Abstract
A major focus of melanoma research continues to be the search for genes/proteins that may be suitable targets for molecular therapy of primary and metastatic melanoma. In line with this effort, the objective of the study presented herein was to determine whether interfering with cell cycle progression and in particular, the expression and function of select cyclin-dependent kinases, would impair the biological features of advanced melanoma. We provide data, which document that unlike nevi and melanoma in situ, primary and metastatic melanomas express high levels of CDK2, CDK1, and CDK5. Furthermore, we present the results of in vitro and preclinical in vivo studies, which demonstrate that treatment with a small-molecule cyclin-dependent kinase inhibitor that selectively blocks the function of CDK2, CDK5, CDK1, and CDK9, leads not only to inhibition of melanoma cell proliferation and apoptosis of melanoma cells, but also impairs the growth of human melanoma xenografts.
Introduction
One of the intrinsic and prevalent characteristics of locally advanced and stage IV melanoma is their extreme resistance to chemotherapy and radiotherapy. In addition, monotherapy, including molecular targeted single-agent therapy, has little impact on the overall survival of patients with advanced melanoma. The RAF/MEK/ERK pathway has recently become a target for molecular therapy of advanced melanomas harboring the BRAFV600E mutation;Citation1 however, not every primary and metastatic melanoma lesion carries this mutation. Thus, one of the focal points of molecular studies regarding this disease continues to be the challenge to identify signaling pathways that are ‘drivers’ rather than ‘passengers’ for this malignancy and to determine, which of the key regulatory genes in these molecular ‘driver’ pathways, that by being dysregulated or upregulated to high levels with progression to advanced melanoma, can be effective targets for combination therapies involving small-molecule inhibitors. In line with this objective, the aim of the study summarized herein, was to determine whether blocking the function of select cyclin-dependent kinases (CDKs) such as CDK2 and CDK1, would be an efficacious approach to interfering with the aggressive biologic features of advanced melanoma.
Studies pertaining to the analysis of cell cycle regulators in advanced melanoma have to date, focused primarily, and to a large extent, upon the characterization of mutations in the p16INK4A/p14ARF proteins, that encoded by the on chromosome 9p21 residing cyclin-dependent kinase inhibitor 2A (CDKN2A) locus, are associated with a high risk for patients with familial melanoma (for recent reviews and references therein, see references Citation2 and Citation3). In contrast, regarding the in 90% of melanoma patients occurring sporadic form of melanoma, significantly less information is available concerning possible aberrant functions of cyclins and/or cyclin-dependent kinases that are central regulators of the mammalian cell cycle and its checkpoints (for review, see reference Citation4). However, a limited number of previous studies have provided evidence that in advanced melanoma compared with nevi, cyclin E and CDK2 are expressed at elevated levels;Citation5,Citation6 CDK2 is a downstream effector of the Microphthalmia-associated transcription factor (MITF);Citation7 and that CDK1 is expressed at higher levels in metastatic compared with primary melanoma.Citation8
Currently being evaluated in preclinical pharmacokinetic/pharmacodynamic (PK/PD) and/or phase I studies are small-molecule inhibitors that with various affinities, target different CDKs. One of these small-molecule agents is SCH 727965 (Dinaciclib), which inhibits the activity of CDK2, CDK5, CDK1, and CDK9. To determine whether, and to what extent, CDK2 and these three other CDKs play a role in the biology of advanced melanoma comprising primary melanoma in the vertical growth phase (VGP melanoma) and melanoma in the metastatic growth phase (MGP melanoma), we performed a series of studies that involved treatment of VGP and MGP melanoma cells with the small-molecule inhibitor SCH 727965.
Specifically, we document that unlike benign and atypical nevi, or melanoma in situ, VGP and MGP melanomas express high levels of CDK2 as well as CDK1, and that in vitro, treatment with SCH 727965 inhibits melanoma cell proliferation, and drives melanoma cells into massive apoptosis. We also present the findings of a preclinical study, which demonstrated that systemic treatment of athymic mice bearing human MGP melanoma xenografts with this small-molecule CDK inhibitor, when administered alone or in combination with the chemotherapeutic drug, Paclitaxel, interfered with the growth of these tumors.
Results
Status of CDK2, CDK1 and CDK5 expression in normal skin, nevus, and melanoma tissues.
To determine the status of CDK2, CDK1 and CDK5 expression in the melanoma progression pathway, tissue samples representing normal skin, benign nevi, atypical nevi, which are the precursors and risk markers of melanoma, melanoma in situ, which although noninvasive is the first stage of melanoma development, VGP melanoma and subcutaneous and visceral MGP melanoma, were probed with antibody to human CDK2, CDK1 and CDK5. The results of this immunohistochemistry analysis () revealed no expression of CDK2, CDK1, or CDK5 in junctional melanocytes of normal skin. Similarly, none of these three CDKs was expressed in nevocytes, which are the clusters of melanocytes in benign and atypical nevi, and they were not expressed in melanoma in situ cells. In contrast, VGP melanomas, and to an even greater extent, MGP melanomas, demonstrated strong expression of CDK2, CDK1 and CDK5. Given the different histologic stages, at least two and in most cases, seven representative tissue samples were probed with the different CDK antibodies. All tissue specimens revealed a similar pattern and status of CDK2, CDK1 and CDK5 expression as the tissue samples depicted in .
Expression of cyclin-dependent kinases in melanoma cells, melanocytes, and skin fibroblasts.
