
Abstract
Ewing sarcoma is a malignant bone cancer that primarily occurs in children and adolescents. Eighty-five percent of Ewing sarcoma is characterized by the presence of the aberrant chimeric EWS/FLI1 fusion gene. Previously, we demonstrated that an interaction between EWS/FLI1 and wild-type EWS led to the inhibition of EWS activity and mitotic dysfunction. Although defective mitosis is considered to be a critical step in cancer initiation, it is unknown how interference with EWS contributes to Ewing sarcoma formation. Here, we demonstrate that EWS/FLI1- and EWS-knockdown cells display a high incidence of defects in the midzone, a midline structure located between segregating chromatids during anaphase. Defects in the midzone can lead to the failure of cytokinesis and can result in the induction of aneuploidy. The similarity among the phenotypes of EWS/FLI1- and EWS siRNA-transfected HeLa cells points to the inhibition of EWS as the key mechanism for the induction of midzone defects. Supporting this observation, the ectopic expression of EWS rescues the high incidence of midzone defects observed in Ewing sarcoma A673 cells. We discovered that EWS interacts with Aurora B kinase, and that EWS is also required for recruiting Aurora B to the midzone. A domain analysis revealed that the R565 in the RGG3 domain of EWS is essential for both Aurora B interaction and the recruitment of Aurora B to the midzone. Here, we propose that the impairment of EWS-dependent midzone formation via the recruitment of Aurora B is a potential mechanism of Ewing sarcoma development.
Keywords: :
Introduction
Ewing sarcoma is the second most common bone tumor in adolescents and exhibits a characteristic small round blue cell morphology. The majority (>85%) of Ewing sarcoma patients have a common molecular abnormality, the t(11; 22) chromosomal translocation, which results in the expression of a chimeric fusion protein containing EWS (also known as EWSR1)-derived sequences at the N-terminus fused to the carboxyl-terminus of the ETS transcription factor FLI1.Citation1,Citation2 Tumors in the remaining Ewing sarcoma patients express proteins that result from the fusion of EWS with other ETS transcription factors (ERG, ETV1, ETV4/EIAF, or FEV).Citation1,Citation3-Citation7 The most well defined function of EWS/FLI1 is the transcriptional misregulation of target genes.Citation2,Citation8-Citation14 The interaction between RNA helicase A and EWS/FLI1 enhances EWS/FLI1-dependent transcriptional activity.Citation15,Citation16 EWS/FLI1 also regulates a subgroup of miRNAs that specifically regulate the IGF pathway.Citation17 Ewing sarcoma patient cells display high rates of aneuploidy.Citation18,Citation19 However, it is currently unknown how aneuploidy is induced in Ewing sarcoma cells.
We previously demonstrated that the expression of both EWS/FLI1- and EWS-knockdown in HeLa cells and in zebrafish embryos led to mitotic dysfunction that was characterized by disorganized mitotic spindles and multipolar cells.Citation20,Citation21 We also demonstrated that a biochemical interaction between EWS/FLI1 and wild-type EWS led to the inhibition of EWS activity in a dominant manner and induced mitotic dysfunction.Citation21 Mitotic dysfunction is often associated with tumorigenesis because it leads to chromosome instability (CIN), which induces mutations including aneuploidy.Citation22-Citation24 CIN is induced by chromosome missegregation due to failures in chromosomal condensation, spindle attachment to the kinetochore, or failure in cytokinesis. Impairment in the midzone formation during anaphase is one of the major causes for the failure in cytokinesis. The midzone is composed of the central spindles (anti-parallel microtubules) that are located between the segregating chromosomes and midzone proteins (Aurora B, Borealin, Survivin, INCENP, PRC1, KIF4, MKLP2, etc), which regulate the elongation of the central spindle and the formation of the cleavage furrow.Citation25-Citation33 Among the midzone proteins, Aurora B is a key factor for regulating early midzone formation. Aurora B forms the chromosomal passenger complex (CPC) with Borealin, Survivin, INCENP, and it localizes at the inner centromere and regulates the mitotic checkpoint by phosphorylating checkpoint molecules during the metaphase. During the metaphase-anaphase transition, Aurora B relocates to the midzone by interacting with MKLP2.Citation27 The Aurora B that was recruited to the central spindle phosphorylates KIF4, thus enhancing the interaction between phosphorylated KIF4 and PRC1. This KIF4-PRC1 complex regulates the length of the central spindle by inhibiting its growth.Citation28
We previously discovered that Aurora B failed to relocalize from the centromere to the midzone at the metaphase-anaphase transition in EWS/FLI1- and EWS siRNA-transfected HeLa cells.Citation20,Citation21 Therefore, we conducted this study to elucidate how EWS/FLI1 and EWS regulate mitosis and how these molecules associate with Aurora B kinase. Here, we demonstrate that EWS regulates midzone formation by relocating Aurora B from the inner centromere to the midzone. The mapping analysis for EWS revealed that the R565 located in the RGG3 domain is the critical amino acid required for both interaction with Aurora B and the recruitment of Aurora B to the midzone. This report may bridge the knowledge gap between the function of EWS and the induction of aneuploidy during Ewing sarcoma formation. Furthermore, this novel mechanism will also provide a platform to study how EWS/FLI1 affects this mechanism.
Results
Expression of EWS/FLI1 and knockdown of EWS lead to defects in midzone formation
Because we previously identified the aberrant localization of Aurora B during anaphase in EWS/FLI1- and EWS siRNA-transfected HeLa cells, we examined the other CPC components (Borealin, INCENP and Survivin) in the same sample groups. The pSG5-2xFLAG-EWS/FLI1 DNA construct was transfected into HeLa cells. Representative images of the expression levels for EWS/FLI1 protein in HeLa cells, which were visualized via anti-FLAG antibody, are shown in Figure S1A. The EWS/FLI1-transfected HeLa cells were subjected to immunocytochemistry using anti-Borealin, anti-INCENP and anti-Survivin antibodies. During anaphase, CPC components normally localize to the central spindles of the midzone, a structure that is composed of antiparallel microtubules and is held together by bundling proteins during anaphase.Citation34 This structure later becomes the midbody and plays a central role in cytokinesis, leading to the formation of two daughter cells. CPC components are normally evenly distributed along the midzone, a process that is essential for cytokinesis. The immunocytochemistry using anti-Borealin, anti-INCENP and anti-Survivin antibodies in the EWS/FLI1-expressing HeLa cells revealed that all of the CPC proteins displayed disorganized localization patterns, including either dense or sparse localization along the plane of the cleavage furrow (). We scored the aberrant localization of CPC components on the midzone. As a result, there were increased percentages of EWS/FLI1-transfected cells with disorganized CPC localization patterns at the midzone during anaphase (’). The results suggest that EWS/FLI1 impairs midzone formation.
