Abstract
REV1 is a Y-family polymerase specialized for replicating across DNA lesions at the stalled replication folk. Due to the high error rate of REV1-dependent translesion DNA synthesis (TLS), tight regulation of REV1 activity is essential. Here, we show that human REV1 undergoes proteosomal degradation mediated by the E3 ubiquitin ligase known as anaphase-promoting complex (APC). REV1 associates with APC. Overexpression of APC coactivator CDH1 or CDC20 promotes polyubiquitination and proteosomal degradation of REV1. Surprisingly, polyubiquitination of REV1 also requires REV7, a TLS accessory protein that interacts with REV1 and other TLS polymerases. The N-terminal region of REV1 contains both the APC degron and an additional REV7-binding domain. Depletion of REV7 by RNA interference stabilizes REV1 by preventing polyubiquitination, whereas overexpression of REV7 augments REV1 degradation. Taken together, our findings suggest a role of REV7 in governing REV1 stability and interplay between TLS and APC-dependent proteolysis.
Introduction
Replicative DNA polymerases are endowed with high processivity and high fidelity. Owing to the stringency of their active sites, their progression at replication fork is blocked at lesions caused by DNA damage. In order to maintain the integrity of genome, cells have evolved mechanisms to repair damaged DNA.Citation1 However, some damage evades repair leading to persistent replication block. Because stalled replication can cause chromosome abnormalities or cell death, cells utilize DNA damage tolerance machinery to allow replication past DNA lesions. Translesion DNA synthesis (TLS) is an extensively studied damage-tolerance mechanism. Specialized DNA polymerases, which possess low processivity and fidelity, are employed in TLS, because their active sites are more relaxed and therefore can accommodate bulky distorted damaged bases.Citation2,Citation3 TLS is evolutionarily conserved from bacteria to eukaryotes and most DNA polymerases involved belong to the Y-family. There are two Y-family polymerases in budding yeast, known as REV1 and polymerase η (Polη), while two additional enzymes, Polι and Polκ, are found in mammals. In eukaryotes, a B-family polymerase Polζ, which is a heterodimer of REV3 and REV7, also participates in TLS.Citation4
Due to its low fidelity, TLS is critically involved in tumorigenesis. On one hand, point mutations seen in cancer cells are largely derived from TLS. In this sense, TLS would affect genome instability that drives tumorigenesis. On the other hand, tolerance of DNA damage mediated by TLS might have protective roles in cancer cells.Citation3 Particularly, loss of Polζ was found to enhance spontaneous tumorigenesis.Citation5 It is not surprising that expression and activity of TLS polymerases in cancer cells would influence cellular sensitivity to anticancer drugs such as cisplatin.Citation6 In addition, many oncogenic and tumor-suppressive proteins might function by targeting TLS polymerases. For instance, Fanconi anemia core complex was recently found to target REV1.Citation7
REV1 is a specialized dCMP transferase whose activity is conserved throughout eukaryotes. However, the essentiality and the exact roles of dCMP transferase activity of REV1 in TLS and DNA damage tolerance remain to be clarified and may vary depending on the type of DNA lesions.Citation8,Citation9 REV1 physically binds to many proteins involved in TLS. REV1 harbors at its N terminus a BRCT domain, which is responsible for binding with proliferating cell nuclear antigen (PCNA).Citation10 REV1, like other Y-family polymerases, uses two ubiquitin-binding motifs (UBMs) to facilitate its interaction with monoubiquitinated PCNA.Citation11 The 100–150 amino acids at the C terminus of mammalian REV1 interact with other Y-family polymerases, including Polη, Polκ and Polι.Citation12,Citation13 The C terminus of human REV1 can also bind PCNA.Citation14 In addition, the C terminus of yeast REV1 binds to the catalytic subunit of Polζ, REV3, and this interaction promotes the proficiency of Polζ for mismatch extension and extension opposite DNA lesions.Citation15 Furthermore, yeast REV1 is capable of binding REV7, the accessory subunit of Polζ, via its BRCT domain, polymerase-associated domain (PAD) and the C-terminal region.Citation16,Citation17 The interaction of REV7 with REV1 and REV3 is conserved in all eukaryotes.Citation18,Citation19 Structural analysis of human REV3-REV7 complex suggests that REV7, upon REV3 binding, undergoes conformation change to provide an interface for REV1 binding.Citation20 Indeed, REV1 works closely with Polζ for most spontaneous and induced mutagenesis.Citation4,Citation21 Given its indispensability in Polζ’s function and its capability to interact with TLS polymerases and PCNA, REV1 is suggested to provide a platform for recruiting and coordinating TLS proteins at DNA lesion sites. It is also thought that the role of human REV1 and Polζ might even be extended to other damage tolerance mechanisms such as homologous recombination repair.Citation19
REV7, the non-catalytic subunit of Polζ, forms a stable complex with REV3 and substantially enhances the catalytic activity of REV3 at least in yeast and Drosophila.Citation22,Citation23 We have previously demonstrated that knockdown of REV7 in human cancer cell lines leads to hypersensitivity to DNA-damaging agents, reduction in spontaneous and drug-induced mutation frequencies and increase in chromosome aberration in response to DNA damage.Citation24 Similar findings were also obtained in REV7-compromised chicken DT40 cells and human foreskin fibroblasts, indicating a conservative role of REV7 in TLS in eukaryotes.Citation25,Citation26 REV7 is also called MAD2B because it shares high sequence homology with MAD2, a key regulator in mitotic checkpoint.Citation27 Although REV7 does not bind MAD1, an anchoring protein for MAD2 at the kinetochore at the onset of checkpoint activation,Citation28 REV7, like MAD2, is capable of binding CDH1 and/or CDC20, which are coactivators of anaphase-promoting complex (APC), to impede APC activation.Citation29,Citation30 APC is a multi-subunit E3 ubiquitin ligase, which targets proteins for proteasome-mediated degradation. CDC20 and CDH1 recognize APC substrates and promote substrate recruitment to APC.Citation31-Citation33 Two major substrate recognition motifs are destruction box (D box) with consensus sequence RxxLxxxxN/D/E and KEN box with sequence KENxxxN/D.Citation34,Citation35 Some substrates, such as ninein-like protein and forkhead box M1,Citation36,Citation37 contain both degrons or multiple copies of one or two degrons. Other non-canonical APC recognition motifs also contribute to substrate recognition.Citation38,Citation39 The roles of REV7 in DNA damage tolerance and ubiquitin-mediated proteolysis suggest that a crosstalk may exist between these two pathways, in which REV7 might act as an orchestrator. Consistent with the presence of a HORMA domain specialized for protein-protein interaction, REV7 has also been shown to interact with several other proteins, including Shigella IpaB protein and adenovirus E3-11.6K.Citation40,Citation41
Emerging evidence indicates that the expression and activity of TLS polymerases and related proteins are regulated by protein modifications. Ubiquitination of PCNA not only enhances its interaction with REV1, but might also activate REV1 and Polη.Citation42 Yeast and mammalian Polη can undergo proteasomal degradation.Citation43-Citation45 Human Polλ, a DNA repair enzyme implicated in base excision repair, non-homologous end joining and TLS, is stabilized by phosphorylation, which prohibits proteasomal degradation in late S and G2 phases.Citation46 Yeast REV1 mRNA and protein levels oscillate in a cell cycle-dependent manner.Citation47 Proteasomal degradation and DNA damage-induced hyperphosphorylation of yeast REV1 have also been reported.Citation48 Monoubiquitination of human REV1 has been shown to stabilize the interaction with a subunit of the Fanconi anemia core complex.Citation7 In another study, human REV1 has been suggested to undergo proteasomal degradation modulated by Hsp90 chaperone.Citation49 However, exactly how stability of human REV1 might be regulated by ubiquitination and proteasome-dependent proteolysis remains elusive. In this study, we characterize proteasomal degradation of human REV1. We further demonstrate that the destruction is mediated by APC. More importantly, we show that REV7 is required for APC-dependent degradation of REV1. We provide the first evidence for the involvement of APC in governing the stability of REV1.
Results
REV1 is degraded by proteasome
To determine whether REV1 is a fast-turnover protein, we first examined the decay of REV1 protein in FLAG-REV1-expressing HEK293T cells in which protein synthesis had been blocked by cycloheximide (CHX). Although a slight drop in REV1 protein amount was observed in control cells incubated with solvent (, lanes 1–4), REV1 level declined more swiftly along the time of incubation with CHX (, lanes 5–8). The half-life of REV1 in the presence of CHX was calculated to be 3.56 h (), which was much shorter than that (6.09 h) in its absence. A similar reduction in cyclin B1 expression in the same cells (, middle panel) not only verified the activity of CHX, but also indicated that both REV1 and cyclin B1 have a fast turnover.