Mammalian CDK2,Citation9 a 33 kD nuclear protein, is inactive until complexed with cyclin E at G1/S-phase entry,Citation10,Citation11 and thereafter with cyclin A for S-phase progression.Citation12,Citation13 Activation of the cyclin/Cdk2 complex requires dephosphorylation of CDK2 at threonine 14 (Thr-14) and tyrosine 15 (Tyr-15) and phosphorylation at threonine (Thr-160).
To obtain information as to whether in cells representing advanced melanoma, CDK2 is present predominantly in its monomeric form or in a complex, we probed total cell lysates, prepared from melanoma cells derived from five different MGP melanomas, with an antibody to total CDK2 and an antibody that recognizes phospho(p)-CDK2 (Thr-14). The data of this immunoblot analysis revealed that all five non-synchronized MGP melanoma cell lines expressed two proteins of approximately 50 and 70 kD, which may represent complexed CDK2 (, lanes c–g). Two of these five MGP melanoma cell lines, WM1158 (, lane c) and WM852 (, lane d), did not show expression of the 33 kD monomeric form of CDK2, but did show strong expression of pCDK2 (Thr-14), while the other three cell lines, WM266-4 (, lane e), SK-MEL-28 (, lane f), WM983-B (, lane g), revealed the opposite. Since the results of the immunohistochemistry analysis () had demonstrated stronger expression of CDK2 in MGP compared with VGP melanoma tissues, we also performed an immunoblot analysis of two cell lines that were established from a VGP and an MGP melanoma of a same patient. This side-by-side comparison showed that unlike the WM983-B MGP melanoma cells (, lane b), the WM983-A VGP melanoma cells (, lane a) did not express the 33 kD monomeric form of CDK2, and the intensity of the 50 and 70 kD proteins was slightly less.
Also depicted in are total cell lysates of human neonatal melanocytes (, lane h), which showed expression of the 70 kD protein, the monomeric 33 kD CDK2 protein and CDK2 phosphorylated at Thr-14. Not present in these melanocytes was the 50 kD protein. In contrast, human neonatal skin fibroblasts demonstrated in addition to the 70 kD protein, strong expression of the 50 kD protein (, lane wi) and the presence of phosphorylated CDK2. The finding that, albeit at low levels, short-term cultures of human neonatal melanocytes express CDK2, but not as shown in , melanocytes residing in the epidermal/dermal junction of normal human skin tissue, may be attributable to the fact that in order for human neonatal melanocytes to proliferate for a few passages in vitro before they undergo senescence, their growth medium has to be supplemented not only with the tumor promoter, TPA, but also with considerably high amounts of basic fibroblast growth factor (bFGF).Citation14
Because of our subsequently performed molecular targeting studies, described below, which involved the use of the small-molecule inhibitor, SCH 727965, which in addition to CDK2, inhibits CDK5, CDK1 and CDK9, we performed as depicted in and , additional immunoblot analyses to determine the status of expression of these latter three CDKs in the different melanoma cell lines. The results demonstrated that all five MGP melanoma cell lines (, lanes c–g) as well as human neonatal melanocytes (, lane h), strongly expressed the 42 kD isoform of CDK9, which shares 47% homology with other CDKs, complexes with cyclin T variants, and functions primarily in the elongation of RNA transcripts.Citation15–Citation17 Comparatively weak was the expression of CDK9 in short-term cultures of human neonatal skin fibroblasts (, lane i).
Unlike CDK2, CDK1 or the other known CDKs, CDK5Citation18,Citation19 does not require for its activation phosphorylation on the T loop, and it is not a checkpoint kinase, but functions as a regulatory kinase in important neuronal processes, including brain development and neuronal stress responses (for review, see reference Citation20). Yet, like the tissue specimens representing advanced melanoma (see ), every one of the five MGP melanoma cell lines expressed clearly detectable levels of CDK5 (, lanes a–e).
CDK1 complexed with cyclin A in G2 and cyclin B in M phase, is the only CDK that can drive mammalian cells through the entire cell cycle (for review, see reference Citation21). Isoform 1 of CDK1 codes for a protein of 34 kD, which was strongly expressed in 2/5 MGP melanoma cell lines (, lane a and e), whereas all five cell lines revealed strong expression of a protein of about 50 kD, which may represent complexed CDK1.
CDK inhibitor treatment of melanoma cells impairs their CDK activity and phosphorylation of the retinoblastoma protein.
Using in vitro kinase assays, it was previously determinedCitation22 that the small-molecule inhibitor, SCH 727965, inhibits CDK2, CDK5, CDK1 and CDK9 activity with IC50 values of 1, 1, 3 and 4 nmol/L, respectively. Given the high expression of CDK2 and the three other CDKs in tissues and cell lines representing advanced melanoma, we projected that it might require a relatively high dose of SCH 727965 to block the function of these CDKs in melanoma cell lines. Thus, given the first experiment, we treated WM1158 MGP melanoma cells for 24, 48 and 72 hr with 1 µM of this small-molecule inhibitor. Total cell lysates from these CDK inhibitor-treated cells were then probed with antibody to pCDK2 (Thr-14), total CDK9, or the 110 kD retinoblastoma protein phosphorylated at Thre-821, the site that is phosphorylated by complexed CDK2/cyclin A and CDK2/ cyclin E. As depicted in , compared with WM1158 MGP melanoma cells that had received only the small-molecule inhibitory delivery vehicle, Dimethyl Sulfoxide (DMSO), WM1158 cells that had been incubated in the presence of SCH 727965 revealed that as early as 24 hr post treatment, CDK2 activity and CDK9 expression were impaired and the retinoblastoma protein was no longer phosphorylated.
