Abstract
The cellular response to hypoxia relies on the activation of a specific transcriptional program. Although, most of the attention is focused on the transcription factor HIF, other transcription factors are also activated in hypoxia. We have recently described the mechanism for hypoxia induced NFκB. We have demonstrated the crucial dependency on the IKK complex as well as in the upstream IKK kinase TAK1. TAK1 and IKK activation is dependent upon the calcium calmodulin kinase, CaMK2 and requires Ubc13 as the E2 ubiquitin conjugation enzyme. We report a role for XIAP as the possible E3-ubiquitin ligase for this system. Interestingly, hypoxia induced IKK mediated phosphorylation of IκBα, does not lead to degradation. Hypoxia prevents IκBα de-sumoylation of Sumo-2/3 chains on critical lysine residues, normally required for K-48 linked polyubiquitination. Our results define a novel pathway regulating NFκB activation.
Hypoxia or lowering of the oxygen tension below that to which a cell or tissue is accustomed to, mounts a variety of cellular and molecular responses.Citation1,Citation2 The majority of studies have focused on the heterodimeric transcription factor, hypoxia inducible factor (HIF). Molecularly, hypoxia is sensed by a class of dioxygenases that catalyse hydroxylation reactions on the HIF-α subunits, rendering these susceptible to degradation by the proteasome.Citation2 However, a number of additional transcription factors are also activated by hypoxia, among these is the major transcription factor NFκB.Citation2,Citation3
NFκB is the collective name for a family of transcription factors that include: RelA (p65), RelB, c-Rel, NFκB1 (p105/p50) and NFκB2 (p100/p52) (reviewed in Oncogene special editionCitation4). Many possible combinations of homo and heterodimers that act to modulate NFκB DNA binding activity and hence achieve target selectivity; however, the p50/RelA heterodimer is the most commonly studied.
NFκB is held inactive in the cytoplasm by binding to a member of the IκB family of inhibitory proteins, including IκBα, β or ε. NFκB can be considered as a stress responsive transcription factor, as its activation occurs in response to a large range of external stimuli. These include, although not exclusively, inflammatory cytokines such as tumor necrosis factorα (TNFα), bacterial components such as LPS and viral infection (reviewed in ref. Citation5).
There are two major pathways leading to NFκB activation, the canonical or the non-canonical pathway.Citation6 The canonical pathway is activated by external ligands binding to a specific membrane bound receptor, resulting in oligomerization, recruitment of a number of adaptor molecules and ubiquitin-dependent activation of the TAK1-IKK complex.Citation7 IKK then mediates the phosphorylation of IκBα at Ser 32 and 36, which signals it for proteasomal degradation. This results in NFκB dimer release and translocation into the nucleus.
In the non-canonical pathway, ligand-binding results in the activation of NIK, leading to the activation of IKKα. IKKα phosphorylates and mediates the processing of p100 to p52. p52/RelB are then able to translocate into the nucleus and activate target genes.Citation6
Aberrant activation of NFκB, characterised by its constitutive nuclear localization, is associated with many diseases.Citation8,Citation9 Furthermore, NFκB is a regulator of many target genes that control cellular responses such as proliferation, apoptosis, angiogenesis and metastasis.Citation4,Citation10
The fact that hypoxia leads to NFκB activation has been known for over a decade, however, due to the unconventional nature of this stimulus, several studies have proposed diverse mechanisms behind this. In early studies, it was reported that hypoxia induced NFκB was mediated by phosphorylation at a tyrosine residue on IκBα and that this activation did not result in IκB degradation, suggesting an IKK-independent mechanism.Citation11
More recently, the dioxygenases responsible for the control of HIFα, PHDs were suggested to act as direct negative regulators of IKK and hence, hypoxia would promote NFκB activation.Citation12 However, this is not the case for all cell types and organisms as, various cell types depleted of PHDs do not result in activation of NFκB.Citation13
We have found that apart for the strict requirement of the IKK complex for hypoxia induced NFκB, the upstream kinase TAK1 is also necessary. Furthermore, increased levels and activity of TAK1 are sufficient to activate NFκB.Citation13
Activation of TAK1 and IKK has been linked with multiple forms of ubiquitin linkages.Citation7 In fact, research into how TNFα induces NFκB lead to the finding of linear ubiquitin chains.Citation14,Citation15 We could not find any role for the enzymes promoting the linear ubiquitin chains in hypoxia induced IKK. However, we found a role for the E2-ubiquitin ligase, Ubc13. This enzyme promotes specifically K63-ubiquitin linkages to substrates. Ubc13 can bind several E3 ligases such as TRAF6 and XIAP.Citation16,Citation17 Since, our results demonstrated that hypoxia induced IKK-NFκB did not require TRAF6,Citation13 we analysed the role of other possible E3 ligases. Given its connection with TAK1-TAB1 complex as well as with NFκB, we decided to focus on XIAP. We found that the E3-ligase XIAP seems to be required for the IKK activation seen following hypoxia ().