Figure 1A–D. Proteasome-dependent degradation of REV1. (A) Fast turnover of REV1 protein. HEK293T cells were transiently transfected with FLAG-REV1-expressing plasmid. Forty-eight hours after transfection, 15 μg/mL cycloheximide (CHX, lanes 5–8) or solvent (ethanol) alone (lanes 1–4) was added to cells, and cells were harvested at indicated time points after treatment. FLAG-REV1 levels were analyzed by western blotting with anti-FLAG antibody. Endogenous cyclin B1 (cyc B1) protein levels were also detected as a control for CHX activity. (B) FLAG-REV1 protein signals in A were quantified and plotted. Its half-lives (t1/2) upon addition of CHX and solvent were calculated as described.Citation74,Citation75 (C) REV1 is a polyubiquitinated protein. HEK293T cells were co-transfected with His-Ub and increasing amount of FLAG-REV1 expressing plasmid (lanes 1–2), or FLAG-REV1 and increasing amount of His-Ub (lanes 3–4). Cell lysates were pulled down with Ni-NTA resin in 6 M guanidine buffer followed by immunoblotting using anti-FLAG to detect ubiquitin-conjugated FLAG-REV1 (polyUb-REV1). One-fifth of harvested cells were lysed with RIPA to detect FLAG-REV1 (middle panel) and β-actin (lower panel). (D) REV1 is stabilized by proteasome inhibitor MG132. HEK293T cells were treated with increasing amount of proteasome inhibitor MG132 (10 and 20 μM) or DMSO. Cells were harvested at 0 h and 12 h after treatment and analyzed for endogenous REV1 protein expression with anti-REV1 antibodies. The membrane was also immunoblotted with anti-cyclin B1 antibody as a control for MG132 activity.
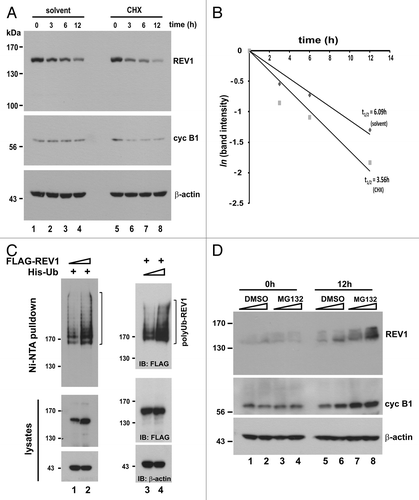
Since the degradation of yeast REV1 is mediated by ubiquitin-dependent proteolysis,Citation50 we next asked whether human REV1 is also ubiquitinated. To address this question, FLAG-REV1 and 6× histidine-tagged ubiquitin (His-Ub) were expressed in HEK293T cells. Ni-NTA pulldown assay was then performed to collect His-tagged proteins, which were subsequently analyzed by immunoblotting with anti-FLAG antibody. We observed a smear of FLAG-reactive signals in Ni-NTA pulldowns (, lanes 1 and 2). These slow migrating bands specifically represented polyubiquitinated REV1 proteins, because no other proteins in the cells were tagged with FLAG. In support of the specificity of this modification of REV1, the polyubiquitination signals were more pronounced when the dose of FLAG-REV1 or His-Ub was escalated individually (, compare lane 2 to lane 1, and lane 4 to lane 3). These results indicated that human cells likely employ ubiquitin-mediated proteolysis to degrade REV1.
To further demonstrate proteasomal degradation of human REV1, we blocked 26S proteasome activity with MG132. As the first step, we investigated how MG132 might affect the steady-state amount of endogenous REV1 in HEK293T cells. Consistent with its fast turnover, REV1 was barely detected by anti-REV1 in untreated and DMSO-treated cells (, lanes 1–6). In contrast, MG132 was found to stabilize both REV1 and cyclin B1 in a dose-dependent manner (, lanes 7 and 8). We next analyzed the stability of transiently expressed FLAG-REV1 in the presence of MG132. A 2.1- to 2.4-fold increase of REV1 level, in comparison with an increase of similar magnitude (1.5- to 1.9-fold) in cyclin B1 level, was observed in MG132-treated cells (, lanes 5–6). Furthermore, REV1 destabilization, seen in the presence of CHX, was reversed with the addition of MG132 (, lane 4 compared with lanes 1–3). That is to say the degradation of polyubiquitinated REV1 was inhibited when proteasomal function was compromised by MG132. Similar findings on REV1 ubiquitination and degradation were also obtained in HeLa and IMR90 cells (data not shown), suggesting that they might not be cell type-specific. Taken together, human REV1 is subject to ubiquitin-mediated proteolysis.
Figure 1E–F. (E) Cells were transiently transfected with same amount of FLAG-REV1 expression plasmid for 48 h. Increasing dose of MG132 (10 and 20 μM; lanes 5–6) or DMSO (lanes 2–3) was then added to cells. Cells were harvested 6 h after treatment and FLAG-REV1 was detected by western blotting with anti-FLAG. Relative levels of REV1 and cyclin B1 were determined by densitometry. (F) Inhibition of proteasome activity with MG132 attenuates REV1 degradation in HEK293T cells. Cells were transfected with FLAG-REV1 plasmid for 36 h. CHX was then added to cells (lanes 2–4). Meanwhile, cells were also supplemented with DMSO (lane 3) and 20 μM MG132 (lane 4), respectively. Cells were analyzed for FLAG-REV1 protein 6 h after drug treatment. Similar findings were also obtained from HeLa and IMR90 cells.
Degradation of REV1 is mediated by APC
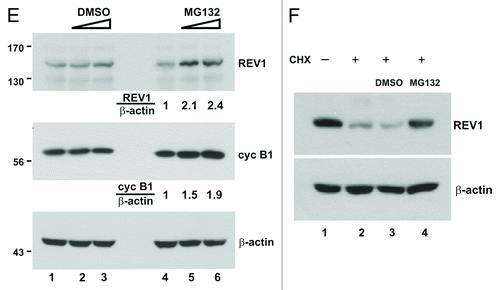
REV1 associates with REV7,Citation18,Citation19 which interacts with CDH1, a coactivator of APC.Citation29,Citation30 To determine whether degradation of human REV1 might be affected by CDH1, we expressed FLAG-REV1 and HA-CDH1 in HEK293T cells. It was found that the steady-state amount of REV1 was progressively reduced with the increase in CDH1 expression (, lanes 2 and 3 compared with lane 1). We next extended our analysis to CDC20, another coactivator of APC. Similar results were obtained which correlated CDC20 expression with diminution of REV1 protein (, lanes 2 and 3 compared with lane 1). To verify that CDH1 and CDC20 promote REV1 degradation by proteasome, MG132 was applied to cells expressing REV1 and CDH1/CDC20. Addition of MG132 reversed the effect of CDH1/CDC20 and stabilized REV1, which escalated to an amount close to the condition without APC coactivator (, compared lanes 4 to lanes 1–3). Hence, degradation of human REV1 is plausibly mediated by APC and proteasome.
Figure 2. CDH1 and CDC20 promote REV1 degradation. (A) HEK293T cells were co-transfected with FLAG-REV1 plus empty vector (lane 1) or plus increasing dose of HA-CDH1-expression plasmids (lanes 2–3) for 48 h. FLAG-REV1 and HA-CDH1 proteins were detected with anti-FLAG and anti-HA antibodies, respectively. (B) Cells were co-transfected with FLAG-REV1 plus empty vector (lane 1) or plus increasing dose of Myc-CDC20 expression plasmids (lanes 2–3). (C) Proteasome inhibitor MG132 prevents CDH1-promoted degradation of REV1. Cells were transfected with FLAG-REV1 and HA-CDH1 (lanes 2–4) for 48 h. DMSO (lane 3) or 20 μM MG132 (lane 4) was added to cells for 6 h before harvest. (D) MG132 prevents CDC20-dependent degradation of REV1. Cells were co-transfected with FLAG-REV1 and HA-CDC20 plasmids and were then treated with MG132.
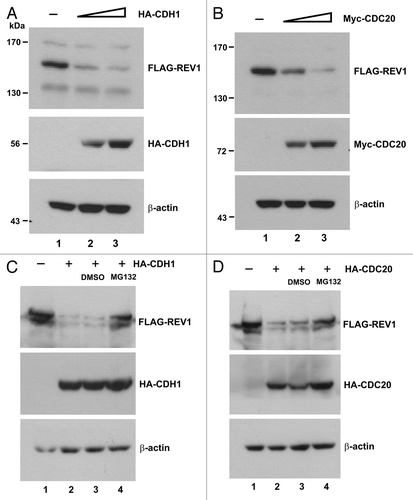
To substantiate our finding on APC-dependent regulation of REV1, we investigated whether polyubiquitination of REV1 () could be regulated by CDH1 and CDC20. We first expressed HA-CDH1 and His-Ub in HEK293T cells and assessed the impact of CDH1 on polyubiquitination of endogenous REV1. When immunoblot of Ni-NTA pulldowns was probed with anti-REV1 antibody, expression of CDH1 was found to augment REV1 polyubiquitination (, lane 2 compared with lane 1). Addition of MG132 further enhanced the effect of CDH1 by stabilizing polyubiquitinated REV1 (, lane 3). A dose-dependence analysis was subsequently performed using HA-CDH1, Myc-CDC20 and FLAG-REV1. We found that the polyubiquitination signal of FLAG-REV1 was considerably more robust when CDH1 or CDC20 was overexpressed (, lanes 2 and 3 compared with lane 1). If APC activated by CDH1 and CDC20 is involved in REV1 ubiquitination, REV1 should be associated with the APC complex. Indeed, a core component of APC termed APC3 (also known as CDC27) was readily detected in the FLAG-REV1-containing protein complex immunoprecipitated from cell lysate with anti-FLAG antibody (, lane 2 compared with lane 1). These results further support APC-mediated proteolysis of human REV1.