Figure 1. EWS/FLI1- and EWS siRNA-transfected HeLa cells display abnormal CPC component localizations at the midzone. (A) Immunocytochemistry: left, untransfected; middle, empty vector; right, pSG5-2xFLAG-EWS/FLI1-transfected HeLa cells using: (a) top, merged images of anti-Borealin antibody (green) and DAPI (blue): bottom: anti-Borealin antibody; (b) top, merged images of anti-INCENP antibody (red) and DAPI (blue); bottom, anti-INCENP antibody; (c) top, merged images of anti-Survivin antibody (green) and DAPI (blue); bottom, anti-Survivin antibody. (A’). The percentages of cells that displayed mislocalized CPC components (31 to 72 anaphase cells per sample) (Experiments were repeated for n = 3 for Borealin, n = 4 for INCENP, n = 3 for Survivin). (B) Immunocytochemistry: left, untransfected; middle, control-siRNA; right, EWS siRNA-transfected HeLa cells using: (a) top, merged images of anti-Borealin antibody (green) and DAPI (blue); bottom, anti-Borealin antibody; (b) top, merged images of anti-INCENP antibody (red) and DAPI (blue); bottom: anti-INCENP antibody; (c) top, merged images of anti-Survivin antibody (green) and DAPI (blue); bottom, anti-Survivin antibody. (B’) The percentages of cells that displayed mislocalized CPC components (26 to 69 anaphase cells per sample) (Experiments were repeated for n = 3 for Borealin, n = 3 for INCENP, n = 4 for Survivin). Vector, empty vector; E/F, EWS/FLI1; Cont si, control-siRNA; EWS si, EWS siRNA-transfected HeLa cells. <, area of midzone with aberrant localization of CPC components; [, area of midzone with localization of CPC components.
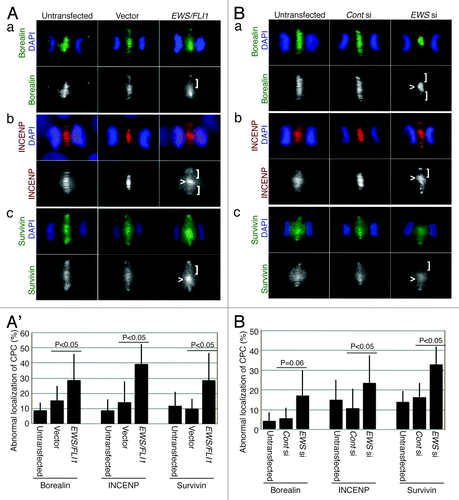
To further examine whether EWS plays a role in the midzone localization of CPC components, EWS siRNA- and control siRNA- (scrambled sequence of siRNA) was transfected into HeLa cells. A reduction in the expression level of EWS protein in the EWS siRNA-transfected cells is shown in Figure S1B. The siRNA-transfected cells were subjected to immunocytochemistry using anti-Borealin, anti-INCENP and anti-Survivin antibodies. As was consistent with the results obtained with EWS/FLI1, the EWS siRNA-transfected HeLa cells also displayed an increased incidence of unevenly distributed CPC components at the midzone (). The numbers of EWS siRNA-transfected HeLa cells that exhibited abnormal localization patterns for the CPC components was significantly higher than that in the controls (untransfected and control siRNA-transfected HeLa cells) (’). The results suggest that EWS knockdown leads to impairment in midzone formation. This result is consistent with our previous finding that EWS/FLI1 induces mitotic dysfunction through the inhibition of EWS function.Citation21 Together, these results suggest that the expression of EWS/FLI1 leads to a mislocalization of the CPC components through a dominant negative effect on endogenous EWS.