Immunofluorescence analysis of WM1158 MGP melanoma cells, treated for 24 hr with 1 µM of the small-molecule inhibitor, also demonstrated significantly less pCDK2 expression compared with WM1158 MGP melanoma that had received only DMSO (). Likewise, immunofluorescence analysis of CDK inhibitor and DMSO-treated WM1158 cells performed with an antibody to (total) CDK2 or CDK9, showed noticeably less CDK2 as well as CDK9 expression in the small-molecule inhibitor treated melanoma cells (data not shown).
CDK inhibitor treatment blocks melanoma cell proliferation and impairs melanoma cell cycle progression.
To determine whether treatment with the SCH 727965 small-molecule agent, which as previously reported is active against a broad spectrum of human cancer cell lines,Citation22 would have an impact on melanoma cell proliferation, we performed an experiment wherein MGP melanoma cells were incubated in the presence of three increasing doses of this CDK inhibitor. Although the results demonstrated that over a period of 96 hr, all three doses impaired melanoma cell proliferation (), the lowest dose of 0.5 µM was not as effective as the two higher doses of 1 µM and 5 µM, and there was not a substantial difference between the latter two. The fact that treatment with the inhibitor severely interfered with the proliferation of melanoma cells became even more apparent in the context of a second experiment (), wherein we compared the proliferation of CDK inhibitor to DMSO-treated WM1158 and likewise, WM852 (data not shown) MGP melanoma cells.
Interestingly, but not entirely unexpected, it was not only the cells' proliferation that was majorly impaired upon treatment with the CDK inhibitor but also their morphology. As depicted in , within 24 hours following addition of the inhibitor, and even more pronounced thereafter, the cells severed their cell-cell contacts while at the same time, a limited but noticeable number of cells that continued to adhere to the surface of the Petri dishes, became overtly round in shape and larger in size—an indication that these cells were unable to progress through the entire cell cycle. To obtain experimental evidence in support of this observation, WM1158 MGP melanoma cells that had been treated with 0.5 µM or 1 µM of the CDK inhibitor, or had received only DMSO, were labeled with Propidium Iodide and analyzed by flow cytometry. In comparison with the DMSO-treated melanoma cells, the CDK inhibitor-treated melanoma cells demonstrated an increased accumulation in S phase and a significantly reduced presence in G2/M phase (), suggesting that their DNA replication and subsequently, cell division were impaired.
CDK inhibitor treatment leads rapidly to massive apoptosis of melanoma cells.
One of the prominent biological characteristics of VGP and MGP melanomas is their extreme resistance to apoptosis. Thus, it was surprising that in addition to being impaired in their proliferation and cell cycle progression, the CDK inhibitor-treated melanoma cells displayed major signs indicative of apoptosis. Following TUNEL as well as Annexin V staining, immunofluorescence analysis revealed significant numbers of apoptotic cells among WM1158 MGP melanoma cells that had been treated with 1 µM of the CDK inhibitor for 48 and 72 hours (), whereas WM1158 melanoma cells that had received only DMSO showed no TUNEL and very little Annexin V staining ().
Furthermore, c-PARP immunoblot analysis documented that WM1158 and likewise, WM852 (data not shown) MGP melanoma cells treated with the CDK inhibitor at a dose of 1 µM or higher, underwent apoptosis within 24 hours following addition of the inhibitor as reflected by cleavage of PARP to cPARP (). The data of these experiments were further supported by the results of an Annexin V/Propidium Iodide-based flow cytometry analysis (), which showed that regardless of whether the CDK inhibitor was present for 24, 48, or 72 hours, it was not stress-induced necrosis but apoptosis that led to melanoma cell death and the cells' inability to resume growth after they had been rinsed and resuspended in medium not containing the inhibitor (data not shown).
In vivo and ex vivo analysis of CDK inhibitor-treated human melanoma xenografts.
Given the extreme resistance of advanced melanoma to standard regimens of treatment, and the fact that, to date, limited information is available regarding genes that might be suitable targets for molecular therapy of advanced melanoma, we next undertook a series of preclinical studies to determine whether CDK inhibitor treatment would have efficacy for human MGP melanoma xenografts grown as subcutaneous tumors in nude mice.
Specifically, these in vivo studies focused upon systemic, intraperitoneal (i.p.) treatment of nude mice bearing WM983-B MGP human melanoma xenografts with 40 mg/kg of the CDK inhibitor first administered on the day (Day 1) a tumor had reached 5 mm in any dimension. Thereafter, the tumor-bearing animals were injected three more times with the same and well-tolerated 40 mg/kg dose of the inhibitor, which was below the as previously in a human lung adenocarcinoma (A549) xenograft model determined maximally tolerated dose of 87 mg/kg.Citation22 Following the last injection on Day 10, tumor sizes were measured for an additional period of two weeks at which point the animals were sacrificed because the tumors in the DMSO control arm had reached the maximum allowable size. A second experimental arm was comprised of WM983-B MGP melanoma xenograft-bearing nude mice that using the same dose of 40 mg/kg, were given i.p. injections of the CDK inhibitor every third or fourth day until Day 17. The results of this series of preclinical studies () revealed that compared with the tumors in the nude mice that had received i.p. injections of only DMSO, the tumors in the animals that had been treated a total of four times with the CDK inhibitor grew noticeably slower as did the tumors in the animals that had received the inhibitor six times.
