Abstract
Neither the molecular mechanisms whereby cancer cells intrinsically are or become resistant to the DNA-damaging agent cisplatin nor the signaling pathways that account for cisplatin cytotoxicity have thus far been characterized in detail. In an attempt to gain further insights into the molecular cascades elicited by cisplatin (leading to resistance or underpinning its antineoplastic properties), we comparatively investigated the ability of cisplatin, C2-ceramide and cadmium dichloride, alone or in the presence of an array of mitochondrion-protective agents, to trigger the permeabilization of purified mitochondria. In addition, we compared the transcriptional response triggered by cisplatin, C2-ceramide and cadmium dichloride in non-small cell lung carcinoma A549 cells. Finally, we assessed the capacity of cisplatin, C2-ceramide and cadmium dichloride to reduce the clonogenic potential of a battery of yeast strains lacking proteins involved in the regulation of cell death, DNA damage signaling and stress management. This multipronged experimental approach revealed that cisplatin elicits signaling pathways that are for the most part “private,” i.e., that manifest limited overlap with the molecular cascades ignited by other inducers of mitochondrial apoptosis, and triggers apoptosis mainly in a transcription-independent fashion. Indeed, bona fide cisplatin-response modifiers that we have recently identified by a functional genome-wide siRNA screen are either not transcriptionally regulated during cisplatin-induced cell death or their transcriptional modulation reflects the activation of an adaptive response promoting cisplatin resistance
Introduction
cis-Diammineplatinum(II) dichloride, best known as cisplatin (CDDP), is a platinum-containing agent currently employed for the treatment of a large array of solid tumors, encompassing head and neck, lung, colorectal, bladder, prostate, ovarian and germ cell cancers.Citation1 For a long time, CDDP has been considered as a pure DNA-damaging agent, promoting the formation of intra- and inter-strand DNA adducts that eventually result in the activation of (often apoptotic) cell death or senescence.Citation2,Citation3 Nowadays, following the demonstration that CDDP also exerts cytotoxic effects on enucleated cells,Citation4,Citation5 and that only 1% of intracellular CDDP binds nuclear DNA,Citation6 it has become clear that the signaling pathways underlying the antineoplastic effects of CDDP are complex and integrate cytoplasmic (post-translational, rapid)Citation7 and nuclear (transcriptional, delayed) mechanisms.Citation8
Testicular germ cell cancer patients receiving CDDP-based chemotherapy undergo complete and durable regressions in a sizeable fraction (> 80%) of cases, and these individuals can de facto be considered as cured.Citation9 In other clinical settings, including colorectal, lung and prostate cancer, the percentage of patients that are intrinsically resistant to CDDP-based chemotherapy is significantly higher. Moreover, a consistent fraction of originally sensitive tumors eventually develop chemoresistance, which rapidly translates into relapse and therapeutic failure.Citation10,Citation11 Besides relatively mild side effects (including nausea, vomiting and appetite loss), CDDP, which is generally administered as a short-term infusion in physiological saline, can be associated with grade I–II nephrotoxicity (in more than 30% of patients), neurotoxicity and ototoxicity (both in 10–30% of patients).Citation1 This said, the most critical limitation to the clinical use of CDDP as an antineoplastic agents is provided by the high incidence of innate and acquired chemoresistance.
During the last two decades, the molecular mechanisms whereby cancer cells elude the antineoplastic activity of CDDP have been the subject of intense investigation. The detailed description of these pathways largely exceeds the scope of this introduction and can be found elsewhere.Citation10,Citation12 Still, it is worth noting that cancer cells can avoid CDDP cytotoxicity at least at four different levels: (1) by avoiding its intracellular accumulation and/or availability (pre-target resistance); (2) by repairing CDDP-induced DNA lesions more proficiently (on-target resistance); (3) by interrupting the signaling cascades that normally bridge DNA damage to the execution of cell death or the activation of cell senescence (post-target resistance) and (4) via alterations in signaling cascades that are not directly elicited by CDDP, yet compensate for CDDP-driven lethal signals.Citation10 Moreover, it should be noted that, in the vast majority of cases, CDDP resistance appears to be multifactorial, i.e., to involve the simultaneous activation of several non-overlapping mechanisms, de facto complicating the development of clinically meaningful strategies of chemosensitization.Citation10
During the last few years, our attention has been centered on the molecular mechanisms that are activated by CDDP in non-small cell lung carcinoma (NSCLC), a frequent and aggressive form of lung cancer that is responsible for more than 1 million deaths worldwide annually.Citation13,Citation14 Here, we report the results of a multipronged experimental approach revealing that CDDP activates signaling pathways that are mainly “private,” i.e., that manifest limited overlap with the molecular cascades elicited by prototypic inducers of mitochondrial apoptosis, such as C2-ceramide (C2-CER) and cadmium dichloride (CdCl2). Moreover, we show that functional modifiers of the cytotoxic response of NSCLC cancer cells to CDDP are generally not subjected to transcriptional regulation during CDDP-induced cell death, and that, when they are, this reflects the activation of an adaptive response to CDDP.
Results and Discussion
Cadmium dichloride, but neither cisplatin nor C2-ceramide, trigger the permeabilization of purified liver rat mitochondria
To get further insights into the intracellular events elicited by CDDP, we investigated whether CDDP is capable of directly promoting the permeabilization of mitochondrial membranes. This coincides with a massive entry of solutes (driven by the anionic milieu generated by intramitochondrial proteins) and water (driven by osmosis) and, in vitro, is reflected by a rapid drop in the absorbance of the mitochondrial suspension (a phenomenon known as large-amplitude swelling).Citation15 Thus, we isolated mitochondria from rat livers according to conventional procedures and exposed them to increasing concentrations of CDDP. Alongside, purified mitochondria were incubated with increasing concentrations of two chemotherapy-unrelated inducers of mitochondrial apoptosis, C2-CERCitation16 and CdCl2,Citation17 as well as with fixed concentrations of GD3 ganglioside and CaCl2, providing two distinct positive control conditions. Thereafter, absorbance was monitored regularly at 1 min intervals over a period of 30 min. Finally, data were analyzed by means of an algorithm that we developed to determine (1) whether or not large-amplitude swelling had taken place and, if so, (2) to what extent and (3) by which kinetics ().
