Abstract
ZBP-89, a zinc finger transcription factor, participates in histone deacetylases inhibitors (HDACi)-mediated growth arrest and apoptosis in cancer cells. p53 mutants may interact with ZBP-89 that transcriptionally regulates p21Waf1 (p21). However, this interaction and its consequence in cancer treatments are poorly understood. In this study, we demonstrate that ZBP‑89 is essentially required in HDACi-mediated p21 upregulation in hepetocellular carcinoma (HCC). Overexpression of ZBP-89 protein enhanced the lethal effectiveness of Trichostatin A (TSA). p53 mutant p53G245D, but not p53R249S, directly bound to ZBP-89 and prevented its translocation from cytoplasm to nucleus. Furthermore, p53G245D was shown to have a similar pattern of subcellular localization to ZBP-89 in tissues of HCC patients in Hong Kong. Functionally, the cytoplasmic accumulation of ZBP-89 by p53G245D significantly abrogated the induction of p21 caused by sodium butyrate (NaB) treatment and protected cells from TSA-induced death. The activations of several apoptotic proteins, such as Bid and PARP, were involved in p53G245D-mediated protection. Moreover, the resistance to HDACi in p53G245D-expressing cells was reversed by overexpression of ZBP-89. Taken together, these data suggest a potential mechanism via which mutant p53 enables tumor cells to resist chemotherapy and, therefore, establish a plausible link between mutant p53 binding to ZBP-89 and a decreased chemosensitivity of HCC cells.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgements
This work was supported by the Research Grants Council of the Hong Kong SAR. No. CUHK4551/05M and CUHK 462009 to P.B.S.L. and G.G.C. and US Public Health Service grant R01-DK055732 to J.L.M.
Figures and Tables
Figure 1 HDACi induces cell cycle arrest and apoptosis in HCC cells. (A) Cytotoxicity of NaB and TSA in HCC cells. Hep3B (A-1 and A-3) and PLC/PRF/5 (A-2 and A-4) cells cultured in 96-well plates were treated with various concentrations of NaB (0–10 mM) or TSA (0–1,000 nM) for indicated periods. The cell viability was determined by MTT assays. (B) HDACi treatments in HCC cells led to p21 induction and PARP activation. Hep3B (B-1) and PLC/PRF/5 (B-2) cells were incubated with either 4 mM NaB or 500 nM for indicated time. The effects of both agents on apoptotic marker PARP (CL-PARP, cleaved form of PARP) and cell cycle inhibitor p21 were examined by western blot. (C) HDACi treatment arrested cells in G1 phase. Cells were treated with either NaB at 4 mM or TSA at 500 nM for 24 h and then stained with propidium iodide. DNA contents were measured by flow cytometry, and the percentages of cells in G1 phase were shown. (D) TSA induced apoptosis in HCC cells. Cells were cultured with either 4 mM of NaB or 500 nM of TSA for 24 h, and then subjected to TUNEL assay to assess apoptosis. Data represent the mean ± SD of three independent experiments. *p < 0.05 and **p < 0.01 vs. the control group.
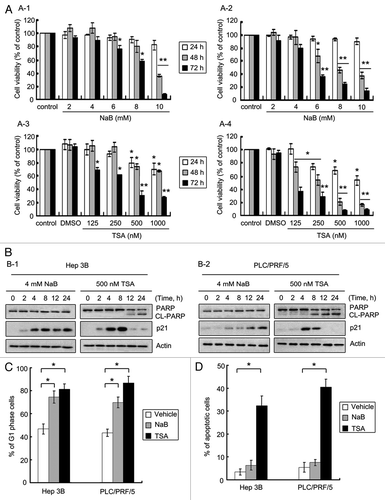
Figure 2 The involvement of ZBP-89 in the effect of HDACi on HCC cells. (A) Diminishment of ZBP-89 expression abrogated NaB-mediated G1 phase arrest. (A-1) Cells were transfected with either siRNA targeted to ZBP-89 mRNA or plasmid-encoded full-length ZBP-89 for 16 h and then cultured with 4 mM NaB for another 24 h. Percentages of cells in G1 phase were determined by flow cytometry. (A-2) HCC cells were first transfected with a mutant (si-control) or wild-type (si-ZBP-89) siRNA oligonucleotide of ZBP-89 for 16 h, and then treated with 4 mM NaB for 24 h. Western blot was performed to examine the expression of ZBP-89, p53, p21 and cyclin D1. GAPDH was used as a loading control. (A) ZBP-89 facilitated TSA-induced apoptosis. (B-1) Cells were first transfected with either siRNA targeted to ZBP-89 mRNA or plasmid-encoded full-length ZBP-89 for 16 h, and then incubated with 500 nM TSA for another 24 h. Apoptosis was assessed by flow cytometry, using TUNEL assay. (B-2) HCC cells treated as described in (B-1) were lysed in RIPA solution. The expression of ZBP-89 and p53 as well as the activation of PARP and caspases 3 were determined by western blot. Actin was used as a loading control. (C) Cellular localization of ZBP-89 and its deletion mutants in Hep3B cells were examined. Hep3B cells were transfected with plasmids encoded ZBP-89 (ZBP-89-FL) or deletion mutants (ZBP-89-Δ6–180 and ZBP-89-Δ298–447) for 24 h. Immunofluorescense was performed to reveal the cytoplasmic distribution of both ZBP-89 deletion mutants. (D) Ectopic expression of both ZBP-89 deletion mutants enhanced activation of PARP. Proteins were collected in TSA-treated cells expressing ZBP-89-FL and ZBP-89 deletion mutants and then subjected to western blot to detect the cleaved form of PARP. (E) Nuclear localization of ZBP-89 might be essential for it to facilitate lethal effect of TSA. Cells expressing ZBP-89-FL and ZBP-89 deletion mutants were treated with 500 nM TSA for 24 h. TUNEL assays were performed to determine apoptosis. Data represent the mean ± SD of three independent experiments. *p < 0.05; ◆ is used to indicate the extra expression of ZBP-89 and its deletion mutants.