The aberrant localization of CPC components with an uneven distribution at the midzone in EWS/FLI1 and EWS siRNA-transfected HeLa cells suggests that the midzone is not properly formed in these cells. To test this, we examined the localization of the midzone protein PRC1, which is the protein that localizes at the antiparallel central spindles by bundling the spindles and by aiding its extension during anaphase.Citation32 Midzone localization of PRC1 relies on the proper localization of Aurora B at the midzone.Citation28 Ewing sarcoma A673 cells, a cell line that expresses EWS/FLI1 endogenously, were subjected to immunocytochemistry using anti-PRC1 antibody (). The A673 cells display a high incidence of an aberrant distribution of PRC1 throughout the midzone (the image is shown in , and the score (46+/−9%, obtained from n = 4 experiments) shown in untransfected lane). The aberrant distribution of PRC1 in the midzone is presumably due to the expression of EWS/FLI1 in A673 cells. To test this, we transfected pSG5-2xFLAG-EWS/FLI1 DNA construct into the HeLa cells and examined the localization of PRC1 by immunocytochemistry. As a result, EWS/FLI1-transfected HeLa cells also displayed significantly a higher incidence of irregular localization of PRC1 at the midzone compared with controls (untransfected and empty vector-transfected HeLa cells) (). Additionally, as is consistent with the irregular localization patterns for the CPC components in the EWS siRNA-transfected HeLa cells, these cells display a significantly high level of abnormal accumulation of PRC1 at the midzone (). Thus, both CPC components and PRC1 failed to form a normal localization pattern in EWS/FLI1 and EWS siRNA-transfected HeLa cells, presumably due to defects in midzone formation ( and ). These results suggest that EWS is required for proper midzone formation and that EWS/FLI1 interference with EWS function through a dominant negative mechanism may lead to these defects. Our results are consistent with the previous study that found that the depletion of PRC1 led to a disorganized central spindle, the absence of a stable midbody, and cytokinesis failure.Citation32
Figure 2. EWS/FLI1-expressing and EWS-knockdown HeLa cells display aberrant localizations of PRC1 at the midzone, and the transfection of Ewing sarcoma A673 cells with EWS rescues the high incidence of aberrant localization of PRC1 at the midzone. (A) Top, merged images, with DNA stained with DAPI (blue) and PRC1 (red) visualized via anti-PRC1 antibody; bottom, PRC1. Untransf., untransfected; vector, empty vector; E/F, human pSG5-2xFLAG-EWS/FLI1-transfected HeLa cells. bottom: PRC1 visualized with anti-PRC1 antibody. (B) The percentages of HeLa cells with mislocalized PRC1. The pSG5-2xFLAG-EWS/FLI-transfected HeLa cells displayed a higher incidence of abnormal PRC1 localization (31 to 72 anaphase cells per sample) (n = 3 experiments). (C) Top, merged images with DNA stained using DAPI (blue), and PRC1 (red) visualized with anti-PRC1 antibody. Untransf, untransfected; CONT si, control-siRNA; EWS si, human EWS siRNA-transfected HeLa cells. Bottom, PRC1 visualized with anti-PRC1 antibody. (D) The percentages of HeLa cells with mislocalized PRC1. The EWS siRNA-transfected HeLa cells display a higher incidence of abnormal PRC1 localization (30 to 56 anaphase were scored for each of the experiment)(n = 4 experiments). (E) Western blotting of A673 cell lysates from untransfected, empty vector-transfected and pSG5-2xFLAG-EWS-transfected cells probed with anti-FLAG antibody to verify transfection, and anti-β-actin antibody control. (F) The percentages of the cells with abnormal localization patterns for PRC1 were scored (n = 3 experiments). The transfection of Ewing sarcoma A673 cells with EWS rescues the high incidence of the aberrant localization of PRC1 at the midzone. (50 to 51 anaphase were scored for each of the experiment)(n = 4 experiments). <, area of midzone with aberrant localization of PRC1; [, area of midzone with localization of PRC1 components.
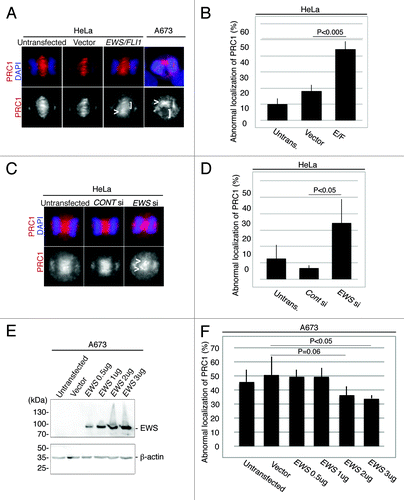
The high incidence of midzone defects in Ewing Sarcoma A673 cells is rescued by the overexpression of EWS
To determine whether the high incidence of midzone defects in Ewing sarcoma A673 cells is due to the inhibition of EWS function, rescue experiments with EWS were performed. A673 cells plated in 6 well plates were transfected with 0.5 µg to 3 µg of pSG5-2xFLAG-EWS DNA construct, and an empty vector transfection was performed as a control. The dose-dependent expression of ectopic EWS was confirmed via western blotting using anti-FLAG antibody (). These cells were also subjected to immunocytochemistry using anti-PRC1 antibody, and the percentages of cells with midzone defects as defined via aberrant PRC1 localization in the midzone were scored for each sample (). The baseline level of midzone defects in the A673 cells is relatively high (). However, when 2 µg and 3ug of pSG5-2xFLAG-EWS DNA construct was overexpressed in A673 cells, the incidence of midzone defects was rescued and became lower compared with that of untransfected and empty vector-transfected A673 cells (). The results indicate that the inhibition of EWS function is sufficient for the induction of aberrant midzone formation, presumably due to the dominant negative effect of EWS/FLI1 on EWS.
EWS colocalizes with Aurora B in A673 and HeLa cells
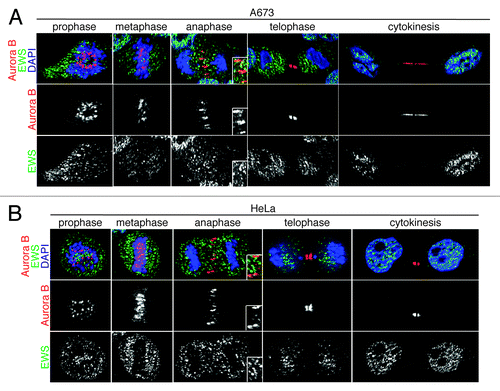
EWS/FLI1-expressing and EWS siRNA-transfected cells display both the aberrant localization of midzone proteins (CPC components and PRC1) and EWS overexpression, which rescues the aberrant localization of PRC1. Because the Aurora B-dependent phosphorylation of KIF4 is required for the interaction and activation of PRC1, we hypothesized that the EWS directly interacts with Aurora B.Citation28 To test this, we first examined the localization of EWS and Aurora B during mitosis via immunocytochemistry in Ewing sarcoma A673 cells; single Z-section images are shown in . The anti-EWS antibody used in this experiment detects only wild type EWS and not EWS/FLI1 because the antigen site is not included in the EWS/FLI1 fusion. Consistent with previous reports, Aurora B displayed dynamic localization throughout the mitosis.Citation35 The localization of EWS was similar to HeLa cells as we described previously.Citation20 Interestingly, the majority of EWS and Aurora B did not co-localize during prophase or metaphase (). However, higher levels of EWS and Aurora B colocalized at the midzone during anaphase, suggesting the involvement of EWS in midzone formation (; anaphase). During telophase and cytokinesis, the localization of Aurora B is limited to the midbody. Some of the EWS proteins also localized at the midbody; however, a majority of the EWS protein localized in the cytoplasm at telophase and in nucleus at cytokinesis. We also examined whether and when the EWS and Aurora B are colocalized in HeLa cells (). Similar to the result obtained from A673 cells, both components showed prominent colocalization at the midzone during anaphase.