Figure 1. Response of isolated mitochondria to cisplatin (CDDP), C2-ceramide (C2-CER) and cadmium dichloride (CdCl2). (A) Large-amplitude swelling categories based on the intensity and kinetics of decreasing absorbance. (B) Direct mitochondrion-permeabilizing effects of 50 μM GD3 ganglioside (GD3) and increasing concentrations (C1-C3) of Ca2+ (500 and 100 μM)CDDP (20, 100 or 200 μM), C2-CER (5, 50 or 100 μM) and CdCl2 (20, 50 or 100 μM). (C) Response of 50 μM Ca2+-, and CdCl2-mediated swelling to increasing concentrations (C1-C2) of the following swelling inhibitors: bongkrekic acid (BA, 10 or 40 μM), cyclosporine A (CsA, 5 or 10 μM), N-acetyl-cysteine (NAC, 2 or 10 mM) and non-oxidized glutathione (GSH, 0.1 or 0.5 mM).
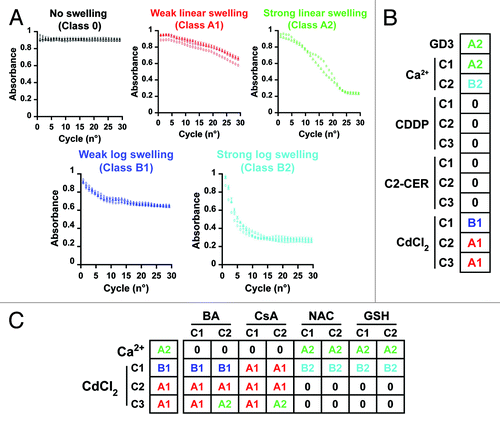
In line with previous reports,Citation18 the absorbance of purified mitochondria exposed to high concentrations of Ca2+ ions (100 μM) decreased to less than half of the initial value and did so in a very rapid fashion (class B2 swelling) (). Lower concentrations of Ca2+ ions (50 μM) induced a response of the same magnitude, yet developing with a slower kinetics (class A2 swelling), as did an equal dose of GD3 ganglioside.Citation16 Mitochondria incubated with increasing concentrations of CDDP or C2-CER failed to undergo the permeability transition (no, or class 0, swelling), while CdCl2 turned out to be capable of triggering mitochondrial membrane permeabilization to a reduced extent (class 1 swelling), whose kinetics (A or B) exhibited a dose-dependent behavior ().
We next tested how CdCl2-triggered large-amplitude swelling is affected by mitochondrion-protective agents encompassing the cyclophilin D inhibitor cyclosporine A (CsA),Citation18 the adenine nucleotide inhibitor bongkrekic acid (BA)Citation19 and two antioxidants, reduced glutathione (GSH) and N-acetyl-cysteine (NAC), all used at two distinct concentrations. In line with previous reports,Citation18,Citation19 the pre-incubation of mitochondria with BA and CsA, but not with GSH and NAC, abolished the ability of Ca2+ ions to trigger large-amplitude swelling (). These data corroborate the notion that large-amplitude swelling as triggered by Ca2+ ions involves components of the permeability transition pore complex (PTPC).Citation15 The mitochondrion-permeabilizing effects of high-dose CdCl2 (class A1 swelling) were not (or only minimally) altered by the presence of BA and CsA, yet were near-to-completely abolished by the pre-incubation of mitochondria with GSH or NAC (), suggesting a prominent role for reactive oxygen species (ROS) in this phenomenon. Unexpectedly, class B1 large-amplitude swelling as induced by low concentrations of CdCl2 was not inhibited by BA, was shifted to an A1 kinetics by CsA and was potentiated in the presence of NAC and GSH ().
Altogether, these observations suggest that, at difference with CdCl2, CDDP and C2-CER fail to directly affect the permeability of purified mitochondria, indicating that their well-established ability to induce the dissipation of the mitochondrial transmembrane potential (Δψm) in cellulaCitation20-Citation22 relies on the engagement of intracellular signaling pathways.
Transcriptional signatures of A549 cells responding to cisplatin, C2-ceramide and cadmium dichloride
To gain further insights into the signaling pathways elicited by CDDP, C2-CER and CdCl2, we determined the transcriptional response elicited by these compounds in human NSCLC A549 cells. Following a strategy that we have recently used to determine the microRNAome of CDDP-treated cells,Citation20 mRNA samples for microarray-based transcriptomic studies were generated from A549 cells that had previously been left untreated or exposed to sub-apoptotic doses of CDDP. In particular, we selected two doses of CDDP that would induce cell death [as determined by cytofluorometry upon co-staining with the exclusion dye propidium iodide and the mitochondrial transmembrane potential-sensitive fluorochrome DiOC6(3)]Citation23,Citation24 in 50% of the cells at 24 and 48 h (i.e., 100 and 50 µM, respectively)Citation20 and collected samples earlier, at 12 and 24 h, respectively. We reasoned that these conditions would allow for the study of transcriptional alterations linked to pre-apoptotic signaling rather than to degradative phenomena that characterize end-stage cell death. The same reasoning was used to obtain the transcriptional signatures elicited in A549 cells by C2-CER and CdCl2. Of note, in this setting, CDDP, C-CER and CdCl2 trigger apoptotic cell death with comparable dose response curves, yet slightly different kinetics.Citation20
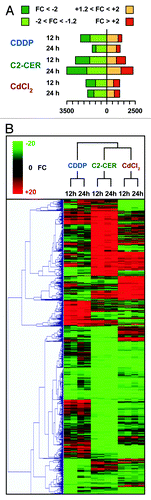
Microarray data were analyzed for statistical significance (threshold level set to p < 10−5) and processed for the computation of fold changes (FC = probe intensity in sample/probe intensity in control conditions, if probe intensity in sample > probe intensity in control conditions; FC = -probe intensity in control conditions/probe intensity in sample, if probe intensity in sample < probe intensity in control conditions). Each of the six experimental conditions analyzed was then associated with a transcriptional signature, including all transcripts associated with a statistically significant FC, as well as with a more “stringent signature,” including transcripts associated with a FC > 2 or < -2 (; Spreadsheet S1).