Since it is unlikely that a small-molecule inhibitor, regardless of its molecular target, when administered as a single agent will ever be effective to the extent that it will be a cure for patients with advanced melanoma, we next determined whether a combination treatment would further enhance the impact of the CDK inhibitor on the growth of human MGP melanoma xenografts. Thus, we administered the CDK inhibitor (40 mg/kg) in combination with 10 mg/kg of the cytotoxic drug Paclitaxel, frequently used as a combination agent in melanoma-based clinical trials. Using a schedule of i.p. injections that spanned Day 1–10, Paclitaxel was administered 24 hr following each injection of the CDK inhibitor. As shown in , compared with the growth rate of the tumors in the nude mice that had been treated with only the inhibitor, the growth of the tumors in the animals that had received the combination treatment was further reduced.
Given the results of these in vivo molecular targeting studies, we next determined the extent to which the systemic i.p. treatment with the small-molecule CDK inhibitor when administered alone or in combination with Paclitaxel had reached and blocked its molecular targets in the MGP melanoma xenografts. For this purpose, we probed tissue sections from several of the resected tumors with antibody to (total) CDK2 as well as CDK5.
As documented in , tumor sections prepared from WM983-B MGP human melanoma xenografts of nude mice that had been treated with only the CDK inhibitor, or a combination of CDK inhibitor and Paclitaxel, exhibited noticeably less CDK2 and likewise, CDK5 expression than the MGP melanoma xenografts from the animals that had been given i.p. injections of only DMSO, or serving as another control arm for the CDKI/Paclitaxel treatment, only Paclitaxel. Furthermore, and similar to the CDK inhibitor-induced morphological cell changes, we had observed in the setting of our in vitro studies (), fluorescent DAPI staining of these different MGP melanoma xenograft tissue sections () revealed that the cells in the CDK inhibitor and even more so the CDK inhibitor/Paclitaxel-treated tumor xenografts had noticeably enlarged nuclei when compared with the nuclei of cells in tumors that represented the DMSO or Paclitaxel control arm. We also probed tissue sections, prepared from the different tumors with antibody to human pRb and pCDK2. However, neither antibody gave consistent and reproducible staining in the case of the DMSO and likewise, Paclitaxel control tumors (data not shown).
Discussion
To date, little information is available regarding a biological link between melanoma progression and possible aberrant cell cycle regulation, and/or dysregulation of cell cycle progression in advanced melanoma. In the study summarized herein, we present evidence that CDK2, CDK1 and CDK5 are expressed at high levels in VGP and MGP melanoma but not in the preceding early stages of melanoma development, and that VGP and MGP melanoma cells and likewise, MGP melanoma xenografts are susceptible to molecular targeting with a small-molecule inhibitor that blocks the function of these CDKs.
Specifically, the data of this study reveal several novel and important findings. First, they demonstrate that it is not only CDK2 and CDK1 that are expressed at high levels in VGP and MGP melanoma tissues and cell lines but also CDK5, which hitherto has not been reported to be expressed in this malignancy. Like CDK9, CDK5 is not a regulator of the cell cycle. Instead, it has essential functions in postmitotic neurons wherein following activation by DNA damage, CDK5 phosphorylation of the Ataxia Telangiectasia Mutated (ATM) protein at Ser-794 precedes and is required for ATM autophosphorylation at Ser-1981.Citation23 In view of the results of a previous study we conducted, which showed that suppressing the high-level expression and function of ATM in advanced-stage melanomas does not sensitize the cells to ionizing radiation,Citation24 it will be of interest to determine whether in VGP and MGP melanomas, CDK5 functions as an upstream regulator of ATM. Furthermore, given the important role CDK5 plays in the pathogenesis of neurodegenerative diseases such as glioblastoma and Alzheimer's, the high-level expression of CDK5 in advanced melanoma is another indicator that as we previously showed in the case of the high-level expression and function of the β-amyloid precursor protein (APP) in advanced melanoma,Citation25 important molecular pathways and aberrant molecular signaling in VGP and MGP melanoma exhibit significant similarities with other neural crest-derived diseases.
The second pivotal point this study raises, pertains to the question is the high-level expression of CDK2, CDK1, CDK5, CDK9 in advanced melanomas merely a result of the increased proliferation of these cells, or is one of these four CDKs a crucial player for VGP and MGP melanomas? Since in the setting of in vitro kinase assays, the CDK inhibitor, SCH 727965, was found to inhibit CDK2 and CDK5 activity with the same IC50 value of 1 nmol/L,Citation22 we performed an experiment in which we transfected the same melanoma cells that we had treated with this small-molecule CDK agent, with a human CDK5-specific four siRNA-containing pool. While this RNAi-based experiment led to clearly detectable downregulation of CDK5, the cells were neither perturbed in their proliferation, nor did they exhibit signs of apoptosis (data not shown). Given its primary function in transcriptional elongation and the fact that SCH 727965 has a four-fold lower and the lowest affinity for CDK9, also makes it unlikely that blocking the function of CDK9 led to the substantial inhibition of melanoma cell proliferation, and the cells undergoing massive apoptosis. Therefore, the most likely possibility is that it is either CDK2 or CDK1, or both that are pertinent cell cycle regulators in advanced melanoma. Although it has been reportedCitation26 that Cdk1 is the only essential cell cycle Cdk, and that in the absence of interphase Cdks, Cdk1 can execute all the events that are required to drive cell division, we believe that in the case of advanced melanoma, CDK2 is the more important player. We did not pursue CDK2 versus CDK1 RNA interference studies to address this specific aspect. However, a previous studyCitation7 did provide data, which demonstrated that when CDK2 was depleted with siRNA or blocked with a dominant-negative mutant, colony-formation in the melanoma cell line A375 was suppressed.