Figure 3. EWS colocalizes with Aurora B at the midzone in A673 and HeLa cells. Single Z-section images of (A) Top, merged images with DNA stained using DAPI (blue), Aurora B (red) visualized using anti-Aurora B antibody, and EWS (green) via anti-EWS antibody; middle, Aurora B visualized with anti-Aurora B antibody; bottom, EWS visualized with anti-EWS antibody in A673 cells; (B) top, merged images with DNA stained with DAPI (blue), Aurora B (red) visualized with anti-Aurora B antibody and EWS (green) with anti-EWS antibody; middle, Aurora B visualized with anti-Aurora B antibody; bottom, EWS visualized with anti-EWS antibody in HeLa cells. Higher magnification images of midzone are shown in the boxed area in anaphase images obtained from both A673 and HeLa cells (A and B).
EWS interacts with Aurora B, and recruits Aurora B to the midzone
Because EWS and Aurora B colocalizes together, we performed co-immunoprecipitation (co-IP) experiments to determine whether EWS interacts with Aurora B. The full-length of pSG5-2xFLAG-EWS DNA construct was transfected into HeLa cells, and the cell lysates were extracted. The endogenous Aurora B was immunoprecipitated with an anti-Aurora B antibody or with an IgG control, and analyzed via western blotting with an antibody recognizing FLAG (Fig. S2B). As a result, the co-IP experiment revealed that full-length EWS interacts with Aurora B in HeLa cells (Fig. S2B). To identify the essential domain of EWS that is required for interaction with Aurora B, we utilized EWS DNA constructs that contained deletions in the C-terminus (delEWS-C) that were tagged with a 2x FLAG tag at the N-terminus (Fig. S2A). The pSG5-2xFLAG-delEWS-C DNA construct lacks the RGG domain (repeats of the RGG sequence required for protein or RNA interaction) and the Nuclear Localization Signal (NLS) is the critical domain for the interaction. The pSG5-2xFLAG-delEWS-C DNA constructs were transfected into HeLa cells, and the cell lysates were extracted from the transfected HeLa cells. The endogenous Aurora B proteins were immunoprecipitated using an anti-Aurora B antibody, which was analyzed via western blotting using an anti-FLAG antibody to confirm the interaction between deletion EWS and Aurora B. Contrary to the interaction between full-length EWS and Aurora B, the delEWS-C failed to bind to Aurora B (Fig. S2B). This result suggests that the fragment containing the RGG domain NLS is the critical domain for the interaction.Citation36
The RGG3 domain has 12 repeats of the RGG sequence. To further identify the single amino acid that is required for the interaction with Aurora B, we introduced a point mutation in Arg at the 565th amino acid that substituted to Ala (EWS-R565A) within the RGG repeat (). To identify whether the EWS R565A mutant could interact with Aurora B, empty vector, the full-length of pSG5-2xFLAG-EWS and pSG5-2xFLAG-EWS-R565A DNA constructs were transfected into HeLa cells, and the cell lysates were extracted. Using the cell lysates, a co-IP experiment was performed by immunoprecipitating the Aurora B proteins and IgG as a control, followed by western blotting using anti-FLAG antibody. As a result, the EWS-R565A failed to interact with Aurora B, whereas the full-length EWS interacted with Aurora B (). This result suggests that the R565 of EWS is the essential amino acid required for interaction with Aurora B.
Figure 4. EWS but not EWS-R565A mutant interacts with Aurora B, and transfection with EWS but not EWS-R565A mutants rescues the high incidence of the aberrant localization of Aurora B in A673 cells. (A) Schematic drawing of EWS and EWS-R565A mutant proteins. (B) Immunoprecipitation of lysates from untransfected, empty vector, pSG5-2xFLAG-EWS- and pSG5-2xFLAG-EWS-R565A mutant-transfected HeLa cells using IgG and anti-Aurora B antibody. Top (left), probing blots with anti-FLAG shows that EWS coimmunoprecipitates with Aurora B. Bottom; left, Probing blots with anti-Aurora B shows immunoprecipitation of endogenous Aurora B; right, input sample (1/50) of cell lysates of untransfected, empty vector, pSG5-2xFLAG-EWS- and pSG5-2xFLAG-EWS-R565A-transfected HeLa cells demonstrated with anti-FLAG antibody (top) and anti-Aurora B antibody (bottom). (C) Top, merged images with DNA stained with DAPI (blue), and Aurora B (red) visualized with anti-Aurora B antibody; bottom, Aurora B visualized with anti-Aurora B antibody in A673 cells. (D) Percentages of cells with abnormal localization patterns for Aurora B were scored in untransfected, empty vector, pSG5-2xFLAG-EWS- and pSG5-2xFLAG-EWS-R565A-transfected A673 cells (50 anaphase were scored for each of the experiment) (n = 3 experiments).
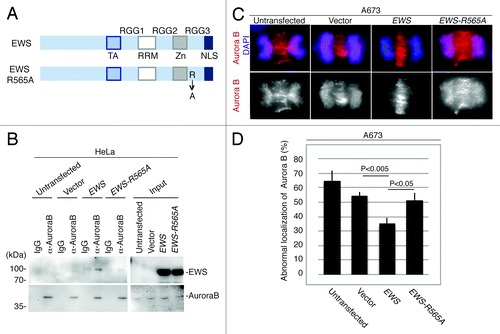
To determine whether a high incidence of midzone defects is linked to Aurora B localization to the midzone in A673 cells, immunocytochemistry was performed using anti-Aurora B. Aurora B kinase failed to localize to the midzone in high numbers of A673 cells (). To further elucidate whether the aberrant localization of Aurora B is due to the inhibition of EWS function, rescue experiments with full-length EWS and EWS-R565A DNA constructs were performed. Because the transfection of 2 µg of pSG5-2xFLAG-EWS DNA construct into the A673 cells was sufficient to rescue the aberrant localization of PRC1 in , the A673 cells were transfected with 2 µg of an pSG5-2xFLAG-EWS, and pSG5-2xFLAG-EWS-R565A DNA constructs along with an empty vector as a control. The expression of EWS and EWS R565A in the A673 cells was confirmed by western blotting (Fig. S3). The transfected A673 cells were also subjected to immunocytochemistry using anti-Aurora B antibody. The percentages of the cells that displayed aberrant Aurora B localization in the midzone were scored for each sample (). pSG5-2xFLAG-EWS rescued the high incidence of aberrant Aurora B localization at the midzone (). This result suggests that the EWS protein plays a role in recruiting Aurora B to the midzone. Additionally, the overexpression of pSG5-2xFLAG-EWS-R565A failed to rescue the high incidence of Aurora B localization in the midzone (). The result suggests that the R565 in the RGG3 domain of EWS plays a critical role in the recruitment of Aurora B to the midzone. Because R565 of EWS is an essential amino acid for both associating with Aurora B and for recruiting Aurora B to the midzone, this may suggest that EWS interacts with Aurora B and that the interaction drives the recruitment of Aurora B to the midzone.