Figure 2. Transcriptional signatures of A549 cells responding to cisplatin (CDDP), C2-ceramide (C2-CER) and cadmium dichloride (CdCl2). (A) Graphical representation of the absolute number of gene products significantly up- or downregulated in A549 cells by the administration of CDDP, C2-CER and CdCl2 for 12 or 24 h. (B) Unsupervised hierarchical clustering of the transcriptional signatures represented in (A). Please note that each experimental condition has been analyzed twice, with dye-swapped samples. FC = fold change, as compared with control conditions (untreated cells). See also, Spreadsheets S1–S3.
To comparatively evaluate the transcriptional signatures associated with the administration of CDDP, C2-CER and CdCl2 to A549 cells, we undertook three distinct approaches. First, we performed a hierarchical cluster analysis based on cosine correlation and on the agglomerative method of the average link (). Second, we identified transcripts whose abundance was altered specifically by CDDP, C2-CER or CdCl2 (at both the 12 h and the 24 h time points) (inducer- and time-specific signatures) (; Spreadsheet S2), as opposed to transcripts whose levels were modified (in the same direction) by more than one of these agents (overlapping, time-specific signatures) (; Spreadsheet S3). Third, we identified the transcriptional alterations (as induced by CDDP, C2-CER or CdCl2), that remained consistent in time, i.e., which could be detected at both the 12 h and 24 h time points. In addition, we deconvoluted such “time-correlated signatures” into an inducer-specific and an overlapping component (; Spreadsheet S4).
Figure 3. Transcriptional signatures of A549 cells responding to cisplatin (CDDP), C2-ceramide (C2-CER) and cadmium dichloride (CdCl2). (A) Graphical representation of time-specific transcriptional signatures exhibited by A549 cells treated with CDDP, C2-CER and CdCl2 for 12 or 24 h. (B) Graphical representation of time-correlated transcriptional signatures exhibited by A549 cells as in (A). See also, Spreadsheets S1–S4.
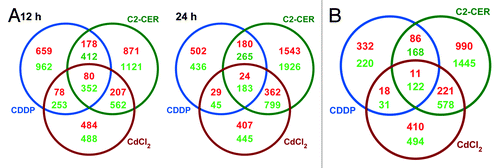
The global transcriptional alterations elicited in A549 cells by CDDP are relatively distinct as compared with those triggered by C2-CER and CdCl2 (). Accordingly, C2-CER and CdCl2 shared with each other a much larger faction of their transcriptional profile than that shared by either of them with CDDP. This held true both when the analysis was performed on the two time points separately () and when time-correlated signatures were considered (). Unexpectedly, CDDP-associated signatures overlapped to a very limited extent with 85 functional CDDP response modifiers (CRMs) that we have recently identified in A549 cells by a genome-wide siRNA-based screening effort.Citation25 Indeed, at 12 and 24 h, transcripts coding for only 15 and 11 out of 85 CRMs were found to be significantly up- or downregulated by the administration of CDDP (; Spreadsheet S5). Among them, however, was the mRNA coding for pyridoxal kinase (PDKX, FC at 12 h = -1.773, FC at 24 h = -1.443), the enzyme that generates the bioactive form of vitamin B6, which is required for optimal CDDP responses, in vitro and in vivo.Citation25 Of note, the CRMs that exhibited the most relevant (and consistent over time) transcriptional alterations in response to CDDP were dehydrogenase/reductase (SDR family) X-linked (DHRSX, FC at 12 h = -4.944, FC at 24 h = -5.395) and Ras-related associated with diabetes (RRAD, FC at 12 h = +6.592, FC at 24 h = +3.754) (; Spreadsheet S5).
Table 1. Transcriptional modulation of cisplatin (CDDP)-response modifiers (excerpt from Spreadsheet S5)
A very limited set of transcripts was consistently up- (11 transcripts) or downregulated (122 transcripts) in all experimental conditions. The former included mRNAs coding for the pro-apoptotic proteins GADD45α and Noxa, whereas the latter encompassed mRNAs coding for anti-apoptotic proteins such as survivin and glutathione reductase, for several members of the minichromosome maintenance deficient (MCM) protein family, i.e., MCM3, MCM5, MCM6 and MCM7 (which are involved in both DNA repair and cell cycle progression) and for transcription factors like E2F1, E2F2, NFIA and PBX1 (; Spreadsheet S4). As these transcriptional changes were always detected, irrespective of cell death inducer and time point, we hypothesize that they may constitute non-causative transcriptional markers of A549 cell death. Further experiments are required to validate this assumption.