In view of the extreme resistance of advanced melanoma to standard therapies, an important question is which combination treatment involving a CDK inhibitor would be most effective? Although the results of our in vivo experiments revealed enhanced anti-tumor activity when the melanoma xenograft-bearing animals were treated with a combination of SCH 727965 and Paclitaxel, or as shown in another study,Citation27 treatment of melanoma cells with the CDK inhibitor PHA-848125 and Temozolomide had synergistic effects, we do not think that combining a CDK inhibitor with a chemotherapeutic agent will have substantial efficacy in patients with advanced melanoma. Instead, we believe that a combination treatment involving a CDK inhibitor with high affinity for CDK2 and a small-molecule inhibitor that targets a gene such as FGFR-1, which as we previously demonstrated,Citation28,Citation29 is essential for the survival of VGP and MGP melanoma cells, may be of greater clinical benefit for patients with locally advanced or stage IV melanoma.
Methods and Materials
Melanoma cell lines and tissues.
VGP (WM983-A) and MGP (WM983-B, WM852, WM266-4, WM1158, SK-MEL-28) human melanoma cell lines were propagated in vitro as previously described.Citation14 Human epidermal melanocytes, purchased from Lifeline Cell Technology (Walkersville, MD), were grown in DermaLife Basal Medium supplemented with LifeFactors (Lifeline Cell Technology). Human foreskin-derived fibroblasts were cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Standard immunohistochemistry of deidentified, post-diagnosis excess cryopreserved human tissue samples representing normal skin, benign and atypical nevi, melanoma in situ, VGP melanoma, and subcutaneous and visceral MGP melanoma was performed with a mouse monoclonal antibody to human (total) CDK2 (Invitrogen, Carlsbad, CA), CDK1 (GeneTex Inc., Irvine, CA), or CDK5 (Epitomics, Inc., Burlingame, CA).
Immunoblot analysis.
Protein lysates (25 µg/sample), separated on sodium dodecyl sulfate-polyacrylamide gels (SDS-PAGE) and transferred onto nylon membrane, were probed with antibody to human CDK2 (Invitrogen; Sigma-Aldrich, St. Louis MO), pCDK2 (Thr-14) (Epitomics), CDK9 (Cell Signaling Technology, Inc., Danvers, MA), CDK5 (Epitomics), CDK1 (GeneTex), pRb (Thr-821) (Epitomics), c-PARP (Cell Signaling Technology), or α-tubulin (Cell Signaling Technology), followed in each case by incubation with a horseradish peroxidase-conjugated secondary antibody and Luminol reagent (Millipore, Billerica, MA).
CDK inhibitor treatment, proliferation, immunofluorescence, flow cytometry, and apoptosis analysis.
Melanoma cells were treated with the CDK inhibitor SCH 727965 (Dinaciclib) (Merck Research Laboratory-Kenilworth, Kenilworth, NJ) solubilized in DMSO, or incubated in the presence of only DMSO.
Melanoma cell proliferation was determined by counting cells with a hemocytometer. At each time point, triplicate samples were analyzed for each dose of CDK inhibitor used. Triplicate samples at each time point were also analyzed in the case of DMSO-only treated melanoma cells.
For immunofluorescence analysis, CDK inhibitor as well as control DMSO-treated melanoma cells fixed with 4% paraformaldehyde, blocked with goat serum, probed with primary antibody (pCDK2, CDK2, or CDK9) followed by an Alexa Fluor 647 or Alexa Fluor 555-conjugated secondary antibody (Invitrogen) and counterstained with fluorescent 40-6-diamidino-2-phenylindole (DAPI) (Invitrogen) were imaged with an inverted, epifluorescent TE2000 Nikon microscope and a charge-coupled device (CCD) camera (Roper Scientific, Inc./ Photometrics, Tucson, AZ).
For flow cytometry-based cell cycle analysis and analysis of apoptosis by flow cytometry, CDK inhibitor as well as control DMSO-treated MGP melanoma were labeled with Propidium Iodide or Alexa Fluor 488 Annexin V/Propidium Iodide (Vybrant Apoptosis Assay Kit #2, Invitrogen Corporation) and analyzed as previously described.Citation30
For immunofluorescence-based determination of apoptosis, cytospin preparations of CDK inhibitor and likewise, DMSO-treated MGP melanoma cells were fixed, permeabilized, labeled with the In Situ Cell Death Detection Kit, TMR red (Roche Applied Science, Indianapolis, IN) or Alexa Fluor 647 conjugate (Invitrogen Corporation), counterstained with fluorescent DAPI, and imaged.
Melanoma xenografts studies.
4-4 week-old, female nude mice (NCr)-Hfh11) (Taconic, Hudson, NY) were injected subcutaneously on their right dorsal site with WM983-B MGP human melanoma cells (8×106 cells per animal). When the tumors had reached a size of 5 mm in any direction, the animals were given i.p. injections of CDK inhibitor (40 mg/kg, dissolved in DMSO), either alone or in combination with Paclitaxel (10mg/kg), dissolved in a formulation of Cremophor-EL (Sigma Aldrich), ethanol, saline at 12.5%/12.5%/75%), administered i.p. 24 hr following injection of the CDK2 inhibitor. Tumor-bearing nude mice that received i.p. injections of only DMSO, or only Paclitaxel (10 mg/kg) served as controls. The number of tumor-bearing animals analyzed in the DMSO control arm was n=3, n=2 in the Paclitaxel control arm and n=4 each in the CDK inhibitor and the CDK/Paclitaxel combination treatment arm. Tumor length and width of the melanoma xenografts were measured twice a week with a caliper and tumor volumes were calculated using the equation v=(lxw2)/2.