Discussion
Identifying the mechanism for EWS/FLI1- or EWS knockdown-dependent mitotic defects may explain how chromosome instability (CIN) is induced in Ewing sarcoma. In comparing the Ewing sarcoma A673 cells with the EWS/FLI1- and EWS siRNA-transfected HeLa cells, it was found that all three experimental cell groups displayed a high incidence of midzone formation defects. Because all samples displayed the same phenotypes, the interaction with EWS/FLI1 may interfere with EWS function during midzone formation. Furthermore, we discovered that EWS interacts with the key mitotic kinase Aurora B and relocates Aurora B from the inner centromere to the midzone. The R565 in the RGG3 domain of EWS is essential for both interaction with Aurora B and the recruitment of Aurora B to the midzone. This EWS-dependent mechanism for midzone formation is a novel mechanism.
Previous reports indicate that the relocation of Aurora B from the inner centromeres to the midzone during the metaphase-anaphase transition is a critical event in deactivating the mitotic checkpoint.Citation37 A high level of Aurora B protein expression can lead to a failure in relocation. AURORA B was among numerous cell cycle regulators that have been identified as EWS/FLI1 target genes, and AURORA B mRNA is upregulated in Ewing sarcoma cells.Citation38 The upregulation of a significant amount of Aurora B protein may also contribute to the failure of its relocalization to the midzone. In addition to the gene regulation of AURORA B, the previous report also showed that the degradation of Aurora B is mediated by Cul3/KLHL9/KLHL13 E3 ligase at the centromere during the metaphase-anaphase transition in HeLa cells.Citation39 In Ewing sarcoma A673 cells, a large number of Aurora B proteins failed to relocate to the midzone and were retained on the centromere. Therefore, it is essential to investigate how and whether EWS and EWS/FLI1 are involved in the degradation mechanism of Aurora B.
In addition to the regulation of AURORA B/Aurora B expression, it is also important to understand how EWS is involved in the midzone formation process. MKLP2 was reported to interact with Aurora B and is required for relocating Aurora B from the inner centromere to the central spindle in the midzone.Citation27 In addition, the Aurora B that was recruited to the central spindle by MKLP2 activates KIF4 and regulates the length of the central spindle by inhibiting its growth.Citation28 It is possible that the EWS is a component of the MKLP2-Aurora B complex. Alternatively, it is possible that the EWS-Aurora B regulates midzone formation independently of MKLP2-Aurora B function. Elucidating how these EWS-complex/dimers activate KIF4 will provide a better understanding of midzone formation. Because EWS was reported to associate with α-tubulin, it is also potentially associated with the central spindle.Citation40 It will be critical to elucidate how this interaction affects the efficiency of polymerization or stability of spindles. In this study, we discovered that the EWS-R565A mutant fails to interact with and to recruit Aurora B to the midzone. Importantly, EWS/FLI1 does not contain this amino acid, which indicates a significant role of EWS during midzone formation. Interestingly, the RGG domain of EWS was reported to be a substrate for methylation catalyzed by protein arginine methyltransferases (PRMT).Citation41-Citation44 It is possible that the interaction between the EWS and Aurora B is regulated by the post transcriptional modification. The EWS R565A mutant is a powerful tool in determining how EWS participates in Aurora B-dependent midzone formation. Because Ewing sarcoma cells display high aneuploidy rates, the impairment of this regulation may directly explain how aneuploidy is induced; knowledge of this would fill the current gap regarding the role of EWS in the induction of mutations in Ewing sarcoma formation.Citation18
Materials and Methods
Gene silencing using siRNA
HeLa cells were plated in 6-well plates and transfected with 70 pmol of EWS-siRNAs (a mixture of siRNAs designed against four independent sites within human EWS) (Santa Cruz, sc-35347) or control- siRNA (siRNA designed against scrambled sequence) (Santa Cruz, sc-37007) in 1 ml of medium as described in the manufacturer’s protocol with minor modifications. The cells were harvested 14 h after transfection and analyzed via western blotting or immunocytochemistry.
Construction of EWS mutant DNA constructs
The human pSG5-2xFLAG-delEWS-C deletion construct was constructed by PCR amplification using primers EcoRI-human EWS1/18F: 5′-GCATGAATTC ATGGCGTCCA CGGATTAC-3′ and Hind III-humanEWS1637R: 5′-GGTAAAGCTT ATGGGGCCTT ACACTGGTTG-3′. PCR products were cloned into pCRII-TOPO (Invitrogen), and the sequence was confirmed by DNA sequencing (ACGT Inc). The pCRII-delEWS-C construct was digested with EcoR I and Hind III cloned into pSG5 digested with EcoR I and Hind III. The human pSG5-2xFLAG-R565A mutant DNA construct was constructed by introducing a mutation in R565 of EWS using QuikChange II Site-Directed Mutagenesis Kit (Agilant Technologies). The following primers were used for the site-directed mutagenesis: human EWS-R565A-F: 5′-GTGGTGATCG TGGCgcaGGT GGCCCTGGTG-3′, human EWS-R565A-R: 5′-CACCAGGGCC ACCtgcGCCA CGATCACCAC-3′.