Gene ontology and KEGG analyses
To better understand the global transcriptomic variations in A549 cells exposed to CDDP, C2-CER and CdCl2, we annotated the transcriptional signatures described above based on two publicly available databases, Gene Ontology (GO, www.geneontology.org) and the Kyoto Encyclopedia of Genes and Genomes PATHWAY (KEGG, www.genome.jp/kegg/). While the former annotates gene products according to general biological processes, molecular functions and cell components,Citation26 the latter provides a more precise annotation, linking gene products to defined biochemical circuitries.Citation27 We then checked whether any of the GO or KEGG terms would be overrepresented in the transcriptional signatures by using the conditional hypergeometric algorithm,Citation28 setting the thresholds for statistically significant overrepresentation at p < 0.01 (for GO) and p < 0.05 (for KEGG). For practical purposes, we limited our analysis to the 10 GO terms and 20 KEGG terms with the highest probability of overrepresentation (associated with the lowest p values). The results of these analyses (Spreadsheet S6 and S7) de facto confirm what we observed by the study of transcriptional signatures: CDDP triggers transcriptomic alterations that involve biological processes, molecular functions and signaling pathway, that are less similar to those elicited by C2-CER and CdCl2 than the latter to each other.
Limited overlap among the signaling pathways leading to cisplatin-, C2-ceramide- and cadmium dichloride-induced cell death
To study functional aspects of the molecular cascades elicited by CDDP, C2-CER and CdCl2 in a genetically tractable organism, we performed clonogenic assays on a panel of Saccharomyces cerevisiae strains (Table S1). These strains were selected as they lack genes coding for proteins that (directly or as orthologs in mammalian cells) are involved in apoptosis (aif1, cyc1, cyc7, mca1, ndi1, nma111, nuc1, aac3, por1, por2, ynl305c), autophagy (atg5, atg10, atg12, vps30, vps34), mitochondrial functions and dynamics (dnm1, fis1, mdm10, mgm1, mir1, oxa1, pcp1, sam37, tim18), cell cycle regulation (chk1, mbp1), DNA replication and repair (mcm2, mcm3, mcm5, mcm6, msh6, psf2), chromatin structure (nhp6a, nhp6b), substrate phosphorylation (adk1, adk2, glk1, hxk1, hxk2, tor1), protein degradation (pep4, rim13, aac1), chaperoning (ecm10, ssq1) and other biological functions related to stress management (cpr3, cyb5, gtt2, nnt1, sod2, tdh1, tdh2, tdh3, trx3). In addition, one yeast strain lacking mitochondrial DNA (rho0) was used.
Conditions were optimized so that the administration of CDDP, C2-CER and CdCl2 would lead to a reduction in the clonogenic potential of the parental strains (haploid BY4741 or diploid BY4743 cells) of approximately 40–60%. We reasoned that, in this way, we would be able to observe both increased and decreased clonogenicity. By this experimental approach, we identified five conditions that aggravate the reduction of clonogenic potential triggered in yeast by CDDP (i.e., the lack of mir1, tdh1, aac3, rim13 and mitochondrial DNA) and one condition that relieves it (i.e., the absence of por2). Along similar lines, we identified Mir1, Tdh1, Por1, Adk1, Fis1, Mdm10 and Vps34 as factors that limit the loss of clonogenicity induced in S. cerevisiae by C2-CER. Finally, Por1 and Rim13 were shown to protect yeast cells against the cytotoxic activity of CdCl2 ().
Figure 4. Response to cisplatin (CDDP), C2-ceramide (C2-CER) and cadmium dichloride (CdCl2) of a panel of Saccharomyces cerevisiae knockout strains. Clonogenic survival of a panel of Saccharomyces cerevisiae strains deficient for the indicated genes upon treatment with CDDP (left panel), C2-CER (middle panel) or CdCl2 (right panel). Data are represented as means ± SEM (n = 3) upon normalization to the clonogenic survival of parental (either haploid BY4741 or diploid BY4743, as appropriate) cells treated with the same compound. *p < 0.05, **p < 0.01 (Student’s t-test), as compared with parental cells.
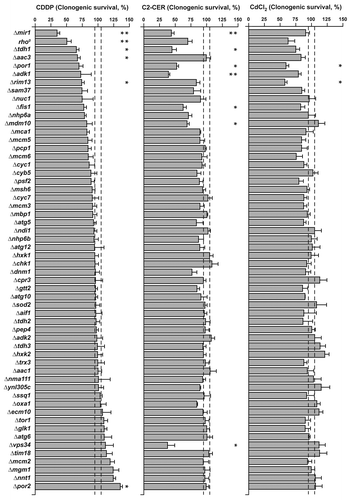
With regards to CDDP-relevant factors, it should be noted that Mir1 is the yeast ortholog of the human mitochondrial phosphate carrier SLC25A3, which has been shown to play a role in mitochondrial apoptosis and fragmentation.Citation29-Citation31 tdh1 codes for the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase, which has been shown to participate in oxidative stress signalingCitation32 and may mediate mitochondrial membrane permeabilization.Citation33 Aac3 is one of the yeast orthologs of mammalian adenine nucleotide translocases, whose role at the interface between energetic metabolism and cell death signaling is well established.Citation34 Rim13 is a calpain-like protease,Citation35 and mammalian calpains have been involved in the execution of apoptotic and necrotic cell death upon lysosomal membrane permeabilization.Citation36,Citation37 Finally, Por2 is one of the yeast orthologs of mammalian voltage-dependent anion channels, which also operate at the frontiers between bioenergetic metabolism and cell death signaling.Citation7,Citation38
In summary, similar to what we observed for the transcriptional response of mammalian A549 cells, CDDP, C2-CER and CdCl2 reduce the clonogenic potential of yeast cells via mechanisms that exhibit limited overlap with each other.
Concluding remarks
Here, we report the results of a multipronged experimental approach demonstrating that the signaling pathways triggered by CDDP (1) do not involve a direct mitochondrion-permeabilizing effect of CDDP; (2) are accompanied by transcriptional alterations that are rather dissimilar to those elicited by chemotherapy-unrelated inducers of mitochondrial apoptosis such as C2-CER and CdCl2 and (3) involve genes and proteins that are not functionally relevant for cell death as induced by C2-CER and CdCl2.