Figure 3. CDH1 and CDC20 enhance polyubiquitination of REV1. (A) Polyubiquitination of endogenous REV1. HEK293T cells were co-transfected with His-Ub plus empty HA (lane 1) or plus HA-CDH1 (lanes 2–3) expression plasmids. DMSO (lane 2) and MG132 (lane 3) were added to cells for 6 h before harvest. Cells were lysed in 6 M guanidine buffer and incubated with Ni-NTA beads. Ni-NTA pulldown assay was performed and polyubiquitinated REV1 was probed with anti-REV1 antibodies. RIPA lysed fractions were immunoblotted to detect HA-CDH1 and endogenous REV1 protein levels. ns, non-specific signal. Cells were co-transfected with FLAG-REV1 and His-Ub, plus increasing amount of HA-CDH1 (B) or Myc-CDC20 (C). Polyubiquitinated REV1 was detected by Ni-NTA pulldown assay and western blotting using anti-FLAG antibody. HA-CDH1 and Myc-CDC20 proteins in cell lysates were probed with anti-HA (B) and anti-MYC (C) antibodies, respectively. (D) REV1 interacts with anaphase APC component APC3. Cells were transfected with empty vector (lane 1) or FLAG-REV1 (lane 2), and the lysates were incubated with anti-FLAG antibody for immunoprecipitation. Precipitates were immunoblotted with anti-FLAG and anti-APC3 antibodies.
Mapping of REV1 degron
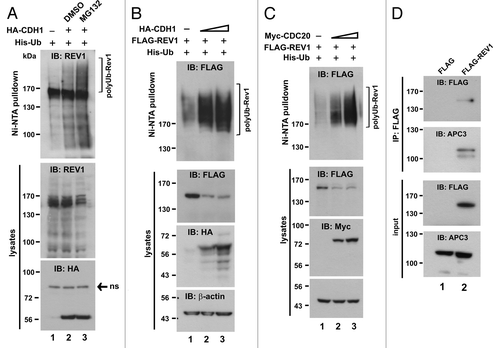
Having identified that APC is responsible for the degradation of human REV1, we attempted to map the REV1 degron recognized by the destruction machinery. Human REV1 contains UBM1 and UBM2 (amino acids 933–962 and 1,011–1,040, respectively) at its C terminus.Citation51 Mutation of a pair of highly conserved leucine-proline residues in UBM1 and UBM2 to alanine (L946A and P947A in UBM1, as well as L1024A and P1025A in UBM2) completely abolished the interaction of UBM with ubiquitin.Citation51 To address whether UBM-mediated ubiquitin binding plays a role in REV1 degradation, we generated a UBM1* and UBM2* double mutant of REV1 (UBM*) as indicated in . A minimal REV7-binding domain was previously mapped to the C terminus (amino acids 1,168–1,217) of REV1.Citation52 Accordingly, a REV1 mutant with this REV7-binding domain deleted (ΔBD) was generated (). Another truncated mutant ΔXbaI, which only contains amino acids 1–884 lacking UBMs and C-terminal REV7-binding domain, was also generated (). In addition, we identified a putative D box in amino acids 454–462 and constructed a mutant (D-box*) in which all three conserved residues in the RxxLxxxxN motif had been changed to alanine ().
Figure 4. Requirement of the N-terminal part for REV1 degradation. (A) Schematic presentation of human REV1 protein (1–1,251 amino acids). D box, UBM1, UBM2, C-terminal REV7-binding domain (REV7 BD) and the two XbaI sites were indicated. Point mutations generated in D box, UBM1 and UBM2 were highlighted with an asterisk (*), and the change of the amino acid residues were specified. Truncated mutant ΔXbaI containing amino acids 1–885 of REV1 was generated by deleting the region between two XbaI sites. ΔBD is a REV1 mutant deleted of REV7 BD. All REV1 wild-type (wt) and mutant proteins were FLAG-tagged at their N termini. (B) Stability of REV1 wild-type and mutant proteins. HEK293T cells were transfected with FLAG-REV1 wild-type (wt) and mutants for 48 h. Cells were treated with 15 μg/mL CHX (lanes 2, 4, 6, 8 and 10) for 12 h and then harvested for western blotting using anti-FLAG antibody. (C) CDH1 promotes degradation of REV1 wild-type and mutant proteins. Cells were transfected with FLAG-REV1 wt and mutants as indicated, in the presence of HA-CDH1 (+) or empty HA-vector (−). FLAG-REV1 and HA-CDH1 proteins were detected by western blotting. (D) C-terminal REV7-binding domain is dispensable for REV1 degradation. Cells were transfected with increasing amounts of FLAG-REV1 ΔBD plasmid (1 μg in lanes 1, 3 and 5; 2 μg in lanes 2, 4 and 6) for 36 h. DMSO or proteasome inhibitor PSI was added to cells for 6 h before harvest. Similar results were also obtained with proteasome inhibitor MG132. (E and F) D-box, UBM and REV7 BD are dispensable for REV1 polyubiquitination. In (E), HEK293T cells were co-transfected with His-Ub plus FLAG-REV1 wt or plus ΔXbaI mutant for 48 h. Cell lysates were incubated with Ni-NTA beads in guanidine buffer to pull down polyubiquitinated REV1 protein, which was detected by western blotting. In (F), cells were co-transfected with FLAG-REV1 wt or mutants as indicated, and His-Ub, in the presence of HA-CDH1 (+) or HA-vector (−). Polyubiquitinated REV1 was pulled down by Ni-NTA beads and analyzed by western blotting using anti-FLAG antibody.
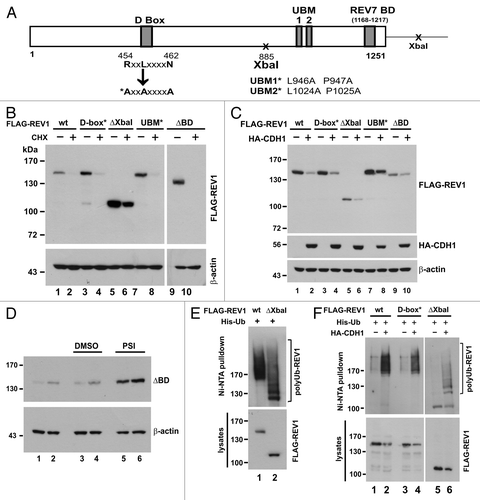
All these mutants along with wild-type REV1 were individually expressed in HEK293T cells. The influence of CHX on their stability was then assessed. It is evident that all four mutants, resembling wild-type REV1, have a fast turnover in CHX-treated cells (). To verify that this decay is mediated by APC, we asked whether their degradation might be stimulated by CDH1. As expected, overexpression of CDH1 abruptly induced degradation of wild-type and mutant REV1 proteins (). Additionally, when ΔBD-expressing cells were treated with proteasome inhibitor PSI, ΔBD mutant protein was found to be stabilized (). Furthermore, ΔXbaI mutant was abundantly polyubiquitinated to an extent similar to wild-type REV1 (). On the other hand, polyubiquitination of both D-box* and ΔXbaI mutants was more pronounced in the presence of CDH1 (). Thus, these mutants were polyubiquitinated and degraded by APC. Consistently, neither D box, nor UBMs, nor C-terminal REV7-binding domain was critically involved in governing REV1 ubiquitination and degradation. Since ΔXbaI mutant, like wild-type REV1, is a target of APC, the N-terminal region (amino acids 1–884) of human REV1 plausibly contains an additional APC recognition motif, other than D-box.
The dispensability of the C-terminal REV7-binding domain to REV1 ubiquitination and degradation () prompted us to further investigate the relevance of REV1-REV7 interaction in the regulation of REV1 stability. As a first step, we assessed the interaction of REV7 with wild-type and mutant REV1 proteins. Here we also generated a mutant, which consists of amino acids 885–1,167 of REV1, excluding the previously identified C-terminal REV7-binding domain.Citation52 To our surprise, all REV1 mutants, except mutant 885–1,167, were able to form a complex with REV7 (, lanes 8–12 compared with lane 13). Noticeably, the expression level of mutant 885–1,167 in cells was considerably high (, lane 6 compared with lanes 1–5). Therefore, a short exposure of this sample in the blot was also provided (, lane 7). The binding of ΔXbaI mutant with REV7, together with the lack of binding between mutant 855–1,167 and REV7, suggested that there would be another REV7-binding domain present in the N-terminal region (amino acids 1–884) of REV1 in addition to the one at the C terminus.
Figure 5. Interaction between REV1 and REV7. (A) Identification of a novel REV7-binding domain. HEK293T cells were co-transfected with FLAG-REV1 wt or mutants as indicated, and V5 His-REV7. FLAG-REV1 885–1,167 truncation mutant contains the C-terminal region (amino acids 885–1,167) of REV1 protein. Cells were lysed 48 h after transfection and incubated with Ni-NTA resin. Both input lysates and Ni-NTA pulldown were analyzed for FLAG-REV1 and V5 His-REV7 proteins by western blotting with antibodies against FLAG and V5. FLAG-REV1 885–1,167 mutant was highly expressed in cells and a short exposure (sh exp) of this lane in input immunoblot was also shown. (B) Stability of FLAG-REV1 885–1,167 mutant. Cells were transfected with FLAG-REV1 wild-type (wt) or 885–1,167 mutant plasmid. Forty-eight hours after transfection, cells were treated with 15 μg/mL cycloheximide (CHX; lanes 2 and 4) for 12 h before harvest. FLAG-REV1 was analyzed by western blotting. Cyclin B1 was also detected to control for CHX activity. (C) Cells were co-transfected with FLAG-REV1 wild-type (wt) or 885–1167 mutant plus HA-CDH1 plasmids (lanes 2 and 4). Forty-eight hours after transfection, cells were lysed, and FLAG-REV1 and HA-CDH1 proteins were analyzed by western blotting.
