is linked to a new form of dominant X-linked chondrodysplasia. Hum Mol Genet 2010; 19:2015 - 27; http://dx.doi.org/10.1093/hmg/ddq083; PMID: 20181727
- Chen Y, Sawyers CL, Scher HI. Targeting the androgen receptor pathway in prostate cancer. Curr Opin Pharmacol 2008; 8:440 - 8; http://dx.doi.org/10.1016/j.coph.2008.07.005; PMID: 18674639
- Riolo MT, Cooper ZA, Holloway MP, Cheng Y, Bianchi C, Yakirevich E, Ma L, Chin YE, Altura RA. Histone deacetylase 6 (HDAC6) deacetylates survivin for its nuclear export in breast cancer. J Biol Chem 2012; 287:10885 - 93; http://dx.doi.org/10.1074/jbc.M111.308791; PMID: 22334690
- Kaluza D, Kroll J, Gesierich S, Yao TP, Boon RA, Hergenreider E, Tjwa M, Rössig L, Seto E, Augustin HG, et al. Class IIb HDAC6 regulates endothelial cell migration and angiogenesis by deacetylation of cortactin. EMBO J 2011; 30:4142 - 56; http://dx.doi.org/10.1038/emboj.2011.298; PMID: 21847094
- Lafarga V, Aymerich I, Tapia O, Mayor F Jr., Penela P. A novel GRK2/HDAC6 interaction modulates cell spreading and motility. EMBO J 2012; 31:856 - 69; http://dx.doi.org/10.1038/emboj.2011.466; PMID: 22193721
- Liu Y, Peng L, Seto E, Huang S, Qiu Y. Modulation of histone deacetylase 6 (HDAC6) nuclear import and tubulin deacetylase activity through acetylation. J Biol Chem 2012; 287:29168 - 74; http://dx.doi.org/10.1074/jbc.M112.371120; PMID: 22778253
- Oehme I, Lodrini M, Brady NR, Witt O. Histone deacetylase 10-promoted autophagy as a druggable point of interference to improve the treatment response of advanced neuroblastomas. Autophagy 2013; 9:2163 - 5; http://dx.doi.org/10.4161/auto.26450; PMID: 24145760
- Song C, Zhu S, Wu C, Kang J. Histone deacetylase (HDAC) 10 suppresses cervical cancer metastasis through inhibition of matrix metalloproteinase (MMP) 2 and 9 expression. J Biol Chem 2013; 288:28021 - 33; http://dx.doi.org/10.1074/jbc.M113.498758; PMID: 23897811
- Van Damme M, Crompot E, Meuleman N, Mineur P, Bron D, Lagneaux L, Stamatopoulos B. HDAC isoenzyme expression is deregulated in chronic lymphocytic leukemia B-cells and has a complex prognostic significance. Epigenetics 2012; 7:1403 - 12; http://dx.doi.org/10.4161/epi.22674; PMID: 23108383
- Wilting RH, Yanover E, Heideman MR, Jacobs H, Horner J, van der Torre J, DePinho RA, Dannenberg JH. Overlapping functions of Hdac1 and Hdac2 in cell cycle regulation and haematopoiesis. EMBO J 2010; 29:2586 - 97; http://dx.doi.org/10.1038/emboj.2010.136; PMID: 20571512
- Oehme I, Linke JP, Böck BC, Milde T, Lodrini M, Hartenstein B, Wiegand I, Eckert C, Roth W, Kool M, et al. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc Natl Acad Sci U S A 2013; 110:E2592 - 601; http://dx.doi.org/10.1073/pnas.1300113110; PMID: 23801752
- Hackanson B, Rimmele L, Benkißer M, Abdelkarim M, Fliegauf M, Jung M, Lübbert M. HDAC6 as a target for antileukemic drugs in acute myeloid leukemia. Leuk Res 2012; 36:1055 - 62; http://dx.doi.org/10.1016/j.leukres.2012.02.026; PMID: 22464548
- Kaliszczak M, Trousil S, Åberg O, Perumal M, Nguyen QD, Aboagye EO. A novel small molecule hydroxamate preferentially inhibits HDAC6 activity and tumour growth. Br J Cancer 2013; 108:342 - 50; http://dx.doi.org/10.1038/bjc.2012.576; PMID: 23322205
- Qin HX, Cui HK, Pan Y, Yang J, Ren YF, Hua CH, Hua FF, Qiao YH. HDAC6 siRNA inhibits proliferation and induces apoptosis of HeLa cells and its related molecular mechanism. Asian Pac J Cancer Prev 2012; 13:3367 - 71; http://dx.doi.org/10.7314/APJCP.2012.13.7.3367; PMID: 22994762
- Subramanian C, Jarzembowski JA, Opipari AW Jr., Castle VP, Kwok RP. HDAC6 deacetylates Ku70 and regulates Ku70-Bax binding in neuroblastoma. Neoplasia 2011; 13:726 - 34; PMID: 21847364
- Zuo Q, Wu W, Li X, Zhao L, Chen W. HDAC6 and SIRT2 promote bladder cancer cell migration and invasion by targeting cortactin. Oncol Rep 2012; 27:819 - 24; PMID: 22089141
- Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang X-F, Yao T-P. HDAC6 is a microtubule-associated deacetylase. Nature 2002; 417:455 - 8; http://dx.doi.org/10.1038/417455a; PMID: 12024216
- Cabrero JR, Serrador JM, Barreiro O, Mittelbrunn M, Naranjo-Suárez S, Martín-Cófreces N, Vicente-Manzanares M, Mazitschek R, Bradner JE, Ávila J, et al. Lymphocyte chemotaxis is regulated by histone deacetylase 6, independently of its deacetylase activity. Mol Biol Cell 2006; 17:3435 - 45; http://dx.doi.org/10.1091/mbc.E06-01-0008; PMID: 16738306
- Valenzuela-Fernández A, Cabrero JR, Serrador JM, Sánchez-Madrid F. HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol 2008; 18:291 - 7; http://dx.doi.org/10.1016/j.tcb.2008.04.003; PMID: 18472263
- Gao YS, Hubbert CC, Lu J, Lee YS, Lee JY, Yao TP. Histone deacetylase 6 regulates growth factor-induced actin remodeling and endocytosis. Mol Cell Biol 2007; 27:8637 - 47; http://dx.doi.org/10.1128/MCB.00393-07; PMID: 17938201
- Hideshima T, Bradner JE, Wong J, Chauhan D, Richardson P, Schreiber SL, Anderson KC. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci U S A 2005; 102:8567 - 72; http://dx.doi.org/10.1073/pnas.0503221102; PMID: 15937109
- Nawrocki ST, Carew JS, Pino MS, Highshaw RA, Andtbacka RH, Dunner K Jr., Pal A, Bornmann WG, Chiao PJ, Huang P, et al. Aggresome disruption: a novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cells. Cancer Res 2006; 66:3773 - 81; http://dx.doi.org/10.1158/0008-5472.CAN-05-2961; PMID: 16585204
- Boyault C, Zhang Y, Fritah S, Caron C, Gilquin B, Kwon SH, Garrido C, Yao TP, Vourc’h C, Matthias P, et al. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev 2007; 21:2172 - 81; http://dx.doi.org/10.1101/gad.436407; PMID: 17785525
- Santo L, Hideshima T, Kung AL, Tseng JC, Tamang D, Yang M, Jarpe M, van Duzer JH, Mazitschek R, Ogier WC, et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012; 119:2579 - 89; http://dx.doi.org/10.1182/blood-2011-10-387365; PMID: 22262760
- Rao R, Fiskus W, Yang Y, Lee P, Joshi R, Fernandez P, Mandawat A, Atadja P, Bradner JE, Bhalla K. HDAC6 inhibition enhances 17-AAG--mediated abrogation of hsp90 chaperone function in human leukemia cells. Blood 2008; 112:1886 - 93; http://dx.doi.org/10.1182/blood-2008-03-143644; PMID: 18591380
- Komatsu S, Moriya S, Che XF, Yokoyama T, Kohno N, Miyazawa K. Combined treatment with SAHA, bortezomib, and clarithromycin for concomitant targeting of aggresome formation and intracellular proteolytic pathways enhances ER stress-mediated cell death in breast cancer cells. Biochem Biophys Res Commun 2013; 437:41 - 7; http://dx.doi.org/10.1016/j.bbrc.2013.06.032; PMID: 23792097
- Hoffmann MJ, Florl AR, Seifert HH, Schulz WA. Multiple mechanisms downregulate CDKN1C in human bladder cancer. Int J Cancer 2005; 114:406 - 13; http://dx.doi.org/10.1002/ijc.20749; PMID: 15551363
- Koch A, Hatina J, Rieder H, Seifert HH, Huckenbeck W, Jankowiak F, Florl AR, Stoehr R, Schulz WA. Discovery of TP53 splice variants in two novel papillary urothelial cancer cell lines. Cell Oncol (Dordr) 2012; 35:243 - 57; http://dx.doi.org/10.1007/s13402-012-0082-8; PMID: 22669776
- Swiatkowski S, Seifert HH, Steinhoff C, Prior A, Thievessen I, Schliess F, Schulz WA. Activities of MAP-kinase pathways in normal uroepithelial cells and urothelial carcinoma cell lines. Exp Cell Res 2003; 282:48 - 57; http://dx.doi.org/10.1006/excr.2002.5647; PMID: 12490193
- Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods 1991; 139:271 - 9; http://dx.doi.org/10.1016/0022-1759(91)90198-O; PMID: 1710634
- Shechter D, Dormann HL, Allis CD, Hake SB. Extraction, purification and analysis of histones. Nat Protoc 2007; 2:1445 - 57; http://dx.doi.org/10.1038/nprot.2007.202; PMID: 17545981