Importantly, the transcriptional signatures of A549 cells treated with CDDP failed to exhibit consistent overlaps with the functional CRMs that have recently been identified in a genome-wide siRNA-based screen, indicating that a significant fraction of CDDP cytotoxicity proceeds via post-transcriptional mechanisms. Of note, however, the mRNA coding for PDXK was downregulated by A549 cells exposed to CDDP at both 12 and 24 h (; Spreadsheet S5). PDXK is required for optimal CDDP responses in vitro and in vivo,Citation25 suggesting that the transcriptional downregulation of PDXK reflects the activation of an adaptive mechanism leading to CDDP resistance rather than underlying its cytotoxicity. In line with this possibility, several cancer cell lines that had been rendered CDDP-resistant by prolonged culture in the presence of low-dose CDDP expressed either decreased levels of PDXK or increased levels of its antagonistic enzyme pyridoxal phosphate (PDXP).Citation25
The CRMs that exhibited the most pronounced transcriptional alterations in A549 cells responding to CDDP were DHRSX (which was consistently downregulated) and RRAD (which was consistently upregulated) (; Spreadsheets S5). The siRNA-mediated knockdown of DHRSX protects A549 cells from the lethal effects of CDDP, similar to the knockdown of PDKX. Conversely, the depletion of RRAD increases the sensitivity of A549 cells to CDDP.Citation25 Thus, the transcriptional alterations of DHRSX and RRAD that we detected in the course of CDDP-induced cell death are also likely to reflect the activation of a cytoprotective response rather than to contribute to CDDP cytotoxicity. Intriguingly, whereas the mRNA coding for RRAD was also upregulated in response to C2-CER (at 24 h) and CdCl2 (at both 12 and 24 h), the abundance of the PDXK-encoding mRNA was unaltered in the presence of CdCl2, and the DHRSX-coding transcript was regulated in an inducer-specific pattern (Spreadsheet S5). This suggests that some CRMs may be implicated in the signaling cascades elicited by chemotherapy-unrelated inducers of mitochondrial apoptosis. In line with this notion, the siRNA-mediated depletion of PDXK has also been shown to protect A549 cells from the cytotoxic effects of C2-CER and CdCl2.Citation25
Altogether, CDDP appears to trigger the apoptotic death of A549 via signaling pathways that are relatively “private,” in that they exhibit limited overlap to those elicited by other inducers of mitochondrial apoptosis. Moreover, CDDP induces apoptosis in a largely transcription-independent fashion, as most CRMs are not transcriptionally modulated during CDDP-induced cell death.
Materials and Methods
Chemicals and human cell cultures
Unless otherwise specified, chemicals were purchased from Sigma-Aldrich, culture media and supplements for cell culture from Gibco-Invitrogen and plasticware from Corning. Human wild-type (WT), non-small cell lung carcinoma (NSCLC) A549 cells were cultured in Glutamax®-containing DMEM/F12 medium supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES buffer, 100 units/mL penicillin G sodium and 100 µg/mL streptomycin sulfate. Cells were routinely maintained at 37°C under 5% CO2 in T175 flasks, and seeded into appropriate supports 24 h before the experiments.
Transcriptome studies
NSCLC A549 cells were treated with 100 µM cisplatin (CDDP), 75 µM C2-ceramide (C2-CER) or 100 µM cadmium dichloride (CdCl2) for 12 h, or with 50 µM CDDP, C2-CER or CdCl2 for 24 h. Thereafter, cells were collected and lysed for RNA extraction, following established procedures.Citation39,Citation40 mRNA expression changes were quantified on G4112A 44k Whole Human Genome Oligo Microarrays (Agilent Technologies), as previously reported.Citation8 mRNA microarrays underwent standardized post-hybridization processing, image acquisition and analysis.Citation8
Data from microarray experiments were analyzed with the Rosetta Resolver® System (Rosetta Biosoftware). The threshold for statistical significance of probe signals (Intensity) was set at p = 10−5. In case of replicate probes for the same gene product, average intensity was calculated for all correlating probes that exhibited p < 10−5. For each mRNA, fold change (FC) was then defined as intensitysample/intensitycontrol when intensitysample > intensitycontrol or as –(intensitycontrol/intensitysample) when intensitysample < intensitycontrol.
Yeast assays
The haploid WT Saccharomyces cerevisiae strain BY4741 (MATa; his3Δ1; leu2Δ0; met15Δ0; ura3Δ0), its diploid counterpart BY4743 (MATa/MATα; his3Δ1/his3Δ1; leu2Δ0/leu2Δ0; MET15/met15Δ0; LYS2/lys2Δ0; ura3Δ0/ura3Δ0) as well as the null-mutants employed in this study were obtained from Euroscarf (Table S1). All yeast experiments were performed by culturing cells on a routine basis at 28°C in synthetic complete medium supplemented with 2% D-glucose, amino acids, adenine and uracil,Citation41 with the exception of large-scale CdCl2 toxicity determinations, for which yeast extract peptone dextrose (YEPD) medium containing 1% yeast extract (Difco Laboratories), 2% bacto peptone (Difco Laboratories) and 4% D-glucose was used. Stationary phase cells from an overnight culture (8–9 × 107 cells/mL) were inoculated in 10 mL of medium at a density of 2.5 × 105 cells/mL, allowed to grow at 28°C for additional 4 h (up to a final concentration of approximately 1 × 106 cells/mL) and treated for further 4 h with 0.5 mM CDDP, 40 μM C2-CER or 0.5 mM CdCl2. Control cultures were treated with an equal volume of solvent: N-N-dimethylformamide (DMF) for CDDP, dimethylsulfoxide (DMSO) for C2-CER and H2O for CdCl2.