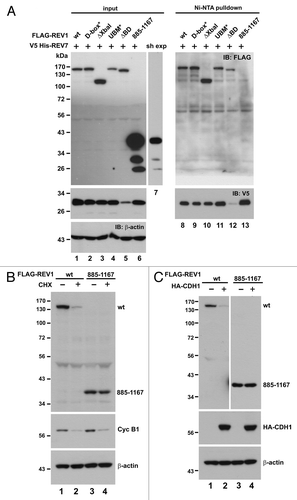
Having obtained and verified a REV1 mutant (885–1,167), which is unable to interact with REV7, we were interested to monitor the stability of this mutant. Consistent with earlier results (), the level of wild-type REV1 protein diminished robustly in the presence of CHX (, lanes 1 and 2). In stark contrast, the amount of mutant 885–1,167 was not significantly different in the presence of CHX (, lanes 3 and 4). This indicates that mutant 885–1,167, incapable of binding with REV7, is a stable protein. This provides a possible explanation for its abundant expression in cells as revealed in (lane 6). To consolidate this, we overexpressed CDH1 in mutant 885–1,167-expressing cells to see whether the degradation of the mutant might be boosted. The protein level of this mutant remained unchanged regardless of the overexpression of CDH1 (, lanes 3–4), while the amount of wild-type REV1 declined substantially when CDH1 was overexpressed (lanes 1–2). These results are compatible with the notion that binding with REV7 is important for REV1 destruction.
REV7 promotes degradation of REV1
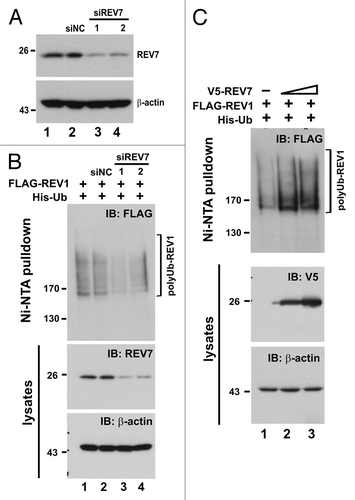
Because REV7 is able to modulate APC activity by binding to CDH1 and CDC20,Citation29,Citation30 we next investigated whether REV7 is required for REV1 degradation mediated by APC. The expression of REV7 was silenced using a short hairpin RNA against REV7 (shREV7), which has been validated to be highly effective in our previous study.Citation24 Although REV7 shares 23% amino acid sequence homology with MAD2,Citation21,Citation28 shREV7 specifically knocked down REV7 but not MAD2 (). To our surprise, the amount of FLAG-REV1 in HEK293T cells progressively increased when the dose of shREV7 was escalated (). In other words, depletion of REV7 with shREV7 correlated with the accumulation of REV1 protein in cells. This stabilization effect was specific to REV1, since the level of cyclin B1 was not affected (). Moreover, shREV7 dampened REV1 polyubiquitination signal in a dose-dependent manner (). To corroborate our finding, we employed two additional small interfering RNAs (siRNAs) against REV7, siREV7-1 and siREV7-2, targeting two different regions in REV7 coding sequence. Both siREV7-1 and siREV7-2 could effectively deplete REV7 (). As expected, polyubiquitination of REV1 was suppressed in the presence of either siREV7, while the negative control siRNA (siNC) had no influence on ubiquitination (). On the contrary, ectopic expression of REV7 enhanced polyubiquitination of REV1 (). The gain-of-function and loss-of-function assays of REV7 consistently supported the notion that Rev7 contributes to the degradation of REV1 plausibly through ubiquitin-mediated proteolysis.
Figure 6. Depletion of REV7 by shRNA stabilizes REV1 protein. (A) Verification of shREV7. HEK293T cells were transfected with increasing amount of plasmid expressing shRNA against REV7 (shREV7, lanes 3–5); a plasmid expressing an irrelevant control shRNA (shCtrl) was used as a negative control. Cells were lysed 48 h after transfection for detection of REV7 and MAD2 protein by western blotting. (B) Cells were co-transfected with FLAG-REV1 plasmid and increasing dose of shREV7 plasmid as indicated for 48 h. Cell lysates were analyzed by western blotting. Expression of FLAG-REV1, cyclin B1 (cyc B1) and REV7 was determined. (C) Knockdown of REV7 prevents REV1 polyubiquitination. Cells were co-transfected with FLAG-REV1 and His-Ub plasmids, plus increasing amount of shREV7 as indicated. Cells transfected with shCtrl was used as a control. Polyubiquitinated REV1 pulled down by Ni-NTA resin was analyzed by western blotting. Expression of REV7 was also determined.
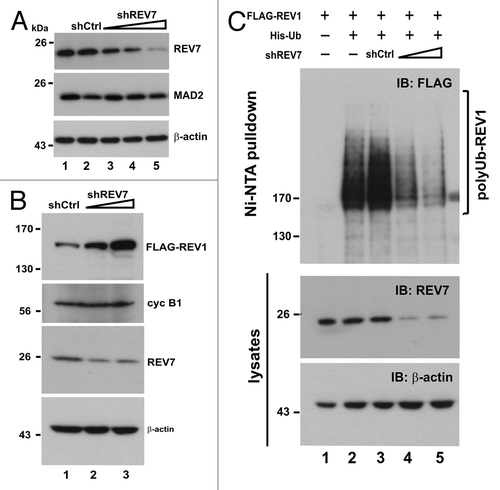
Figure 7. Depletion of REV7 by siRNA stabilizes REV1 protein. (A) Verification of siREV7. Two siRNAs (siREV7-1, -2) targeting two different regions of REV7 were transfected into HEK293T cells for 36 h. siNC, negative control siRNA provided by the manufacturer. (B) Cells were transfected with two siREV7s and siNC separately for 36 h. Then cells were co-transfected with FLAG-REV1 and His-Ub plasmids for 48 h. Ni-NTA resin was added to lysates to pull down polyubiquitinated REV1, which was then analyzed by western blotting. Cells were also lysed with RIPA to detect REV7 protein. (C) Overexpression of REV7 enhances REV1 polyubiquitination. Cells were co-transfected with FLAG-REV1 and His-Ub plasmids, plus increasing amount of V5-REV7 plasmid. Empty V5 vector was also transfected as control (lane 1). Cell lysates were incubated with Ni-NTA resin to pull down polyubiquitinated REV1.
Discussion
APC-mediated degradation of REV1
REV1 is a key protein in TLS.Citation53 The first finding we presented in this study was that human REV1 undergoes polyubiquitination and proteasomal degradation (Fig. 1). This agrees with a recent study, which documents proteasomal degradation of yeast REV1.Citation50 Therefore, proteasome-dependent destruction is a conserved mechanism in eukaryotes for regulation of REV1. The stability of human REV1 has also been shown to be regulated by other proteins, such as Hsp90 and the Fanconi anemia core complex.Citation7,Citation49 It remains to be seen whether these regulators of REV1 might also function by modulating REV1 polyubiquitination. It will also be of interest to determine the ubiquitin linkage type and polyubiquitination sites in REV1.
We also provided three lines of evidence to support that human REV1 is a target of APC. First, overexpression of CDH1 and CDC20 resulted in considerable degradation of REV1 mediated by the proteasome (). Second, overexpression of CDH1 and CDC20 promoted polyubiquitination of REV1 (). Finally, REV1 was associated with APC core component APC3 (). Although APC activated by CDC20 and CDH1 is one of the major E3 ubiquitin ligases governing the progression of cell cycle, APC is also critically involved in the destruction of various substrate proteins that function beyond the cell cycle,Citation33 for example SnoN, which regulates TGF-β signaling.Citation54 Some APC substrates are targeted specifically by either CDH1 or CDC20. For instance, forkhead box M1,Citation37 JNK1,Citation55 dynamin-related protein 1Citation56 and RAD17,Citation57 are degraded specifically by APC-CDH1 complex, but not APC-CDC20. On the other hand, there also exist substrates targeted by both CDH1 and CDC20, such as monopolar spindle 1 and ninein-like protein.Citation36,Citation58 However, the molecular basis of APC coactivator specificity remains elusive. In the case of REV1, both CDH1 and CDC20 induced polyubiquitination and degradation of REV1 to similar extents ( and ). Exactly when, where and how REV1 could be targeted by APC-CDC20 and APC-CDH1 require further investigations. Since yeast REV1 also undergoes proteasomal degradation,Citation47,Citation48,Citation50 it will be of great interest to see whether degradation of yeast REV1 is also mediated by APC.
APC can target its substrates through coactivators CDH1 and CDC20, which directly interact with the recognition motifs on substrates.Citation59 Prototypic substrate recognition motifs include D box and KEN box.Citation34,Citation35 In the present study, although we identified a putative D box at the N terminus of REV1, its disruption did not stabilize REV1 (). This suggests that human REV1 might harbor a noncanonical sequence for CDH1 and CDC20 recognition. This is not uncommon in APC substrates. One example is ubiquitin-specific protease 1, in which three putative D box and KEN box sequences are found to be irrelevant to protein stability.Citation60 We have also searched the REV1 sequence for other APC substrate recognition motifs, including A-box,Citation38 CRY box,Citation39 GxEN box,Citation61 O boxCitation62 and TEK box,Citation63 but none is present. In some proteins targeted by APC, as long as the conserved arginine and leucine residues in a relaxed version of the D box (RxxL) were mutated, protein degradation was abolished.Citation36,Citation58,Citation64,Citation65 If we take this into account, there would be six more copies of D box, five of which reside in the ΔXbaI mutant. A more comprehensive mutation analysis would therefore be required to determine whether they are indeed APC degrons, yet the presence of a noncanonical APC recognition motif in REV1 is possible. All members of Y-family polymerases are endowed with ubiquitin-binding domains, which are required for efficient interaction with monoubiquitinated PCNA in response to DNA damage. REV1 contains two UBMs,Citation11,Citation51 but mutations of them could not stabilize REV1 (). Furthermore, mutant ΔXbaI, in which both UBMs were deleted, could still be polyubiquitinated (). Conceivably, these motifs are not involved in the regulation of REV1 stability.