Transfection using DNA constructs
The transfection experiments were performed using Lipofectamine 2000 transfection reagent (Invitrogen, Cat: 11668–019) following the manufacturer’s protocol. The HeLa cells were grown on coverslips in 6-well plates, followed by transfection with a total of 4 µg of pSG5-2xFLAG-EWS/FLI1 and pSG5-2xFLAG (empty vector). The A673 cells were also grown on coverslips and were transfected with 3 µg of pSG5-2xFLAG-EWS/FLI1 and pSG5-2xFLAG (empty vector) for , and 2 μg of same DNA constructs for . Both HeLa and A673 cells were harvested 15 h after transfection and analyzed via western blotting or immunocytochemistry. For Co-IP experiments, the HeLa cells were plated on 9 cm dishes and were transfected with 28 µg of pSG5, pSG5-EWS, pSG5-delEWS-C and pSG5-EWS-R565A DNA constructs using Lipofectamine 2000 (Invitrogen, Cat:11668–019) following the manufacturer’s protocol.
Immunocytochemistry and western blotting
Both the A673 cells and HeLa cells were fixed with 4% paraformaldehyde for 10 min, washed in PBS, and permeabilized with methanol for 10 min at −20 °C. The cells were washed in PBS, incubated in blocking solution (1% fetal bovine serum in PBS) for 1 h followed by 1 h incubation with primary antibodies: anti-Borealin (1:1000 dilution)(Abcam, Cat:ab70910), anti-INCENP (1:1000 dilution)(Abcam, Cat:ab23956), anti-Survivin (1:1000 dilution)(R&D Systems, Cat:AF886), anti-EWS antibody (1:1000 dilution)(Sigma, Cat:E4533), anti-Aurora B (1:500 dilution)(BD Biosciences, Cat:611082) and anti-PRC1 (1:100 dilution)(BioLegend, Cat:629002). After three 5 min washes in PBS, the cells were incubated in both Alexa Fluor 594 goat anti-mouse IgG (1:500 dilution)(Invitrogen, Cat:A11032) and Alexa Fluor 488 goat anti-rabbit IgG (1:500 dilution) (Invitrogen, Cat:A11034) for 1 h. After the PBS washes, the coverslips were mounted with DAPI/ProLong Gold (Invitrogen, Cat:P36935) and visualized at 1000× magnification on a Nikon Ti Eclipse microscope; the images were captured using MetaMorph imaging software. The single Z-section images were documented using OptiGrid Structured Illumination microscopy (Nikon).
Both the A673 cells transfected with pSG5-EWS DNA constructs and the HeLa cells transfected with pSG5-EWS/FLI1 DNA constructs or EWS siRNA were subjected to western blotting using a 1:1000 dilution of anti-mouse FLAG (Agilent Technology, Cat:200472–21), 1:1000 dilution of anti-EWS (C-19) antibody (Santa Cruz), or anti-β-actin (1:2500 dilution)(Sigma, Cat:A2228) followed by HRP-linked anti-mouse secondary antibody (1:100 000 dilution) or HRP-linked anti-rabbit secondary antibody (1:100 000 dilution). The proteins were visualized using SuperSignal West Femto Chemiluminescent Substrate (Thermo Scientific, Cat: 34095).
Co-Immunoprecipitation (co-IP) experiment
The co-IP experiments were performed using the same protocol described previously with minor modifications.Citation21 Cell lysates were immunoprecipitated using anti-Aurora B antibody (AbCam: ab2254) for 4 °C 30 min, and the samples were washed three times for 15 min. The samples were subjected to western blotting visualized via anti-FLAG antibody (1:1000 dilution)(Agilent Technology, Cat:200472–21) and anti-Aurora B antibody (1:500 dilution)(BD biosciences, Cat:611082).
| Abbreviations: | ||
| CIN | = | chromosome instability |
| CPC | = | chromosomal passenger complex |
Additional material
Download Zip (316.8 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Lisa Embree (National Cancer Institute / National Institutes of Health) for her important comments. This manuscript was supported by the Massman Family Ewing Sarcoma Research Fund, the Sarcoma Foundation of America, P20RR016475 / P20GM103418, and P20 RR032682-01.
References
- Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, Kovar H, Joubert I, de Jong P, Rouleau G, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992; 359:162 - 5; http://dx.doi.org/10.1038/359162a0; PMID: 1522903
- Toomey EC, Schiffman JD, Lessnick SL. Recent advances in the molecular pathogenesis of Ewing’s sarcoma. Oncogene 2010; 29:4504 - 16; http://dx.doi.org/10.1038/onc.2010.205; PMID: 20543858
- Sorensen PH, Lessnick SL, Lopez-Terrada D, Liu XF, Triche TJ, Denny CT. A second Ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat Genet 1994; 6:146 - 51; http://dx.doi.org/10.1038/ng0294-146; PMID: 8162068
- Jeon IS, Davis JN, Braun BS, Sublett JE, Roussel MF, Denny CT, Shapiro DN. A variant Ewing’s sarcoma translocation (7;22) fuses the EWS gene to the ETS gene ETV1. Oncogene 1995; 10:1229 - 34; PMID: 7700648
- Kaneko Y, Yoshida K, Handa M, Toyoda Y, Nishihira H, Tanaka Y, Sasaki Y, Ishida S, Higashino F, Fujinaga K. Fusion of an ETS-family gene, EIAF, to EWS by t(17;22)(q12;q12) chromosome translocation in an undifferentiated sarcoma of infancy. Genes Chromosomes Cancer 1996; 15:115 - 21; http://dx.doi.org/10.1002/(SICI)1098-2264(199602)15:2<115::AID-GCC6>3.0.CO;2-6; PMID: 8834175