Cell number was then quantified by means of a CASY® cell counter (Schärfe System), and 500 cells from each experimental condition were seeded on three parallel YEPD plates. Following 3 d of incubation at 28°C, plates were finally subjected to the quantification of colony-forming units (CFUs). For each genotype and experimental condition, clonogenic survival was calculated as the ratio between the number of CFUs observed upon the administration of CDDP, C2-CER or CdCl2 in knockout cells and the number of CFUs observed upon the administration of CDDP, C2-CER or CdCl2 in parental (either haploid BY4741 or diploid BY4743, appropriate) cells.
Isolation of rat liver mitochondria and assessment of mitochondrial swelling
Mitochondria were isolated from the liver of male Wistar Kyoto rats (Charles River Laboratories Inc.) by differential centrifugation, as previously described.Citation42 Briefly, immediately after organ collection, livers were homogenized with a glass teflon homogenizer in an isolation buffer containing 10 mM triethanolamine, 10 mM acetic acid, 280 mM sucrose, 0.2 mM EGTA (pH adjusted to 7.4 with KOH). Homogenates were cleared from debris and nuclei by two rounds of centrifugation at 750 G (10 min, 4°C). Mitochondria then were pelleted by centrifugation at 9,000 G (10 min, 4°C), washed three times in isolation buffer (once at 9,000 G, then at 15,000 G, 10 min, 4°C) and resuspended in EGTA-free isolation buffer. The integrity and functionality of isolated mitochondria was routinely checked by standard respiratory measurements.Citation42
For large-amplitude swelling determinations, mitochondria were resuspended in swelling buffer (10 mM Tris-MOPS pH 7.4, 200 mM sucrose, 5 mM succinate, 1 mM phosphate, 10 µM EGTA and 2 µM rotenone) and pre-incubated for 5 min at RT with swelling buffer (negative control) or either of the following permeability transition inhibitors: 10 or 40 μM bongkrekic acid (BA), 5 or 10 μM cyclosporine A (CsA); 0.1 or 0.5 mM reduced glutathione (GSH) or 2 or 10 mM N-acetyl-cysteine (NAC). Then, swelling buffer (negative control) or either of the following chemicals were added: 50 or 100 μM Ca2+ (positive control), 50 μM GD3 ganglioside, 20, 100 or 200 μM CDDP, 5, 50 or 100 μM C2-CER or 20, 50 or 100 μM CdCl2. The final assay volume was 200 µL, containing 0.5 mg/mL mitochondria. Permeability transition-induced large-amplitude swelling was assessed by measuring light scattering of mitochondrial suspensions at 540 nm on a standard microplate absorbance reader (µ-Quant™, Bio-Tek) every minute over a total assay time of 30 min (at RT), as previously described.Citation42
The following mathematical model has been generated from the entire absorbance data set: ABSijt = μ + αi + repij + βi × t + γi × √t + εijt where i = experiment number, j = replicate number, t = time and εijt = model residues, leading to the definition of the following swelling classes (to which each inhibitor/inducer combination has been assigned):
Class 0 (no swelling) = γi + 6.5 x βi > -0.01
Class A (linear decrease in absorbance) = γi + 6.5 x βi ≥ +0.01 and -0.05 ≤ γi ≤ 0.03
Class B (logarithmic decrease in absorbance) =γi + 6.5 x βi ≥ +0.01 and -0.15 ≤ γi ≤ 0.05
Class A and B have been further subdivided according to the amplitude of absorbance decrease (ξ = (ABSmax - ABSmin) / ABSmax) into:
Subclass 1 = ξ < 0.35
Subclass 2 = ξ ≥ 0.35.
Common statistical procedures
Unless otherwise specified, all experiments were performed in triplicate and repeated at least twice.Citation43,Citation44 Data are reported as means ± SEM and were analyzed with Microsoft Excel (Microsoft Co.) or with the free, open-source R Studio integrated development environment (www.rstudio.org). Statistical significance was assessed by means of two-tailed unpaired Student’s t-tests. Unless otherwise specified, p values < 0.05 were considered statistically significant.
| Abbreviations: | ||
| BA | = | bongkrekic acid |
| C2-CER | = | C2-ceramide |
| CDDP | = | cisplatin |
| CFU | = | colony-forming unit |
| CRM | = | CDDP response modifier |
| CsA | = | cyclosporine A |
| ∆ψm | = | mitochondrial transmembrane potential |
| DiOC6(3) | = | 3,3′dihexiloxalocarbocyanine iodide |
| DMF | = | N-N-dimethylformamide |
| DMSO | = | dimethylsulfoxide |
| FC | = | fold change |
| GO | = | Gene Ontology |
| GSH | = | reduced glutathione |
| KEGG | = | Kyoto Encyclopedia of Genes and Genomes |
| NAC | = | N-acetyl-cysteine |
| NSCLC | = | non-small cell lung carcinoma |
| PTPC | = | permeability transition pore complex |
| ROS | = | reactive oxygen species |
| SEM | = | standard error of the mean |
| YEPD | = | yeast extract peptone dextrose |
Additional material
Download Zip (3.5 MB)Acknowledgments
L.G. is funded by the LabEx ImmunoOncology while D.C.G. receives a Lipotoxicity grant from the Austrian FWF. F.M. is grateful to the FWF (grants LIPOTOX, P23490-B12, P24381-B20, DK MCD W 1226-B18) and to the EC for grant Apo-Sys. G.K. is supported by the Ligue Nationale contre le Cancer (Equipe labelisée), Agence Nationale pour la Recherche (ANR), European Commission (Active p53, Apo-Sys, Artforce, ChemoRes, ApopTrain), Fondation pour la Recherche Médicale (FRM), Institut National du Cancer (INCa), Cancéropôle Ile-de-France, AXA Research Fund and the Labex Immuno-Oncology.