Role of REV7 in REV1 destruction
Our work pointed to a new role of REV7 in adapting REV1 to APC-dependent ubiquitination and proteolysis (–). First, we found that mutant ΔXbaI, which could be polyubiquitinated by APC, was able to interact with REV7 through a newly identified REV7-binding domain at the N terminus (amino acids 1–884) of REV1 (). Second, depletion of REV7 stabilized REV1 by preventing polyubiquitination ( and ). Third, overexpression of REV7 promoted REV1 polyubiquitination (). Collectively, our findings suggest a new function of REV7 in the regulation of REV1 stability.
The interaction between ΔXbaI mutant and REV7 suggests an alternative REV7-binding domain at the N terminus of REV1 (). It is noteworthy that BRCT domain, PAD and C-terminal region of yeast REV1 are required for binding with REV7.Citation17 It is therefore not surprising that human REV1 may also possess multiple REV7-binding domains. A detailed mapping of REV7-binding domain in this N-terminal region of human REV1 is warranted. Given the large size of human REV1 (1,251 residues) compared with its yeast homolog (985 residues),Citation21 it is possible that human REV1 allows Polζ and other Y-family polymerases to bind simultaneously, and this supports the notion that REV1 provides a docking surface for TLS proteins at the DNA lesion sites.
It has been reported that binding of REV7 to REV1 did not affect stability and activity of REV1 in reconstitution assays using purified proteins.Citation66 One possible interpretation to those results is that in the absence of APC-proteasome machinery, REV1 is stable, although REV7 is present. REV7 binds REV3 and REV1 separately in vitro, but a heterotrimeric complex of three proteins could not be detected.Citation67 However, a recent structural study showed that upon binding to REV3, REV7 undergoes a conformational change, which favors REV1 interaction.Citation20 In line with this, REV7 was found to bridge REV3 and REV1 in two recent studies.Citation19,Citation68 It is not known whether REV7 might stabilize or destabilize REV3, or whether the interaction between REV3 and REV7 could impact REV1 stability and function. Further investigations are required to address these important questions.
REV7, which is a paralog of mitotic checkpoint protein MAD2, is able to bind and inhibit CDH1 and/or CDC20 in vitro.Citation29,Citation30 In sharp contrast to this previous finding, here we demonstrated the facilitation of REV1 ubiquitination and degradation by REV7. This raises the interesting question concerning the net effect of REV7 on APC activation. In this regard, it is particularly noteworthy that MAD2 has also found to exhibit two opposite activities on APC in its open and closed conformations.Citation32 It remains to be further investigated whether active and inactive conformers of REV7 might exist and how they could convert between each other, but it is not too surprising that REV7 could also inhibit APC in one context and activate it in another.
We and others have previously showed that depletion of REV7 by shRNA in human cancer cell lines or by gene disruption in chicken DT40 cells had no effect on cell cycle progression or mitotic checkpoint activation.Citation24,Citation25,Citation28 Meanwhile REV7-depleted cells were hypersensitive to DNA damaging agents, such as cisplatin and γ-irradiation. These findings have implicated REV7 in the regulation of cellular response to DNA damage, but not mitotic perturbation. Consistent with this, compromising REV7 did not significantly affect cyclin B1 degradation in our experimental setting (). Our results further suggested that REV7 could not only bind and inhibit CDH1 and CDC20, but might also serve as a substrate recruitment protein for APC to facilitate the degradation of particular regulatory proteins involved in DNA damage response, such as REV1. Because both REV7-binding domain and APC degron reside in the N-terminal region of REV1 ( and ), we hypothesize that binding of REV7 promotes recognition of REV1 by APC via CDH1 or CDC20, because REV7 brings APC coactivators in close vicinity. Such spatial coordination renders REV1 more readily to be ubiquitinated and degraded by APC. Considering that human REV7 interacts with various protein partners, such as REV3 and Shigella IpaB,Citation40 it will also be of interest to determine whether REV7 might adapt any of its interaction partners to APC-dependent proteolysis. Analysis of the dynamic protein interactome of REV1, REV7, REV3 and APC would help us to comprehend the underlying mechanism for APC-mediated degradation of REV1 and other APC substrates.
The exact roles of REV7 in TLS and DNA repair remain to be fully understood. Yeast rev7 mutants are substantially deficient for DNA damage-induced mutagenesis.Citation69 Whereas overexpression of both REV3 and REV7 in yeast led to an increase in UV-induced mutagenesis, this could not alleviate the defect in UV-induced mutagenesis in rev1 mutant.Citation70 Further analyses are required to clarify whether yeast REV7 might also promote REV1 degradation and how this might relate to the phenotype of cells that are deficient of REV7 or overexpress REV7.
Taken together, our study provides the first evidence that human REV1 is degraded by APC-proteasome machinery, and REV7 promotes degradation of REV1. We propose that REV7 functions to adapt REV1 to APC and proteasome, thus coordinating a crosstalk between TLS and APC-proteasome degradation machinery.
Materials and Methods
Plasmids, mutagenesis, shRNA and siRNAs
pcDNA3.1(+)/FLAG-hREV1 plasmid (FLAG-REV1) expressing full-length human REV1 with an N-terminal FLAG tag was kindly provided by Dr. Yoshiki Murakumo.Citation67 His6-Ub expression vector (His-Ub) consisting of eight ubiquitin units, each with an N-terminal 6 × His-tag, was a gift from Dr. Dirk Bohmann.Citation71 Expression plasmid HA-CDH1, which contains human CDH1 has been described previously.Citation72 Myc-CDC20 expression plasmid was constructed by inserting full-length human CDC20 into pCS2+MT vector containing 6 × Myc tag, which was a gift from Dr. David Turner.Citation73 HA-CDC20 was constructed by sub-cloning CDC20 from Myc-CDC20 plasmid to pCMV-HA expression vector (Clontech).
FLAG-REV1 D-box point mutant (D-box*), in which three conserved RxxLxxxxN residues were changed to alanine (R454A, L457A, N462A), was generated by two rounds of PCR using primer pairs 5′-GTAACAGAGG CACAGGAGCG GCACCTGCAC GTCCTGGCGC TA-3′ and 5′-TAGCGCCAGG ACGTGCAGGT GCCGCTCCTG TGCCTCTGTT AC-3′; and then 5′-ACCTTTACGT CCTGGCGCTG CCCCCAGCT G-3′ and 5′-CAGCTGGGGG GCAGCGCCAG GACGTAAAGG T-3′. FLAG-REV1 UBM1 and UBM2 double point mutant (UBM*) was generated by first preparing UBM1 mutant using primers 5′-GATCAGTCTG TTTTAGAAGC AGCTGCACCT GATCTCCGGG AACAAG-3′ and 5′-CTTGTTCCG GAGATCAGGT GCAGCTGCTT CTAAAACAGA CTGATC-3′. UBM1 mutant was then used as a template to generate UBM* using primers 5′-CCTGAGGTAT TTGCTGCCGC TGCTGCTGAA CTTCAGAGGG-3′ and 5′-CCCTCTGAAG TTCAGCAGCA GCGGCAGCAA ATACCTCAGG-3′. FLAG-REV1 with C-terminal REV7-binding domain deleted (∆BD, deletion of amino acids 1168–1217) was generated using primers 5′-CCTGAGGTAT TTGCTGCCGC TGCTGCTGAA CTTCAGAGGG-3′ and 5′-CCCTCTGAAG TTCAGCAGCA GCGGCAGCAA ATACCTCAGG-3′. The above point and deletion mutants were generated by using QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies). FLAG-REV1 885–1,167 truncation mutant containing amino acids 885–1,167 of REV1 was generated by PCR using primers 5′-ACTTAAGCTT CTACCATGGA CTACAAAGAC GATGACGACA AGTCTAGAAC TTGCACTTTC TTGCCA-3′ and 5′-AGACTCGAGT TAATCATTGA ATTCAACAGC TCCAGCTAG-3′ and the PCR fragment containing a FLAG-tag at its N terminus was then ligated into pcDNA3.1(+) vector (Invitrogen). FLAG-REV1 with deletion between XbaI site at amino acid 885 of REV1 and XbaI site in the multiple cloning sites of pcDNA3.1(+), located at the 3′ of REV1 insert in FLAG-REV1 plasmid (∆XbaI) was generated by cleaving FLAG-REV1 plasmid with XbaI restriction endonuclease, followed by re-ligating the cleaved plasmid. Plasmid expressing N-terminal V5-tagged REV7 (V5-REV7) was prepared by ligating PCR amplified REV7 with V5-tag into pcDNA3.1(+). V5 His-REV7 expression plasmid was generated by cloning PCR amplified REV7 into pcDNA3.1/V5-His B vector (Invitrogen). All constructs were verified by DNA sequencing.
Short hairpin interfering RNA against REV7 (shREV7; 5′-GAUGCAGCUU UACGUGGAAG A-3′) has been described previously.Citation24 It was constructed into a lentiviral vector using BLOCK-iT Lentiviral RNAi Expression System (Invitrogen). Two small interfering RNA (siRNA) oligonucleotides targeting human REV7 were siREV7–1: 5′-GAUGCAGCUU UACGUGGAATT-3′ and siREV7–2: 5′-GGAAGAGCGC GCUCAUAAATT-3′ (GenePharma). A negative control siRNA (siNC) was provided by the manufacturer.