siRNA depletion of XIAP severely inhibits hypoxia induced IKK (). XIAP has previously been shown to bind to TAK1-TAB1 complex and hence promote NFκB activation.Citation18 Gain of function experiments, where XIAP was overexpressed led to increased IKK phosphorylation in normoxia and hypoxia (). Furthermore, using XIAP null mouse embryonic fibroblasts, we observed a severe reduction in the levels of phosphorylated IKK and IκBα (). These results suggest that XIAP is one of the possible E3-ubiquitin ligases activated by hypoxia to induce the NFκB pathway.
We have shown that hypoxia induced NFκB both activates and represses specific target genes, which contribute to the cellular response to hypoxia. Our studies were focused mostly on RelA, but we have also found c-Rel and p52 in DNA binding assays.Citation13 However, at present we have no information on c-Rel or p52 specific target genes activated by hypoxia. Since c-Rel and p52 have the closest links to cancer,Citation19,Citation20 it would therefore be of interest to determine how do these transcription factors contribute to the pathology associated with hypoxia in cancer. Future studies directing at dissecting the role of c-Rel and p52 should provide novel and important information regarding the function of these transcription factors in hypoxia.
Perhaps one of our most intriguing findings was the fact that in hypoxia, IκBα is not ubiquitinated even though, the recognition motif for the SCF-ubiquitin conjugation complex is present. We demonstrated that the lack of IκBα degradation following hypoxia, is due to sumoylation of key ubiquitin accepting lysine residues by Sumo-2/3. Furthermore, modulation of Sumo-2/3 conjugation/deconjugation has a direct impact on NFκB activity. Our data suggests that rather than an active sumo-2/3 conjugation in hypoxia, there is an inhibition of de-sumoylation of IκBα. As such, this implies that hypoxia prevents sumo proteases activity.
The biological functions of the six known sumo proteases (Senps), are only now being uncovered. Thus far only Senp1 and Senp2 deletion in mice have been published. Senp1 null mice have severe defects in the production of red blood cells,Citation21 while Senp2 null mice have defects in heart formation.Citation22 Senp5 and Senp6 reduction by siRNA have showed their requirement for normal cell cycle progression,Citation23,Citation24 while Senp3 function has been associated with rDNA processing and nucleolar function.Citation25 Senp7 is a sumo-2/3 specific peptidase but its function has not been delineated thus far.Citation26
Assessing the activity of sumo peptidases in vivo is at present not possible. However, it would be of interest to determine what is the substrate specificity of these enzymes and in particular how it is changed during the hypoxia response. Future studies investigating the dynamics of these enzymes might prove informative for the elucidation of the biological functions of Senps in hypoxia.