Related Research Data
References
- Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer 2007; 7:573 - 84; http://dx.doi.org/10.1038/nrc2167; PMID: 17625587
- Nardella C, Clohessy JG, Alimonti A, Pandolfi PP. Pro-senescence therapy for cancer treatment. Nat Rev Cancer 2011; 11:503 - 11; http://dx.doi.org/10.1038/nrc3057; PMID: 21701512
- Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev 2007; 87:99 - 163; http://dx.doi.org/10.1152/physrev.00013.2006; PMID: 17237344
- Mandic A, Hansson J, Linder S, Shoshan MC. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J Biol Chem 2003; 278:9100 - 6; http://dx.doi.org/10.1074/jbc.M210284200; PMID: 12509415
- Berndtsson M, Hägg M, Panaretakis T, Havelka AM, Shoshan MC, Linder S. Acute apoptosis by cisplatin requires induction of reactive oxygen species but is not associated with damage to nuclear DNA. Int J Cancer 2007; 120:175 - 80; http://dx.doi.org/10.1002/ijc.22132; PMID: 17044026
- Gonzalez VM, Fuertes MA, Alonso C, Perez JM. Is cisplatin-induced cell death always produced by apoptosis?. Mol Pharmacol 2001; 59:657 - 63; PMID: 11259608
- Tajeddine N, Galluzzi L, Kepp O, Hangen E, Morselli E, Senovilla L, et al. Hierarchical involvement of Bak, VDAC1 and Bax in cisplatin-induced cell death. Oncogene 2008; 27:4221 - 32; http://dx.doi.org/10.1038/onc.2008.63; PMID: 18362892
- Castedo M, Coquelle A, Vivet S, Vitale I, Kauffmann A, Dessen P, et al. Apoptosis regulation in tetraploid cancer cells. EMBO J 2006; 25:2584 - 95; http://dx.doi.org/10.1038/sj.emboj.7601127; PMID: 16675948
- Winter C, Albers P. Testicular germ cell tumors: pathogenesis, diagnosis and treatment. Nat Rev Endocrinol 2011; 7:43 - 53; http://dx.doi.org/10.1038/nrendo.2010.196; PMID: 21116298
- Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, et al. Molecular mechanisms of cisplatin resistance. Oncogene 2012; 31:1869 - 83; http://dx.doi.org/10.1038/onc.2011.384; PMID: 21892204
- Köberle B, Tomicic MT, Usanova S, Kaina B. Cisplatin resistance: preclinical findings and clinical implications. Biochim Biophys Acta 2010; 1806:172 - 82; PMID: 20647037
- Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene 2003; 22:7265 - 79; http://dx.doi.org/10.1038/sj.onc.1206933; PMID: 14576837
- Beers MH. Lung carcinoma. In: Porter RS, Jones TV, eds. The Merck manual of diagnosis and therapy. Rahway: Merck & Co., Inc., 2008:2992.
- Jemal A, Thun MJ, Ries LA, Howe HL, Weir HK, Center MM, et al. Annual report to the nation on the status of cancer, 1975-2005, featuring trends in lung cancer, tobacco use, and tobacco control. J Natl Cancer Inst 2008; 100:1672 - 94; http://dx.doi.org/10.1093/jnci/djn389; PMID: 19033571
- Brenner C, Grimm S. The permeability transition pore complex in cancer cell death. Oncogene 2006; 25:4744 - 56; http://dx.doi.org/10.1038/sj.onc.1209609; PMID: 16892087
- Rippo MR, Malisan F, Ravagnan L, Tomassini B, Condo I, Costantini P, et al. GD3 ganglioside directly targets mitochondria in a bcl-2-controlled fashion. FASEB J 2000; 14:2047 - 54; http://dx.doi.org/10.1096/fj.99-1028com; PMID: 11023989
- Jimi S, Uchiyama M, Takaki A, Suzumiya J, Hara S. Mechanisms of cell death induced by cadmium and arsenic. Ann NY Acad Sci 2004; 1011:325 - 31; http://dx.doi.org/10.1196/annals.1293.032; PMID: 15126309
- Novgorodov SA, Gudz TI, Milgrom YM, Brierley GP. The permeability transition in heart mitochondria is regulated synergistically by ADP and cyclosporin A. J Biol Chem 1992; 267:16274 - 82; PMID: 1644813
- Lê Quôc K, Lê Quôc D. Involvement of the ADP/ATP carrier in calcium-induced perturbations of the mitochondrial inner membrane permeability: importance of the orientation of the nucleotide binding site. Arch Biochem Biophys 1988; 265:249 - 57; http://dx.doi.org/10.1016/0003-9861(88)90125-7; PMID: 2844116
- Galluzzi L, Morselli E, Vitale I, Kepp O, Senovilla L, Criollo A, et al. miR-181a and miR-630 regulate cisplatin-induced cancer cell death. Cancer Res 2010; 70:1793 - 803; http://dx.doi.org/10.1158/0008-5472.CAN-09-3112; PMID: 20145152
- Kepp O, Galluzzi L, Lipinski M, Yuan J, Kroemer G. Cell death assays for drug discovery. Nat Rev Drug Discov 2011; 10:221 - 37; http://dx.doi.org/10.1038/nrd3373; PMID: 21358741
- Stoica BA, Movsesyan VA, Lea PM 4th, Faden AI. Ceramide-induced neuronal apoptosis is associated with dephosphorylation of Akt, BAD, FKHR, GSK-3beta, and induction of the mitochondrial-dependent intrinsic caspase pathway. Mol Cell Neurosci 2003; 22:365 - 82; http://dx.doi.org/10.1016/S1044-7431(02)00028-3; PMID: 12691738
- Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ 2009; 16:1093 - 107; http://dx.doi.org/10.1038/cdd.2009.44; PMID: 19373242