Cell culture, transient transfection and drug treatment
Human embryonic kidney 293T (HEK293T), cervical carcinoma HeLa and normal embryonic fibroblasts IMR90 cells were grown in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum, 100 units/ml penicillin-G and 100 μg/ml streptomycin and maintained at 37°C in a humidified atmosphere at 5% CO2. Cells were transfected with expression plasmids and shRNA using PolyJet DNA In Vitro Transfection Reagent (SignaGen Laboratories) for 36–48 h before harvest. siRNAs were introduced into cells using X-tremeGENE siRNA Transfection Reagent (Roche). For proteasome inhibition experiments, proteasome inhibitor carbobenzoxy-L-leucyl-L-leucyl-L-leucinal (MG132) or proteasome inhibitor 1 (PSI) (Calbiochem) was added to transfected cells for 6 h before harvest. For protein turnover assay, 15 μg/mL cycloheximide (CHX, Sigma-Aldrich) was added to transfected cells for 12 h before harvest.
Ni-NTA pulldown assay and immunoprecipitation
For ubiquitination assays, one-fifth of harvested cells that served as input were lysed with RIPA buffer (50 mM Tris-Cl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 1% sodium deoxycholate, 1% Nonidet P-40), supplemented with complete protease inhibitor cocktail (Roche), 2 mM PMSF, 5 mM NaF, 0.5 mM sodium orthovanadate, 10 μM MG132 and 5 mM N-ethylmaleimide (NEM). The remaining cells were analyzed for protein ubiquitination. Cells were lysed in guanidine lysis buffer (6 M guanidine hydrogen chloride, 0.1 M sodium phosphate, 0.01 M TRIS-HCl, pH 8.0, 10 mM β-mercaptoethanol) on ice for 30 min, followed by passing through syringe with 23G needle (BD Biosciences) to reduce viscosity. Lysates were incubated with Ni-NTA Superflow resin (Qiagen) overnight at 4°C to pull down His-ubiquitin-conjugated proteins, followed by flushing with urea washing buffer (8 M urea, 0.1 M sodium phosphate, 0.01 M TRIS-HCl, pH 8.0, 10 mM β-mercaptoethanol, 0.2% Triton X-100) and histidine washing buffer (20 mM sodium phosphate, pH 8.0, 0.2 M NaCl) at room temperature. To reduce non-specific binding on Ni-NTA resin, 20 mM imidazole was added to guanidine lysis buffer and urea washing buffer. The bound proteins were eluted from resin with elution buffer (0.15 M TRIS-HCl, pH 6.7, 30% glycerol, 0.72 M β-mercaptoethanol, 5% SDS, 200 mM imidazole). Ten μM MG132, 5 mM NEM and protease-inhibitor cocktail were also added to the buffers above. For V5-REV7 and REV1 protein-binding assay, cells were lysed in lysis buffer (50 mM TRIS-HCl, pH 7.4, 150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, protease inhibitor cocktail, 5 mM NaF, 0.5 mM sodium orthovanadate, 10 μM MG132, 5 mM NEM) with 50 mM imidazole. Cell extracts were clarified by centrifugation at 14,000 rpm for 20 min at 4°C. Protein concentration was determined with Bio-Rad Protein Assay Reagent (Bio-Rad). Equal amount of lysates were incubated with Ni-NTA resin overnight at 4°C and resins were then washed five times with lysis buffer. Proteins were eluted with resin volume of lysis buffer containing 700 mM imidazole.
For immunoprecipitation (IP) assay, cells were lysed in IP buffer (50 mM TRIS-HCl, pH 7.4, 150 mM NaCl, 1% NP40, protease inhibitor cocktail, 5 mM NaF, 0.5 mM sodium orthovanadate, 10 μM MG132, 5 mM NEM). Lysates were incubated with mouse anti-FLAG antibody (M2, Sigma) for 2 h before overnight incubation with rProtein G agarose (Invitrogen) at 4°C. The agarose was washed four times with IP buffer and resuspended in SDS protein sample buffer.
Western blotting and antibodies
Cell lysates and eluted proteins were mixed with 5 × SDS protein sampling buffer [250 mM Tris-Cl, pH6.8, 2% (v/v) β-mercaptoethanol, 10% (w/v) SDS, 0.5% bromophenol blue, 50% (v/v) glycerol] and boiled for 10 min. Samples containing equal amounts of protein were separated by SDS-PAGE and electroblotted onto Immobilon-P membranes (Millipore) using Trans-Blot cell (Bio-Rad). Blots were blocked with 5% skim milk in TBS-T, followed by incubation with primary antibodies overnight at 4°C. Blots were then incubated with goat anti-rabbit or anti-mouse secondary antibody conjugated to horseradish peroxidase (GE Healthcare) and visualized by enhanced chemiluminescence (ECL, GE Healthcare) as described previously.Citation72,Citation74 The primary antibodies used for western blot analysis were mouse anti-FLAG (M2) and mouse anti-β-actin (AC-40) from Sigma; mouse anti-cyclin B1 (D11), rabbit anti-REV1 (H-300), rabbit anti-HA (Y-11) and mouse anti-c-Myc (9E10) from Santa Cruz; mouse anti-MAD2, mouse anti-MAD2B/REV7 and mouse anti-CDC27/APC3 from BD Transduction Laboratories and mouse anti-V5 from Invitrogen.
Acknowledgments
This work was supported by Hong Kong Research Grants Council (HKU7/CRF/09 and HKU1/CRF/11G) and SK Yee Medical Research Fund (2011). We thank Drs. Yoshiki Murakumo, Dirk Bohmann and David Turner for gifts of plasmids; Chi-Ming Wong and Vincent Tang for expert help with some experiments; and Pak-Yin Lui, James Ng, Kam-Leung Siu, Vincent Tang and Sam Yuen for critical reading of the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author Contributions
A.C.-S.C., K.-H.K. and D.-Y.J. designed the study. A.C.-S.C. performed all experiments. A.C.-S.C., K.-H.K. and D.-Y.J. analyzed the data. A.C.-S.C. and D.-Y.J. wrote the paper.
References
- McCulloch SD, Kunkel TA. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res 2008; 18:148 - 61; http://dx.doi.org/10.1038/cr.2008.4; PMID: 18166979
- Prakash S, Johnson RE, Prakash L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem 2005; 74:317 - 53; http://dx.doi.org/10.1146/annurev.biochem.74.082803.133250; PMID: 15952890
- Lange SS, Takata K, Wood RD. DNA polymerases and cancer. Nat Rev Cancer 2011; 11:96 - 110; http://dx.doi.org/10.1038/nrc2998; PMID: 21258395
- Waters LS, Minesinger BK, Wiltrout ME, D’Souza S, Woodruff RV, Walker GC. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol Mol Biol Rev 2009; 73:134 - 54; http://dx.doi.org/10.1128/MMBR.00034-08; PMID: 19258535
- Wittschieben JP, Patil V, Glushets V, Robinson LJ, Kusewitt DF, Wood RD. Loss of DNA polymerase ζ enhances spontaneous tumorigenesis. Cancer Res 2010; 70:2770 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-09-4267; PMID: 20215524
- Doles J, Oliver TG, Cameron ER, Hsu G, Jacks T, Walker GC, et al. Suppression of Rev3, the catalytic subunit of Polζ, sensitizes drug-resistant lung tumors to chemotherapy. Proc Natl Acad Sci USA 2010; 107:20786 - 91; http://dx.doi.org/10.1073/pnas.1011409107; PMID: 21068376
- Kim H, Yang K, Dejsuphong D, D’Andrea AD. Regulation of Rev1 by the Fanconi anemia core complex. Nat Struct Mol Biol 2012; 19:164 - 70; http://dx.doi.org/10.1038/nsmb.2222; PMID: 22266823
- Kim N, Mudrak SV, Jinks-Robertson S. The dCMP transferase activity of yeast Rev1 is biologically relevant during the bypass of endogenously generated AP sites. DNA Repair (Amst) 2011; 10:1262 - 71; http://dx.doi.org/10.1016/j.dnarep.2011.09.017; PMID: 22024240
- Wiltrout ME, Walker GC. The DNA polymerase activity of Saccharomyces cerevisiae Rev1 is biologically significant. Genetics 2011; 187:21 - 35; http://dx.doi.org/10.1534/genetics.110.124172; PMID: 20980236
- Guo C, Sonoda E, Tang TS, Parker JL, Bielen AB, Takeda S, et al. REV1 protein interacts with PCNA: significance of the REV1 BRCT domain in vitro and in vivo.. Mol Cell 2006; 23:265 - 71; http://dx.doi.org/10.1016/j.molcel.2006.05.038; PMID: 16857592