Tissue sections, prepared from the WM983-B MGP melanoma xenografts, were fixed with paraformaldehyde, treated with Rodent Block M (Biocare Medical, Concord, CA), probed with antibody to human CDK2 (Sigma-Aldrich) or CDK5 (Epitomics), incubated with Rabbit-on-Rodent Polymer (for AP) (Biocare Medical) and counterstained with hematoxylin. Adjacent tissue sections were stained with fluorescent DAPI, and imaged.
Figures and Tables
Figure 1 Expression of CDK2, CDK1, and CDK5 in the melanoma progression pathway. Five-µm sections, prepared from cryopreserved tissue specimens representing normal skin, benign nevus, atypical nevus, melanoma in situ and VGP and MGP melanoma were probed with antibody to (A) CDK2, (B) CDK1 or (C) CDK5 and counterstained with hematoxylin.
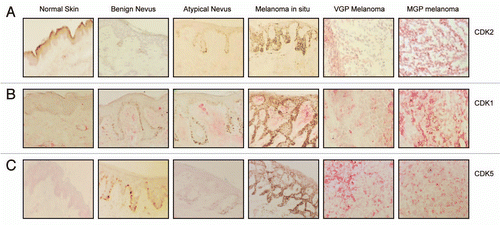
Figure 2 Expression of CDK2, pCDK2, CDK9, CDK5 and CDK1 in VGP and MGP melanoma cell lines, and in human neonatal melanocytes and skin fibroblasts. (A) Immunoblot analysis of CDK2, pCDK2, and CDK9 expression in the VGP melanoma cell line WM983-A (a), in MGP melanoma cell lines WM983-B (b), WM1158 (c), WM852 (d), WM266-4 (e), SK-MEL-28 (f), WM983-B (g), and in human foreskin-derived melanocytes (h), and human foreskin-derived fibroblasts (i). (B) Expression of CDK5 and CDK1 in MGP melanoma cell lines WM1158 (a), WM852 (b), WM266-4 (c), SK-MEL-28 (d), and WM983-B (e). For loading control, the immunoblots were probed with an antibody to α-tubulin.
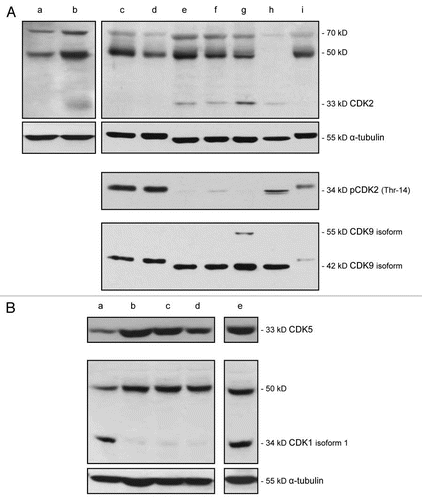
Figure 3 Impact of CDK inhibitor treatment of melanoma cells upon the expression of pCDK2, CDK9 and pRb. (A) pCDK2 (Thr-14), CDK9 and pRb (THR-821) immunoblot analysis of WM1158 MGP melanoma cells that received only DMSO (−) or were treated with 1 µM of the CDK inhibitor (+), SCH 727965 (Dinaciclib), for 24, 48, or 72 hr. Probing of the immunoblots with an antibody to α-tubulin served as loading control. (B) pCDK2 immunofluorescence analysis of MGP melanoma cells (WM1158) incubated in the presence of DMSO or treated with 1 µM of the CDK inhibitor (CDKI) for 24 hr. WM1158 cells that showed expression of pCDK2 are pseudocolored red. Nuclei, counterstained with fluorescent DAPI, are pseudocolored blue.
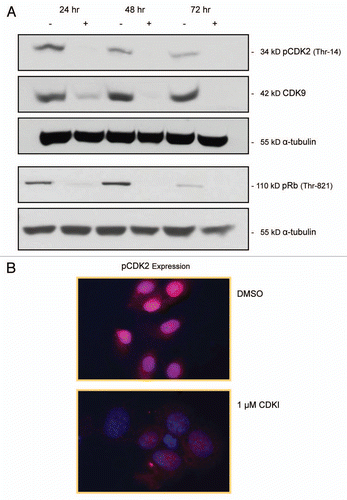
Figure 4 CDK inhibitor treatment of melanoma cells leads to inhibition of melanoma cell proliferation and dysregulation of melanoma cell cycle progression. (A) Proliferation of MGP melanoma cells (WM1158) incubated in the presence of 0.5 µM, 1 µM, or 5 µM of the CDK inhibitor (CDKI) for 24, 48, 72, or 96 hr. Depicted for each dose of CDK inhibitor at each time point, is the mean of triplicate samples analyzed. (B) Proliferation of MGP melanoma cells (WM1158) that received only DMSO or were treated with 1 µM of the CDK inhibitor (CDKI) for 24, 48, 72, or 96 hr. Displayed in each case is the mean of triplicate samples analyzed. (C) Phase contrast images, captured at 10X magnification, depicting the morphology of WM1158 MGP melanoma cells that received only DMSO or were treated with 1 µM of the CDK inhibitor (CDKI) for 24, 48, or 72 hr. (D) Flow cytometry-based cell cycle analysis of WM1158 MGP melanoma cells treated with 0.5 µM or 1 µM of the CDK inhibitor (CDKI) for 24 hr, followed by labeling of the cells with Propidium Iodide. WM1158 MGP melanoma cells that received only DMSO served as the control.