- Peter M, Couturier J, Pacquement H, Michon J, Thomas G, Magdelenat H, Delattre O. A new member of the ETS family fused to EWS in Ewing tumors. Oncogene 1997; 14:1159 - 64; http://dx.doi.org/10.1038/sj.onc.1200933; PMID: 9121764
- Ng TL, O’Sullivan MJ, Pallen CJ, Hayes M, Clarkson PW, Winstanley M, Sorensen PH, Nielsen TO, Horsman DE. Ewing sarcoma with novel translocation t(2;16) producing an in-frame fusion of FUS and FEV. J Mol Diagn 2007; 9:459 - 63; http://dx.doi.org/10.2353/jmoldx.2007.070009; PMID: 17620387
- Abaan OD, Levenson A, Khan O, Furth PA, Uren A, Toretsky JA. PTPL1 is a direct transcriptional target of EWS-FLI1 and modulates Ewing’s Sarcoma tumorigenesis. Oncogene 2005; 24:2715 - 22; http://dx.doi.org/10.1038/sj.onc.1208247; PMID: 15782144
- Smith R, Owen LA, Trem DJ, Wong JS, Whangbo JS, Golub TR, Lessnick SL. Expression profiling of EWS/FLI identifies NKX2.2 as a critical target gene in Ewing’s sarcoma. Cancer Cell 2006; 9:405 - 16; http://dx.doi.org/10.1016/j.ccr.2006.04.004; PMID: 16697960
- Joo J, Christensen L, Warner K, States L, Kang HG, Vo K, Lawlor ER, May WA. GLI1 is a central mediator of EWS/FLI1 signaling in Ewing tumors. PLoS One 2009; 4:e7608; http://dx.doi.org/10.1371/journal.pone.0007608; PMID: 19859563
- Beauchamp E, Bulut G, Abaan O, Chen K, Merchant A, Matsui W, Endo Y, Rubin JS, Toretsky J, Uren A. GLI1 is a direct transcriptional target of EWS-FLI1 oncoprotein. J Biol Chem 2009; 284:9074 - 82; http://dx.doi.org/10.1074/jbc.M806233200; PMID: 19189974
- Zwerner JP, Joo J, Warner KL, Christensen L, Hu-Lieskovan S, Triche TJ, May WA. The EWS/FLI1 oncogenic transcription factor deregulates GLI1. Oncogene 2008; 27:3282 - 91; http://dx.doi.org/10.1038/sj.onc.1210991; PMID: 18084326
- Kinsey M, Smith R, Iyer AK, McCabe ER, Lessnick SL. EWS/FLI and its downstream target NR0B1 interact directly to modulate transcription and oncogenesis in Ewing’s sarcoma. Cancer Res 2009; 69:9047 - 55; http://dx.doi.org/10.1158/0008-5472.CAN-09-1540; PMID: 19920188
- Sohn EJ, Li H, Reidy K, Beers LF, Christensen BL, Lee SB. EWS/FLI1 oncogene activates caspase 3 transcription and triggers apoptosis in vivo. Cancer Res 2010; 70:1154 - 63; http://dx.doi.org/10.1158/0008-5472.CAN-09-1993; PMID: 20103643
- Erkizan HV, Kong Y, Merchant M, Schlottmann S, Barber-Rotenberg JS, Yuan L, Abaan OD, Chou TH, Dakshanamurthy S, Brown ML, et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat Med 2009; 15:750 - 6; http://dx.doi.org/10.1038/nm.1983; PMID: 19584866
- Toretsky JA, Erkizan V, Levenson A, Abaan OD, Parvin JD, Cripe TP, Rice AM, Lee SB, Uren A. Oncoprotein EWS-FLI1 activity is enhanced by RNA helicase A. Cancer Res 2006; 66:5574 - 81; http://dx.doi.org/10.1158/0008-5472.CAN-05-3293; PMID: 16740692
- McKinsey EL, Parrish JK, Irwin AE, Niemeyer BF, Kern HB, Birks DK, Jedlicka P. A novel oncogenic mechanism in Ewing sarcoma involving IGF pathway targeting by EWS/Fli1-regulated microRNAs. Oncogene 2011; 30:4910 - 20; http://dx.doi.org/10.1038/onc.2011.197; PMID: 21643012
- Ozaki T, Paulussen M, Poremba C, Brinkschmidt C, Rerin J, Ahrens S, Hoffmann C, Hillmann A, Wai D, Schaefer KL, et al. Genetic imbalances revealed by comparative genomic hybridization in Ewing tumors. Genes Chromosomes Cancer 2001; 32:164 - 71; http://dx.doi.org/10.1002/gcc.1178; PMID: 11550284
- Bown NP, Reid MM, Malcolm AJ, Davison EV, Craft AW, Pearson AD. Cytogenetic abnormalities of small round cell tumours. Med Pediatr Oncol 1994; 23:124 - 9; http://dx.doi.org/10.1002/mpo.2950230210; PMID: 8202034
- Azuma M, Embree LJ, Sabaawy H, Hickstein DD. Ewing sarcoma protein ewsr1 maintains mitotic integrity and proneural cell survival in the zebrafish embryo. PLoS One 2007; 2:e979; http://dx.doi.org/10.1371/journal.pone.0000979; PMID: 17912356
- Embree LJ, Azuma M, Hickstein DD. Ewing sarcoma fusion protein EWSR1/FLI1 interacts with EWSR1 leading to mitotic defects in zebrafish embryos and human cell lines. Cancer Res 2009; 69:4363 - 71; http://dx.doi.org/10.1158/0008-5472.CAN-08-3229; PMID: 19417137
- Heng HH, Bremer SW, Stevens JB, Horne SD, Liu G, Abdallah BY, Ye KJ, Ye CJ. Chromosomal instability (CIN): what it is and why it is crucial to cancer evolution. Cancer Metastasis Rev 2013; 32:325 - 40; http://dx.doi.org/10.1007/s10555-013-9427-7; PMID: 23605440
- Thompson SL, Bakhoum SF, Compton DA. Mechanisms of chromosomal instability. Curr Biol 2010; 20:R285 - 95; http://dx.doi.org/10.1016/j.cub.2010.01.034; PMID: 20334839
- Holland AJ, Cleveland DW. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol 2009; 10:478 - 87; http://dx.doi.org/10.1038/nrm2718; PMID: 19546858