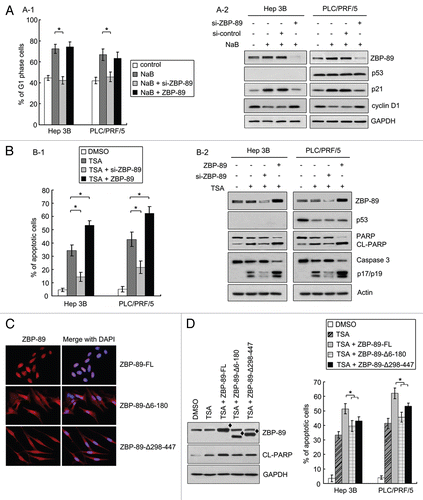
Figure 3 p53G245D interacts with ZBP-89 and retains it in cytoplasm. (A) p53G245D directly bound to ZBP-89. (A-1) Hep3B cells were transfected with pcDNA 3.1-p53G245D or pcDNA 3.1 for 24 h and then selected by antibiotic G418 for another 21 d to establish a stable cell line which was named Hep3B-G245D. Total proteins of Hep3B, Hep3B-G245D, PLC/PRF/5 cells were extracted by RIPA plus protease cocktail and incubated with a primary antibody that recognizes ZBP-89 or p53 mutants. After being incubated overnight at 4°C, the protein-antibody complex was deposited by Protein A/G-agarose. The whole complex was boiled and then subjected to western blot to examine the interaction between ZBP-89 and p53 mutants. (A-2) p53G245D and p53R249S tagged with HA, and ZBP-89 fused to FLAG plasmids were transfected into Hep3B cells. The immunoprecipitated FLAG-ZBP-89 complexes were separated on a sodium dodecyl sulfate-polyacrylamide electrophoresis gel, transferred to membrane and immunoblotted with anti-FLAG or anti-HA antibody. IgG was served as a negative control for antibodies used in immunoprecipitation. (B) p53G245D co-localized with ZBP-89 in cytoplasm. Hep3B, Hep3B-G245D, PLC/PRF/5 cells were fixed by 4% PFA and incubated with p53 and ZBP-89 antibodies overnight at 4°C. After staining with fluorescence secondary antibodies and DAPI, cells were observed under fluorescence microscope. (C) p53G245D affected the cellular localization of ZBP-89. Cytoplasmic and nuclear proteins were isolated. Levels of p53 and ZBP-89 in each fraction were detected. β-actin and Lamin B were used as the cytoplasmic and the nuclear markers, respectively. (D) p53G245D was identified in HCC tissues. DNAs extracted from HCC tissues were subjected to sequencing analysis. The sequencing diagram was shown. Nucleotide and amino acid sequences of both wild-type (case #122) and mutant p53 (case #44) were also presented. (E) ZBP-89 was found in cytoplasm in p53G245D-expressing samples. HCC tissues were fixed in 4% PFA and then subjected to immunohistochemistry to detect the expressions of p53 mutants and ZBP-89. The representative microphotographs of p53 and ZBP-89 immunohistochemical staining in HCC tissues were shown (original magnification, 400x).