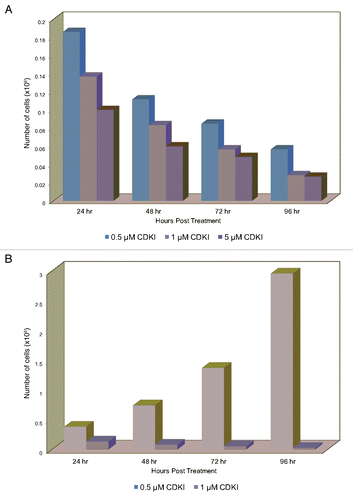
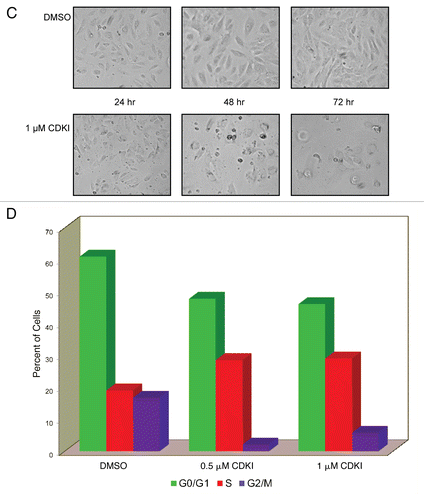
Figure 5 Inhibiting the function of CDKs in melanoma cells leads to melanoma cell apoptosis. (A) WM1158 MGP melanoma cells, treated with 1 µM of the CDK inhibitor (CDKI) for 48 or 72 hr, were analyzed by immunofluorescence-based TUNEL and likewise, Annexin V staining. WM1158 MGP melanoma cells that received only DMSO for 72 hr served as the control. Melanoma cells that had undergone apoptosis are pseudocolored red (TUNEL staining) or pseudocolored yellow (Annexin V staining). Fluorescent DAPI-counterstained nuclei are pseudocolored blue. (B) Immunoblot analysis of WM1158 MGP melanoma cells, treated with 1 µM, 2.5 µM, or 5 µM of the CDK inhibitor (CDKI) for 24, 48, or 72 hr, were probed with an antibody to c-PARP. WM1158 cells that received DMSO for 24, 48, and 72 hr served as the control. (C) After 24, 48, as well as 72 hr following treatment with 1 µM of the CDK inhibitor (CDKI, blue-colored bars), WM1158 MGP melanoma cells were labeled with Annexin V/Propidium Iodide and analyzed by flow cytometry. WM1158 cells that had received only DMSO (DMSO, grey-colored bars) served as the control. At each time point, the light-colored bars represent the percent of melanoma cells in the early stage of apoptosis, medium-colored bars the percent of melanoma cells in late-stage apoptosis and dark-colored bars the percent of necrotic melanoma cells.
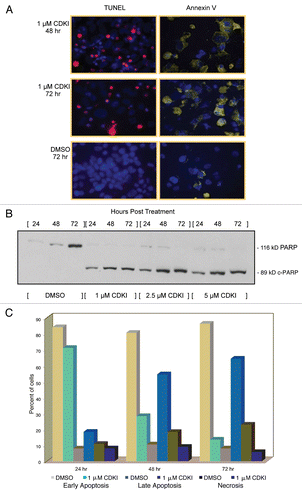
Figure 6 CDK inhibitor treatment of human melanoma xenograft-bearing nude mice. (A) Representative, individual examples of the growth rate of subcutaneous WM983-B MGP melanoma xenografts in nude mice that received the first i.p. injection of the CDK inhibitor (40 mg/kg) (CDKI; light blue-colored line; dark blue-colored line); CDK inhibitor (CDKI) (40 mg/kg) followed 24 hr later by i.p. injection of Paclitaxel (10 mg/kg) (CDKI/Paclitaxel: green-colored line); or only the CDK inhibitor delivery vehicle DMSO (DMSO: black-colored line), on the day (Day 1) a tumor had reached 5 mm in any direction. Subsequent i.p. injections were given on Day 3, 7 and 10. Following the last i.p. injections on Day 10 (indicated by the triangle), tumor volumes in these animals were further recorded on Day 14, 17, 21 and 24. The red-colored line depicts the growth rate of a WM983-B MGP melanoma xenograft in an animal that had been treated i.p. with 40 mg/kg of the CDK inhibitor on Day 1, 3, 7, 10, 14 and 17. (B) CDK2 and CDK5 immunohistochemical analysis of tissue sections prepared from WM983-B MGP melanoma xenografts that until Day 10 had been injected with only DMSO or only Paclitaxel, the CDK inhibitor (CDKI), or the CDK inhibitor combined with Paclitaxel (CDKI/Paclitaxel). Images of the fluorescent DAPI-stained or hematoxylin-counterstained and CDK2 or CDK5-stained tumor sections are depicted at 40X and 20X magnification, respectively.
Acknowledgements
This work was supported by funds from the Merck Research Laboratory-Kenilworth, Kenilworth, NJ, and a grant from the National Institutes of Health to D.B. The authors declare that they have no competing financial interests.