- Wood A, Garg P, Burgers PM. A ubiquitin-binding motif in the translesion DNA polymerase Rev1 mediates its essential functional interaction with ubiquitinated proliferating cell nuclear antigen in response to DNA damage. J Biol Chem 2007; 282:20256 - 63; http://dx.doi.org/10.1074/jbc.M702366200; PMID: 17517887
- Guo C, Fischhaber PL, Luk-Paszyc MJ, Masuda Y, Zhou J, Kamiya K, et al. Mouse Rev1 protein interacts with multiple DNA polymerases involved in translesion DNA synthesis. EMBO J 2003; 22:6621 - 30; http://dx.doi.org/10.1093/emboj/cdg626; PMID: 14657033
- Ohashi E, Hanafusa T, Kamei K, Song I, Tomida J, Hashimoto H, et al. Identification of a novel REV1-interacting motif necessary for DNA polymerase κ function. Genes Cells 2009; 14:101 - 11; http://dx.doi.org/10.1111/j.1365-2443.2008.01255.x; PMID: 19170759
- Ross AL, Simpson LJ, Sale JE. Vertebrate DNA damage tolerance requires the C-terminus but not BRCT or transferase domains of REV1. Nucleic Acids Res 2005; 33:1280 - 9; http://dx.doi.org/10.1093/nar/gki279; PMID: 15741181
- Acharya N, Johnson RE, Prakash S, Prakash L. Complex formation with Rev1 enhances the proficiency of Saccharomyces cerevisiae DNA polymerase ζ for mismatch extension and for extension opposite from DNA lesions. Mol Cell Biol 2006; 26:9555 - 63; http://dx.doi.org/10.1128/MCB.01671-06; PMID: 17030609
- Acharya N, Haracska L, Johnson RE, Unk I, Prakash S, Prakash L. Complex formation of yeast Rev1 and Rev7 proteins: a novel role for the polymerase-associated domain. Mol Cell Biol 2005; 25:9734 - 40; http://dx.doi.org/10.1128/MCB.25.21.9734-9740.2005; PMID: 16227619
- D’Souza S, Walker GC. Novel role for the C terminus of Saccharomyces cerevisiae Rev1 in mediating protein-protein interactions. Mol Cell Biol 2006; 26:8173 - 82; http://dx.doi.org/10.1128/MCB.00202-06; PMID: 16923957
- Kosarek JN, Woodruff RV, Rivera-Begeman A, Guo C, D’Souza S, Koonin EV, et al. Comparative analysis of in vivo interactions between Rev1 protein and other Y-family DNA polymerases in animals and yeasts. DNA Repair (Amst) 2008; 7:439 - 51; http://dx.doi.org/10.1016/j.dnarep.2007.11.016; PMID: 18242152
- Sharma S, Hicks JK, Chute CL, Brennan JR, Ahn JY, Glover TW, et al. REV1 and polymerase ζ facilitate homologous recombination repair. Nucleic Acids Res 2012; 40:682 - 91; http://dx.doi.org/10.1093/nar/gkr769; PMID: 21926160
- Hara K, Hashimoto H, Murakumo Y, Kobayashi S, Kogame T, Unzai S, et al. Crystal structure of human REV7 in complex with a human REV3 fragment and structural implication of the interaction between DNA polymerase ζ and REV1. J Biol Chem 2010; 285:12299 - 307; http://dx.doi.org/10.1074/jbc.M109.092403; PMID: 20164194
- Gan GN, Wittschieben JP, Wittschieben BO, Wood RD. DNA polymerase zeta (pol zeta) in higher eukaryotes. Cell Res 2008; 18:174 - 83; http://dx.doi.org/10.1038/cr.2007.117; PMID: 18157155
- Nelson JR, Lawrence CW, Hinkle DC. Thymine-thymine dimer bypass by yeast DNA polymerase ζ. Science 1996; 272:1646 - 9; http://dx.doi.org/10.1126/science.272.5268.1646; PMID: 8658138
- Takeuchi R, Oshige M, Uchida M, Ishikawa G, Takata K, Shimanouchi K, et al. Purification of Drosophila DNA polymerase ζ by REV1 protein-affinity chromatography. Biochem J 2004; 382:535 - 43; http://dx.doi.org/10.1042/BJ20031833; PMID: 15175013
- Cheung HW, Chun AC, Wang Q, Deng W, Hu L, Guan XY, et al. Inactivation of human MAD2B in nasopharyngeal carcinoma cells leads to chemosensitization to DNA-damaging agents. Cancer Res 2006; 66:4357 - 67; http://dx.doi.org/10.1158/0008-5472.CAN-05-3602; PMID: 16618761
- Okada T, Sonoda E, Yoshimura M, Kawano Y, Saya H, Kohzaki M, et al. Multiple roles of vertebrate REV genes in DNA repair and recombination. Mol Cell Biol 2005; 25:6103 - 11; http://dx.doi.org/10.1128/MCB.25.14.6103-6111.2005; PMID: 15988022
- McNally K, Neal JA, McManus TP, McCormick JJ, Maher VM. hRev7, putative subunit of hPolzeta, plays a critical role in survival, induction of mutations, and progression through S-phase, of UV((254nm))-irradiated human fibroblasts. DNA Repair (Amst) 2008; 7:597 - 604; http://dx.doi.org/10.1016/j.dnarep.2007.12.013; PMID: 18295554
- Cahill DP, da Costa LT, Carson-Walter EB, Kinzler KW, Vogelstein B, Lengauer C. Characterization of MAD2B and other mitotic spindle checkpoint genes. Genomics 1999; 58:181 - 7; http://dx.doi.org/10.1006/geno.1999.5831; PMID: 10366450
- Murakumo Y, Roth T, Ishii H, Rasio D, Numata S, Croce CM, et al. A human REV7 homolog that interacts with the polymerase ζ catalytic subunit hREV3 and the spindle assembly checkpoint protein hMAD2. J Biol Chem 2000; 275:4391 - 7; http://dx.doi.org/10.1074/jbc.275.6.4391; PMID: 10660610
- Chen J, Fang G. MAD2B is an inhibitor of the anaphase-promoting complex. Genes Dev 2001; 15:1765 - 70; http://dx.doi.org/10.1101/gad.898701; PMID: 11459826
- Pfleger CM, Salic A, Lee E, Kirschner MW. Inhibition of Cdh1-APC by the MAD2-related protein MAD2L2: a novel mechanism for regulating Cdh1. Genes Dev 2001; 15:1759 - 64; http://dx.doi.org/10.1101/gad.897901; PMID: 11459825
- Eytan E, Moshe Y, Braunstein I, Hershko A. Roles of the anaphase-promoting complex/cyclosome and of its activator Cdc20 in functional substrate binding. Proc Natl Acad Sci USA 2006; 103:2081 - 6; http://dx.doi.org/10.1073/pnas.0510695103; PMID: 16455800
- Yu H. Cdc20: a WD40 activator for a cell cycle degradation machine. Mol Cell 2007; 27:3 - 16; http://dx.doi.org/10.1016/j.molcel.2007.06.009; PMID: 17612486
- Qiao X, Zhang L, Gamper AM, Fujita T, Wan Y. APC/C-Cdh1: from cell cycle to cellular differentiation and genomic integrity. Cell Cycle 2010; 9:3904 - 12; http://dx.doi.org/10.4161/cc.9.19.13585; PMID: 20935501
- Glotzer M, Murray AW, Kirschner MW. Cyclin is degraded by the ubiquitin pathway. Nature 1991; 349:132 - 8; http://dx.doi.org/10.1038/349132a0; PMID: 1846030
- Pfleger CM, Kirschner MW. The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev 2000; 14:655 - 65; PMID: 10733526
- Wang Y, Zhan Q. Cell cycle-dependent expression of centrosomal ninein-like protein in human cells is regulated by the anaphase-promoting complex. J Biol Chem 2007; 282:17712 - 9; http://dx.doi.org/10.1074/jbc.M701350200; PMID: 17403670
- Park HJ, Costa RH, Lau LF, Tyner AL, Raychaudhuri P. Anaphase-promoting complex/cyclosome-CDH1-mediated proteolysis of the forkhead box M1 transcription factor is critical for regulated entry into S phase. Mol Cell Biol 2008; 28:5162 - 71; http://dx.doi.org/10.1128/MCB.00387-08; PMID: 18573889
- Littlepage LE, Ruderman JV. Identification of a new APC/C recognition domain, the A box, which is required for the Cdh1-dependent destruction of the kinase Aurora-A during mitotic exit. Genes Dev 2002; 16:2274 - 85; http://dx.doi.org/10.1101/gad.1007302; PMID: 12208850
- Reis A, Levasseur M, Chang HY, Elliott DJ, Jones KT. The CRY box: a second APCcdh1-dependent degron in mammalian cdc20. EMBO Rep 2006; 7:1040 - 5; http://dx.doi.org/10.1038/sj.embor.7400772; PMID: 16878123
- Iwai H, Kim M, Yoshikawa Y, Ashida H, Ogawa M, Fujita Y, et al. A bacterial effector targets Mad2L2, an APC inhibitor, to modulate host cell cycling. Cell 2007; 130:611 - 23; http://dx.doi.org/10.1016/j.cell.2007.06.043; PMID: 17719540
- Ying B, Wold WS. Adenovirus ADP protein (E3-11.6K), which is required for efficient cell lysis and virus release, interacts with human MAD2B. Virology 2003; 313:224 - 34; http://dx.doi.org/10.1016/S0042-6822(03)00287-3; PMID: 12951035
- Garg P, Burgers PM. Ubiquitinated proliferating cell nuclear antigen activates translesion DNA polymerases η and REV1. Proc Natl Acad Sci USA 2005; 102:18361 - 6; http://dx.doi.org/10.1073/pnas.0505949102; PMID: 16344468