- Galluzzi L, Zamzami N, de La Motte Rouge T, Lemaire C, Brenner C, Kroemer G. Methods for the assessment of mitochondrial membrane permeabilization in apoptosis. Apoptosis 2007; 12:803 - 13; http://dx.doi.org/10.1007/s10495-007-0720-1; PMID: 17294081
- Galluzzi L, Vitale I, Senovilla L, Olaussen KA, Pinna G, Eisenberg T, et al. Prognostic Impact of Vitamin B6 Metabolism in Lung Cancer. Cell Rep 2012; In press http://dx.doi.org/10.1016/j.celrep.2012.06.017; PMID: 22854025
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al, The Gene Ontology Consortium. Gene ontology: tool for the unification of biology. Nat Genet 2000; 25:25 - 9; http://dx.doi.org/10.1038/75556; PMID: 10802651
- Kanehisa M. A database for post-genome analysis. Trends Genet 1997; 13:375 - 6; http://dx.doi.org/10.1016/S0168-9525(97)01223-7; PMID: 9287494
- Alexa A, Rahnenführer J, Lengauer T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 2006; 22:1600 - 7; http://dx.doi.org/10.1093/bioinformatics/btl140; PMID: 16606683
- Pauleau AL, Galluzzi L, Scholz SR, Larochette N, Kepp O, Kroemer G. Unexpected role of the phosphate carrier in mitochondrial fragmentation. Cell Death Differ 2008; 15:616 - 8; http://dx.doi.org/10.1038/sj.cdd.4402295; PMID: 18174900
- Poncet D, Pauleau AL, Szabadkai G, Vozza A, Scholz SR, Le Bras M, et al. Cytopathic effects of the cytomegalovirus-encoded apoptosis inhibitory protein vMIA. J Cell Biol 2006; 174:985 - 96; http://dx.doi.org/10.1083/jcb.200604069; PMID: 16982800
- Büttner S, Ruli D, Vögtle FN, Galluzzi L, Moitzi B, Eisenberg T, et al. A yeast BH3-only protein mediates the mitochondrial pathway of apoptosis. EMBO J 2011; 30:2779 - 92; http://dx.doi.org/10.1038/emboj.2011.197; PMID: 21673659
- Morigasaki S, Shimada K, Ikner A, Yanagida M, Shiozaki K. Glycolytic enzyme GAPDH promotes peroxide stress signaling through multistep phosphorelay to a MAPK cascade. Mol Cell 2008; 30:108 - 13; http://dx.doi.org/10.1016/j.molcel.2008.01.017; PMID: 18406331
- Tarze A, Deniaud A, Le Bras M, Maillier E, Molle D, Larochette N, et al. GAPDH, a novel regulator of the pro-apoptotic mitochondrial membrane permeabilization. Oncogene 2007; 26:2606 - 20; http://dx.doi.org/10.1038/sj.onc.1210074; PMID: 17072346
- Zhivotovsky B, Galluzzi L, Kepp O, Kroemer G. Adenine nucleotide translocase: a component of the phylogenetically conserved cell death machinery. Cell Death Differ 2009; 16:1419 - 25; http://dx.doi.org/10.1038/cdd.2009.118; PMID: 19696789
- Futai E, Maeda T, Sorimachi H, Kitamoto K, Ishiura S, Suzuki K. The protease activity of a calpain-like cysteine protease in Saccharomyces cerevisiae is required for alkaline adaptation and sporulation. Mol Gen Genet 1999; 260:559 - 68; http://dx.doi.org/10.1007/s004380050929; PMID: 9928935
- Kroemer G, Jäättelä M. Lysosomes and autophagy in cell death control. Nat Rev Cancer 2005; 5:886 - 97; http://dx.doi.org/10.1038/nrc1738; PMID: 16239905
- Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 2010; 11:700 - 14; http://dx.doi.org/10.1038/nrm2970; PMID: 20823910
- Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 2003; 301:513 - 7; http://dx.doi.org/10.1126/science.1083995; PMID: 12881569
- de La Motte Rouge T, Galluzzi L, Olaussen KA, Zermati Y, Tasdemir E, Robert T, et al. A novel epidermal growth factor receptor inhibitor promotes apoptosis in non-small cell lung cancer cells resistant to erlotinib. Cancer Res 2007; 67:6253 - 62; http://dx.doi.org/10.1158/0008-5472.CAN-07-0538; PMID: 17616683
- Boehrer S, Adès L, Braun T, Galluzzi L, Grosjean J, Fabre C, et al. Erlotinib exhibits antineoplastic off-target effects in AML and MDS: a preclinical study. Blood 2008; 111:2170 - 80; http://dx.doi.org/10.1182/blood-2007-07-100362; PMID: 17925489
- Madeo F, Herker E, Maldener C, Wissing S, Lächelt S, Herlan M, et al. A caspase-related protease regulates apoptosis in yeast. Mol Cell 2002; 9:911 - 7; http://dx.doi.org/10.1016/S1097-2765(02)00501-4; PMID: 11983181
- Zischka H, Larochette N, Hoffmann F, Hamöller D, Jägemann N, Lichtmannegger J, et al. Electrophoretic analysis of the mitochondrial outer membrane rupture induced by permeability transition. Anal Chem 2008; 80:5051 - 8; http://dx.doi.org/10.1021/ac800173r; PMID: 18510346
- Morselli E, Maiuri MC, Markaki M, Megalou E, Pasparaki A, Palikaras K, et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis 2010; 1:e10; http://dx.doi.org/10.1038/cddis.2009.8; PMID: 21364612
- Vitale I, Senovilla L, Jemaà M, Michaud M, Galluzzi L, Kepp O, et al. Multipolar mitosis of tetraploid cells: inhibition by p53 and dependency on Mos. EMBO J 2010; 29:1272 - 84; http://dx.doi.org/10.1038/emboj.2010.11; PMID: 20186124