- Carmena M, Wheelock M, Funabiki H, Earnshaw WC. The chromosomal passenger complex (CPC): from easy rider to the godfather of mitosis. Nat Rev Mol Cell Biol 2012; 13:789 - 803; http://dx.doi.org/10.1038/nrm3474; PMID: 23175282
- Lee YM, Kim W. Kinesin superfamily protein member 4 (KIF4) is localized to midzone and midbody in dividing cells. Exp Mol Med 2004; 36:93 - 7; http://dx.doi.org/10.1038/emm.2004.13; PMID: 15031677
- Gruneberg U, Neef R, Honda R, Nigg EA, Barr FA. Relocation of Aurora B from centromeres to the central spindle at the metaphase to anaphase transition requires MKlp2. J Cell Biol 2004; 166:167 - 72; http://dx.doi.org/10.1083/jcb.200403084; PMID: 15263015
- Nunes Bastos R, Gandhi SR, Baron RD, Gruneberg U, Nigg EA, Barr FA. Aurora B suppresses microtubule dynamics and limits central spindle size by locally activating KIF4A. J Cell Biol 2013; 202:605 - 21; http://dx.doi.org/10.1083/jcb.201301094; PMID: 23940115
- Douglas ME, Mishima M. Still entangled: assembly of the central spindle by multiple microtubule modulators. Semin Cell Dev Biol 2010; 21:899 - 908; http://dx.doi.org/10.1016/j.semcdb.2010.08.005; PMID: 20732438
- Speliotes EK, Uren A, Vaux D, Horvitz HR. The survivin-like C. elegans BIR-1 protein acts with the Aurora-like kinase AIR-2 to affect chromosomes and the spindle midzone. Mol Cell 2000; 6:211 - 23; http://dx.doi.org/10.1016/S1097-2765(00)00023-X; PMID: 10983970
- Eckley DM, Ainsztein AM, Mackay AM, Goldberg IG, Earnshaw WC. Chromosomal proteins and cytokinesis: patterns of cleavage furrow formation and inner centromere protein positioning in mitotic heterokaryons and mid-anaphase cells. J Cell Biol 1997; 136:1169 - 83; http://dx.doi.org/10.1083/jcb.136.6.1169; PMID: 9087435
- Mollinari C, Kleman JP, Jiang W, Schoehn G, Hunter T, Margolis RL. PRC1 is a microtubule binding and bundling protein essential to maintain the mitotic spindle midzone. J Cell Biol 2002; 157:1175 - 86; http://dx.doi.org/10.1083/jcb.200111052; PMID: 12082078
- Cesario JM, Jang JK, Redding B, Shah N, Rahman T, McKim KS. Kinesin 6 family member Subito participates in mitotic spindle assembly and interacts with mitotic regulators. J Cell Sci 2006; 119:4770 - 80; http://dx.doi.org/10.1242/jcs.03235; PMID: 17077127
- Khmelinskii A, Schiebel E. Assembling the spindle midzone in the right place at the right time. Cell Cycle 2008; 7:283 - 6; http://dx.doi.org/10.4161/cc.7.3.5349; PMID: 18235228
- Murata-Hori M, Tatsuka M, Wang YL. Probing the dynamics and functions of aurora B kinase in living cells during mitosis and cytokinesis. Mol Biol Cell 2002; 13:1099 - 108; http://dx.doi.org/10.1091/mbc.01-09-0467; PMID: 11950924
- Thandapani P, O’Connor TR, Bailey TL, Richard S. Defining the RGG/RG motif. Mol Cell 2013; 50:613 - 23; http://dx.doi.org/10.1016/j.molcel.2013.05.021; PMID: 23746349
- Vázquez-Novelle MD, Petronczki M. Relocation of the chromosomal passenger complex prevents mitotic checkpoint engagement at anaphase. Curr Biol 2010; 20:1402 - 7; http://dx.doi.org/10.1016/j.cub.2010.06.036; PMID: 20619651
- Wakahara K, Ohno T, Kimura M, Masuda T, Nozawa S, Dohjima T, Yamamoto T, Nagano A, Kawai G, Matsuhashi A, et al. EWS-Fli1 up-regulates expression of the Aurora A and Aurora B kinases. Mol Cancer Res 2008; 6:1937 - 45; http://dx.doi.org/10.1158/1541-7786.MCR-08-0054; PMID: 19074838
- Sumara I, Quadroni M, Frei C, Olma MH, Sumara G, Ricci R, Peter M. A Cul3-based E3 ligase removes Aurora B from mitotic chromosomes, regulating mitotic progression and completion of cytokinesis in human cells. Dev Cell 2007; 12:887 - 900; http://dx.doi.org/10.1016/j.devcel.2007.03.019; PMID: 17543862
- Leemann-Zakaryan RP, Pahlich S, Sedda MJ, Quero L, Grossenbacher D, Gehring H. Dynamic subcellular localization of the Ewing sarcoma proto-oncoprotein and its association with and stabilization of microtubules. J Mol Biol 2009; 386:1 - 13; http://dx.doi.org/10.1016/j.jmb.2008.12.039; PMID: 19133275
- Kim JD, Kako K, Kakiuchi M, Park GG, Fukamizu A. EWS is a substrate of type I protein arginine methyltransferase, PRMT8. Int J Mol Med 2008; 22:309 - 15; PMID: 18698489
- Araya N, Hiraga H, Kako K, Arao Y, Kato S, Fukamizu A. Transcriptional down-regulation through nuclear exclusion of EWS methylated by PRMT1. Biochem Biophys Res Commun 2005; 329:653 - 60; http://dx.doi.org/10.1016/j.bbrc.2005.02.018; PMID: 15737635
- Pahlich S, Zakaryan RP, Gehring H. Identification of proteins interacting with protein arginine methyltransferase 8: the Ewing sarcoma (EWS) protein binds independent of its methylation state. Proteins 2008; 72:1125 - 37; http://dx.doi.org/10.1002/prot.22004; PMID: 18320585
- Pahlich S, Bschir K, Chiavi C, Belyanskaya L, Gehring H. Different methylation characteristics of protein arginine methyltransferase 1 and 3 toward the Ewing Sarcoma protein and a peptide. Proteins 2005; 61:164 - 75; http://dx.doi.org/10.1002/prot.20579; PMID: 16044463