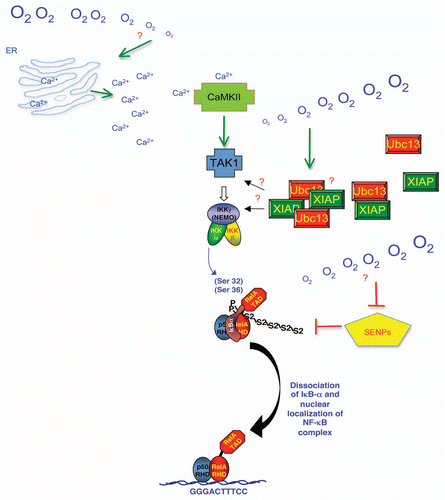
Overall, the mechanism by which hypoxia activates NFκB is very complex (). It involves many of the known players of the NFκB pathway, but also has very many differences. Activation of CamK2 and the K-63 ubiquitin systems are part of the upstream pathway leading to IKK and NFκB activation. Furthermore, inhibition of specific Senps contributes to the lack of IκBα degradation and hence distinguishing hypoxia from cytokine signalling (). Whether Senp inhibition changes the levels of intracellular calcium or leads to activation of the K63-ubiquitin systems is unknown. Further studies are therefore necessary to investigate the connection between all of the pathways activated by hypoxia in the cell.
Abbreviations
| HIF | = | hypoxia inducible factor |
| NFκB | = | nuclear factor kappaB |
| IκBα | = | inhibitor of κBα |
| IKK | = | inhibitor of κBα kinase |
| TAK1 | = | transforming growth factorβ activated kinase 1 |
| TAB1 | = | TAK1 binding protein 1 |
| CamK2 | = | calcium calmodulin-dependent kinase 2 |
| PHDs | = | prolyl-hydroxylases |
| TNFα | = | tumour necrosis factorα |
| siRNA | = | small interfering RNAs |
| XIAP-X | = | linked inhibitor of apoptosis |
| TRAF6 | = | TNF-receptor associated factor 6 |
| Senps | = | sumo/sentrin specific proteases |
Figures and Tables
Figure 1 XIAP is necessary for hypoxia induced IKK activity. (A) U2OS cells were transfected with control and XIAP siRNA oligonucleotides for 48 hours prior to exposure to 1% O2 for the indicated periods of time. Whole cell lysates were analysed by western blot for the levels of the depicted proteins. (B) U2OS cells were transfected with 1 µg of Control and Flag-XIAP expression constructs for 48 hours prior to exposure to 1% O2 for 30 minutes. Whole cell lysates were processes as in (A). (C) Wildtype and XIAP null mouse embryo fibroblasts (MEFs) were exposed to 1% O2 for the indicated periods of time prior to lysis. Whole cell lysates were processed as in (A).
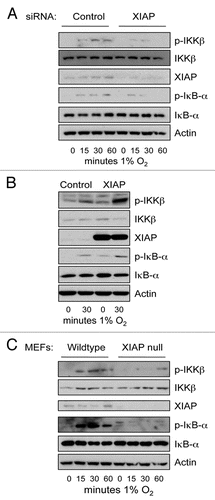
Figure 2 Mechanism of hypoxia induced NFκB. Schematic diagram depicting a model based on our results for the mechanism of hypoxia induced NFκB activity in cells. Upon hypoxia exposure Calcium is released from cellular compartments such as the Endoplasmic Reticulum (ER). This Calcium increase activates CamK2, which leads to the activation of TAK1. TAK-TAB complex are brought to the IKK complex by the K63-ubiquitin chains promoted by Ubc13-XIAP complex leading to IKK activation and hence phosphorylation of IκBα. Hypoxia also promotes the inhibition of specific Senps (5, 6 and 7) leading to increased Sumo-2/3 (S2) chains on IκBα, which promote RelA release, nuclear accumulation and target gene activation and repression. Question marks indicate areas where mechanistic insights are still missing.
Acknowledgements
We would like to thank Prof. Colin Duckett (Michigan, USA) for providing us with the XIAP wildtype and null MEFs as well as the XIAP expression constructs. This study was funded by the Wellcome Trust, the MRC and by a Tenovus Small grant.