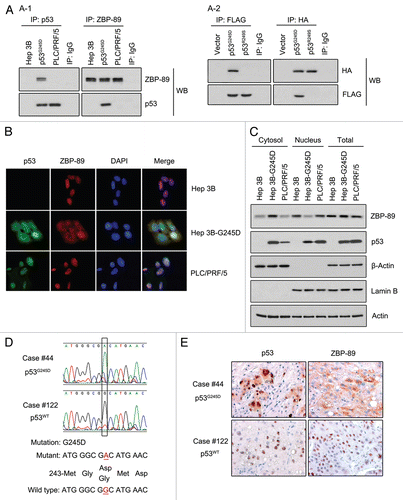
Figure 4 NaB-mediated p21 induction is abrogated in p53G245D-expressing cells. (A) G1-phase arrest induced by NaB treatment was diminished in Hep3B-G245D cells. Hep3B, Hep3B-G245D, PLC/PRF/5 cells were treated with NaB at 4 mM for 24 h. After being stained with propidium iodide, cells were subjected to flow cytometry to measure DNA contents. The percentages of cells in G1 phase were shown. (B) Cells were treated with 4 mM NaB for 24 h. Total RNA was extracted by Trizol reagent. RT-PCR was performed to determine the expression of ZBP-89, p53 and p21. (Upper part: relative expression of p21 mRNA. Bottom part: representative result of three independent experiments). (C) Proteins extracted from cells treated as described in (A) were used in western blot. Levels of ZBP-89, p53 and p21 proteins were examined. (Upper part: relative expression of p21 protein. Bottom part: representative results of three independent experiments). (D) Cells were treated with 4 mM NaB for 3 h and soluble chromatin was prepared. ChIP assay was performed with anti-ZBP-89 antibody. The final DNA extracted was amplified using primers that span from −246 to +46 of the p21 promoter followed by ethidium bromide staining. The input was 1% of the soluble chromatin used for immunoprecipitation. IgG was used as antibody control.
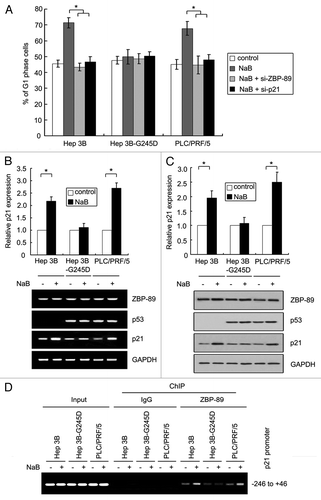
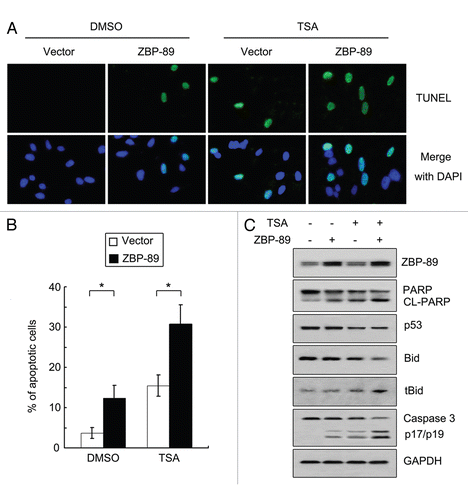
Figure 5 TSA-mediated apoptosis is attenuated in p53G245D-expressing cells. (A) Cells were cultured with 500 nM TSA or DMSO for 24 h. TUNEL assay using In Situ Cell Death Detection Kit was performed to determine apoptosis. (B) Cells treated with 500 nM TSA or DMSO for 24 h were subjected to flow cytometry to measure apoptosis. The percentages of apoptotic cells were shown. Data represent the mean ± SD of three independent experiments. *p < 0.05. (C) Cells cultured in 96-well plates were treated with 500 nM TSA for 24 h. The cell viabilities were determined by MTT assays. Data represent the mean ± SD of three independent experiments. *p < 0.05. (D) Proteins were obtained in cells treated with 500 nM TSA or DMSO for 24 h and then subjected to western blot to examine the expression of ZBP-89, cleaved form of PARP, p53, Bid and truncated Bid (tBid).
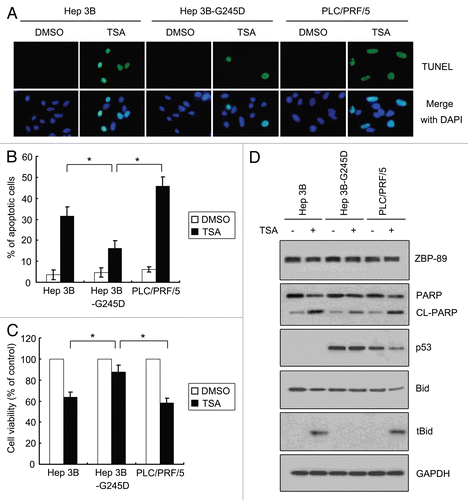
Figure 6 Overexpression of ZBP-89 reverses resistance to TSA-induced cell death in p53G245D-expressing cells. (A) Hep3B-G245D cells were first transfected with pcDNA 3.1, pcDNA 3.1-ZBP-89-FL for 16 h and then cultured with 500 nM TSA or DMSO for 24 h. TUNEL assay using In Situ Cell Death Detection Kit was performed to determine apoptosis. (B) Hep3B-G245D cells treated as described in (A) were subjected to flow cytometry to measure apoptosis. The percentages of apoptotic cells were shown. Data represent the mean ± SD of three independent experiments. *p < 0.05. (C) Proteins were obtained in cells treated as described in (A) and then subjected to western blot to examine the expression of ZBP-89, cleaved form of PARP, cleaved form of caspases 3, p53, Bid and tBid.