- Skoneczna A, McIntyre J, Skoneczny M, Policinska Z, Sledziewska-Gojska E. Polymerase η is a short-lived, proteasomally degraded protein that is temporarily stabilized following UV irradiation in Saccharomyces cerevisiae.. J Mol Biol 2007; 366:1074 - 86; http://dx.doi.org/10.1016/j.jmb.2006.11.093; PMID: 17198712
- Jung YS, Liu G, Chen X. Pirh2 E3 ubiquitin ligase targets DNA polymerase η for 20S proteasomal degradation. Mol Cell Biol 2010; 30:1041 - 8; http://dx.doi.org/10.1128/MCB.01198-09; PMID: 20008555
- Jung YS, Qian Y, Chen X. DNA polymerase η is targeted by Mdm2 for polyubiquitination and proteasomal degradation in response to ultraviolet irradiation. DNA Repair (Amst) 2012; 11:177 - 84; http://dx.doi.org/10.1016/j.dnarep.2011.10.017; PMID: 22056306
- Wimmer U, Ferrari E, Hunziker P, Hübscher U. Control of DNA polymerase λ stability by phosphorylation and ubiquitination during the cell cycle. EMBO Rep 2008; 9:1027 - 33; http://dx.doi.org/10.1038/embor.2008.148; PMID: 18688254
- Waters LS, Walker GC. The critical mutagenic translesion DNA polymerase Rev1 is highly expressed during G(2)/M phase rather than S phase. Proc Natl Acad Sci USA 2006; 103:8971 - 6; http://dx.doi.org/10.1073/pnas.0510167103; PMID: 16751278
- Sabbioneda S, Bortolomai I, Giannattasio M, Plevani P, Muzi-Falconi M. Yeast Rev1 is cell cycle regulated, phosphorylated in response to DNA damage and its binding to chromosomes is dependent upon MEC1. DNA Repair (Amst) 2007; 6:121 - 7; http://dx.doi.org/10.1016/j.dnarep.2006.09.002; PMID: 17035102
- Pozo FM, Oda T, Sekimoto T, Murakumo Y, Masutani C, Hanaoka F, et al. Molecular chaperone Hsp90 regulates REV1-mediated mutagenesis. Mol Cell Biol 2011; 31:3396 - 409; http://dx.doi.org/10.1128/MCB.05117-11; PMID: 21690293
- Wiltrout ME, Walker GC. Proteasomal regulation of the mutagenic translesion DNA polymerase, Saccharomyces cerevisiae Rev1. DNA Repair (Amst) 2011; 10:169 - 75; http://dx.doi.org/10.1016/j.dnarep.2010.10.008; PMID: 21227758
- Guo C, Tang TS, Bienko M, Parker JL, Bielen AB, Sonoda E, et al. Ubiquitin-binding motifs in REV1 protein are required for its role in the tolerance of DNA damage. Mol Cell Biol 2006; 26:8892 - 900; http://dx.doi.org/10.1128/MCB.01118-06; PMID: 16982685
- D’Souza S, Waters LS, Walker GC. Novel conserved motifs in Rev1 C-terminus are required for mutagenic DNA damage tolerance. DNA Repair (Amst) 2008; 7:1455 - 70; http://dx.doi.org/10.1016/j.dnarep.2008.05.009; PMID: 18603483
- Chun AC, Jin DY. Ubiquitin-dependent regulation of translesion polymerases. Biochem Soc Trans 2010; 38:110 - 5; http://dx.doi.org/10.1042/BST0380110; PMID: 20074045
- Wan Y, Liu X, Kirschner MW. The anaphase-promoting complex mediates TGF-β signaling by targeting SnoN for destruction. Mol Cell 2001; 8:1027 - 39; http://dx.doi.org/10.1016/S1097-2765(01)00382-3; PMID: 11741538
- Gutierrez GJ, Tsuji T, Chen M, Jiang W, Ronai ZA. Interplay between Cdh1 and JNK activity during the cell cycle. Nat Cell Biol 2010; 12:686 - 95; http://dx.doi.org/10.1038/ncb2071; PMID: 20581839
- Horn SR, Thomenius MJ, Johnson ES, Freel CD, Wu JQ, Coloff JL, et al. Regulation of mitochondrial morphology by APC/CCdh1-mediated control of Drp1 stability. Mol Biol Cell 2011; 22:1207 - 16; http://dx.doi.org/10.1091/mbc.E10-07-0567; PMID: 21325626
- Zhang L, Park CH, Wu J, Kim H, Liu W, Fujita T, et al. Proteolysis of Rad17 by Cdh1/APC regulates checkpoint termination and recovery from genotoxic stress. EMBO J 2010; 29:1726 - 37; http://dx.doi.org/10.1038/emboj.2010.55; PMID: 20424596
- Cui Y, Cheng X, Zhang C, Zhang Y, Li S, Wang C, et al. Degradation of the human mitotic checkpoint kinase Mps1 is cell cycle-regulated by APC-cCdc20 and APC-cCdh1 ubiquitin ligases. J Biol Chem 2010; 285:32988 - 98; http://dx.doi.org/10.1074/jbc.M110.140905; PMID: 20729194
- Izawa D, Pines J. How APC/C-Cdc20 changes its substrate specificity in mitosis. Nat Cell Biol 2011; 13:223 - 33; http://dx.doi.org/10.1038/ncb2165; PMID: 21336306
- Cotto-Rios XM, Jones MJ, Busino L, Pagano M, Huang TT. APC/CCdh1-dependent proteolysis of USP1 regulates the response to UV-mediated DNA damage. J Cell Biol 2011; 194:177 - 86; http://dx.doi.org/10.1083/jcb.201101062; PMID: 21768287
- Castro A, Vigneron S, Bernis C, Labbé JC, Lorca T. Xkid is degraded in a D-box, KEN-box, and A-box-independent pathway. Mol Cell Biol 2003; 23:4126 - 38; http://dx.doi.org/10.1128/MCB.23.12.4126-4138.2003; PMID: 12773557
- Araki M, Yu H, Asano M. A novel motif governs APC-dependent degradation of Drosophila ORC1 in vivo.. Genes Dev 2005; 19:2458 - 65; http://dx.doi.org/10.1101/gad.1361905; PMID: 16195415
- Jin L, Williamson A, Banerjee S, Philipp I, Rape M. Mechanism of ubiquitin-chain formation by the human anaphase-promoting complex. Cell 2008; 133:653 - 65; http://dx.doi.org/10.1016/j.cell.2008.04.012; PMID: 18485873
- Kraft C, Vodermaier HC, Maurer-Stroh S, Eisenhaber F, Peters JM. The WD40 propeller domain of Cdh1 functions as a destruction box receptor for APC/C substrates. Mol Cell 2005; 18:543 - 53; http://dx.doi.org/10.1016/j.molcel.2005.04.023; PMID: 15916961
- Chow C, Wong N, Pagano M, Lun SW, Nakayama KI, Nakayama K, et al. Regulation of APC/CCdc20 activity by RASSF1A-APC/CCdc20 circuitry. Oncogene 2012; 31:1975 - 87; http://dx.doi.org/10.1038/onc.2011.372; PMID: 21874044
- Masuda Y, Ohmae M, Masuda K, Kamiya K. Structure and enzymatic properties of a stable complex of the human REV1 and REV7 proteins. J Biol Chem 2003; 278:12356 - 60; http://dx.doi.org/10.1074/jbc.M211765200; PMID: 12529368
- Murakumo Y, Ogura Y, Ishii H, Numata S, Ichihara M, Croce CM, et al. Interactions in the error-prone postreplication repair proteins hREV1, hREV3, and hREV7. J Biol Chem 2001; 276:35644 - 51; http://dx.doi.org/10.1074/jbc.M102051200; PMID: 11485998
- Hashimoto K, Cho Y, Yang IY, Akagi J, Ohashi E, Tateishi S, et al. The vital role of polymerase ζ and REV1 in mutagenic, but not correct, DNA synthesis across benzo[a]pyrene-dG and recruitment of polymerase ζ by REV1 to replication-stalled site. J Biol Chem 2012; 287:9613 - 22; http://dx.doi.org/10.1074/jbc.M111.331728; PMID: 22303021
- Lawrence CW, Das G, Christensen RB. REV7, a new gene concerned with UV mutagenesis in yeast. Mol Gen Genet 1985; 200:80 - 5; http://dx.doi.org/10.1007/BF00383316; PMID: 3897794
- Rajpal DK, Wu X, Wang Z. Alteration of ultraviolet-induced mutagenesis in yeast through molecular modulation of the REV3 and REV7 gene expression. Mutat Res 2000; 461:133 - 43; http://dx.doi.org/10.1016/S0921-8777(00)00047-1; PMID: 11018586
- Treier M, Staszewski LM, Bohmann D. Ubiquitin-dependent c-Jun degradation in vivo is mediated by the δ domain. Cell 1994; 78:787 - 98; http://dx.doi.org/10.1016/S0092-8674(94)90502-9; PMID: 8087846
- Zhou Y, Ching YP, Chun AC, Jin DY. Nuclear localization of the cell cycle regulator CDH1 and its regulation by phosphorylation. J Biol Chem 2003; 278:12530 - 6; http://dx.doi.org/10.1074/jbc.M212853200; PMID: 12560341
- Turner DL, Weintraub H. Expression of achaete-scute homolog 3 in Xenopus embryos converts ectodermal cells to a neural fate. Genes Dev 1994; 8:1434 - 47; http://dx.doi.org/10.1101/gad.8.12.1434; PMID: 7926743
- Chan CP, Mak TY, Chin KT, Ng IOL, Jin DY. N-linked glycosylation is required for optimal proteolytic activation of membrane-bound transcription factor CREB-H. J Cell Sci 2010; 123:1438 - 48; http://dx.doi.org/10.1242/jcs.067819; PMID: 20356926
- Bachmair A, Finley D, Varshavsky A. In vivo half-life of a protein is a function of its amino-terminal residue. Science 1986; 234:179 - 86; http://dx.doi.org/10.1126/science.3018930; PMID: 3018930