References
- Rocha S. Gene regulation under low oxygen: holding your breath for transcription. Trends Biochem Sci 2007; 32:389 - 397
- Kenneth NS, Rocha S. Regulation of gene expression by hypoxia. Biochem J 2008; 414:19 - 29
- Cummins EP, Taylor CT. Hypoxia-responsive transcription factors. Pflugers Arch 2005; 450:363 - 371
- Gilmore TD. Introduction to NFkappaB: players, pathways, perspectives. Oncogene 2006; 25:6680 - 6684
- Pahl HL. Activators and target genes of Rel/NFkappaB transcription factors. Oncogene 1999; 18:6853 - 6866
- Scheidereit C. IkappaB kinase complexes: gateways to NFkappaB activation and transcription. Oncogene 2006; 25:6685 - 6705
- Adhikari A, Xu M, Chen ZJ. Ubiquitin-mediated activation of TAK1 and IKK. Oncogene 2007; 26:3214 - 3226
- Perkins ND. The Rel/NFkappaB family: friend and foe. Trends Biochem Sci 2000; 25:434 - 440
- Perkins ND, Gilmore TD. Good cop, bad cop: the different faces of NFkappaB. Cell Death Differ 2006; 13:759 - 772
- Gilmore TD. The Rel/NFkappaB signal transduction pathway: introduction. Oncogene 1999; 18:6842 - 6844
- Koong AC, Chen EY, Giaccia AJ. Hypoxia causes the activation of nuclear factor kappaB through the phosphorylation of IkappaB alpha on tyrosine residues. Cancer Res 1994; 54:1425 - 1430
- Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F, et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci USA 2006; 103:18154 - 18159
- Culver C, Sundqvist A, Mudie S, Melvin A, Xirodimas D, Rocha S. Mechanism of hypoxiainduced NFkappaB. Mol Cell Biol 2010; 30:4901 - 4921
- Iwai K, Tokunaga F. Linear polyubiquitination: a new regulator of NFkappaB activation. EMBO Rep 2009; 10:706 - 713
- Haas AL. Linear polyubiquitylation: the missing link in NFkappaB signalling. Nat Cell Biol 2009; 11:116 - 118
- Lamothe B, Besse A, Campos AD, Webster WK, Wu H, Darnay BG. Site-specific Lys-63-linked tumor necrosis factor receptor-associated factor 6 autoubiquitination is a critical determinant of IkappaB kinase activation. J Biol Chem 2007; 282:4102 - 4112
- Wu ZH, Wong ET, Shi Y, Niu J, Chen Z, Miyamoto S, Tergaonkar V. ATM- and NEMO-dependent ELKS ubiquitination coordinates TAK1-mediated IKK activation in response to genotoxic stress. Mol Cell 2010; 40:75 - 86
- Lu M, Lin SC, Huang Y, Kang YJ, Rich R, Lo YC, et al. XIAP induces NFkappaB activation via the BIR1/TAB1 interaction and BIR1 dimerization. Mol Cell 2007; 26:689 - 702
- Kenneth NS, Mudie S, Rocha S. IKK and NFkappaB-mediated regulation of Claspin impacts on ATR checkpoint function. EMBO J 2010; 29:2966 - 2978
- Schumm K, Rocha S, Caamano J, Perkins ND. Regulation of p53 tumour suppressor target gene expression by the p52 NFkappaB subunit. EMBO J 2006; 25:4820 - 4832
- Cheng J, Kang X, Zhang S, Yeh ET. SUMO-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell 2007; 131:584 - 595
- Kang X, Qi Y, Zuo Y, Wang Q, Zou Y, Schwartz RJ, et al. SUMO-specific protease 2 is essential for suppression of polycomb group protein-mediated gene silencing during embryonic development. Mol Cell 2010; 38:191 - 201
- Di Bacco A, Ouyang J, Lee HY, Catic A, Ploegh H, Gill G. The SUMO-specific protease SENP5 is required for cell division. Mol Cell Biol 2006; 26:4489 - 4498
- Mukhopadhyay D, Arnaoutov A, Dasso M. The SUMO protease SENP6 is essential for inner kinetochore assembly. J Cell Biol 2010; 188:681 - 692
- Haindl M, Harasim T, Eick D, Muller S. The nucleolar SUMO-specific protease SENP3 reverses SUMO modification of nucleophosmin and is required for rRNA processing. EMBO Rep 2008; 9:273 - 279
- Shen LN, Geoffroy MC, Jaffray EG, Hay RT. Characterization of SENP7, a SUMO-2/3-specific isopeptidase. Biochem J 2009; 421:223 - 230