References
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417:949 - 954
- McKenzie HA, Fung C, Becker TM, Irvine M, Mann GJ, Kefford RF, et al. Predicting functional significance of cancer-associated p16(INK4a) mutations in CDKN2A. Hum Mutat 2010; 31:692 - 701
- Leachman SA, Carucci J, Kohlmann W, Banks KC, Asgari MM, Bergman W, et al. Selection criteria for genetic assessment of patients with familial melanoma. J Am Acad Dermatol 2009; 61:677 e1-14
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: A changing paradigm. Nat Rev Cancer 2009; 9:153 - 166
- Bales ES, Dietrich C, Bandyopadhyay D, Schwahn DJ, Xu W, Didenko V, et al. High levels of expression of p27KIP1 and cyclin E in invasive primary malignant melanomas. J Invest Dermatol 1999; 113:1039 - 1046
- Georgieva J, Sinha P, Schadendorf D. Expression of cyclins and cyclin dependent kinases in human benign and malignant melanocytic lesions. J Clin Pathol 2001; 54:229 - 235
- Du J, Widlund HR, Horstmann MA, Ramaswamy S, Ross K, Huber WE, et al. Critical role of CDK2 for melanoma growth linked to its melanocyte-specific transcriptional regulation by MITF. Cancer Cell 2004; 6:565 - 576
- Jaeger J, Koczan D, Thiesen HJ, Ibrahim SM, Gross G, Spang R, et al. Gene expression signatures for tumor progression, tumor subtype, and tumor thickness in laser-microdissected melanoma tissues. Clin Cancer Res 2007; 13:806 - 815
- Elledge SJ, Spottswood MR. A new human p34 protein kinase, CDK2, identified by complementation of a cdc28 mutation in Saccharomyces cerevisiae, is a homolog of Xenopus Eg1. EMBO J 1991; 10:2653 - 2659
- Koff A, Giordano A, Desai D, Yamashita K, Harper JW, Elledge S, et al. Formation and activation of a cyclin E-cdk2 complex during the G1 phase of the human cell cycle. Science 1992; 257:1689 - 1694
- Dulic V, Lees E, Reed SI. Association of human cyclin E with a periodic G1-S phase protein kinase. Science 1992; 257:1958 - 1961
- Krude T, Jackman M, Pines J, Laskey RA. Cyclin/Cdk-dependent initiation of DNA replication in a human cell-free system. Cell 1997; 88:109 - 119
- Coverley D, Laman H, Laskey RA. Distinct roles for cyclins E and A during DNA replication complex assembly and activation. Nat Cell Biol 2002; 4:523 - 528
- Becker D, Meier CB, Herlyn M. Proliferation of human malignant melanomas is inhibited by antisense oligodeoxynucleotides targeted against basic fibroblast growth factor. EMBO J 1989; 8:3685 - 3691
- Marshall NF, Price DH. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J Biol Chem 1995; 270:12335 - 12338
- Marshall NF, Peng J, Xie Z, Price DH. Control of RNA polymerase II elongation potential by a novel carboxyl-terminal domain kinase. J Biol Chem 1996; 271:27176 - 27183
- Marshall RM, Grana X. Mechanisms controlling CDK9 activity. Front Biosci 2006; 11:2598 - 2613
- Hellmich MR, Pant HC, Wada E, Battey JF. Neuronal cdc2-like kinase: A cdc2-related protein kinase with predominantly neuronal expression. Proc Natl Acad Sci USA 1992; 89:10867 - 10871
- Meyerson M, Enders GH, Wu CL, Su LK, Gorka C, Nelson C, et al. A family of human cdc2-related protein kinases. EMBO J 1992; 11:2909 - 2917
- Lalioti V, Pulido D, Sandoval IV. Cdk5, the multifunctional surveyor. Cell Cycle 2010; 9:284 - 311
- Satyanarayana A, Kaldis P. Mammalian cell-cycle regulation: Several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 2009; 28:2925 - 2939
- Parry D, Guzi T, Shanahan F, Davis N, Prabhavalkar D, Wiswell D, et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol Cancer Ther 2010; 9:2344 - 2353
- Tian B, Yang Q, Mao Z. Phosphorylation of ATM by Cdk5 mediates DNA damage signalling and regulates neuronal death. Nat Cell Biol 2009; 11:211 - 218
- Moschos SJ, Dodd NR, Jukic DM, Fayewicz SL, Wang X, Becker D. Suppressing the high-level expression and function of ATM in advanced-stage melanomas does not sensitize the cells to ionizing radiation. Cancer Biol Ther 2009; 8:1815 - 1825
- Botelho MG, Wang X, Arndt-Jovin DJ, Becker D, Jovin TM. Induction of terminal differentiation in melanoma cells on downregulation of beta-amyloid precursor protein. J Invest Dermatol 2010; 130:1400 - 1410
- Santamaria D, Barriere C, Cerqueira A, Hunt S, Tardy C, Newton K, et al. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2007; 448:811 - 815
- Caporali S, Alvino E, Starace G, Ciomei M, Brasca MG, Levati L, et al. The cyclin-dependent kinase inhibitor PHA-848125 suppresses the in vitro growth of human melanomas sensitive or resistant to temozolomide, and shows synergistic effects in combination with this triazene compound. Pharmacol Res 2010; 61:437 - 448
- Wang Y, Becker D. Antisense targeting of basic fibroblast growth factor and fibroblast growth factor receptor-1 in human melanomas blocks intratumoral angiogenesis and tumor growth. Nat Med 1997; 3:887 - 893
- Valesky M, Spang AJ, Fisher GW, Farkas DL, Becker D. Noninvasive dynamic fluorescence imaging of human melanomas reveals that targeted inhibition of bFGF or FGFR-1 in melanoma cells blocks tumor growth by apoptosis. Mol Med 2002; 8:103 - 112
- Moschos SJ, Smith AP, Mandic M, Athanassiou C, Watson-Hurst K, Jukic DM, et al. SAGE and antibody array analysis of melanoma-infiltrated lymph nodes: Identification of Ubc9 as an important molecule in advanced-stage melanomas. Oncogene 2007; 26:4216 - 4225