Abstract
Background: In a prior study, a DNA prime / adenovirus boost vaccine (DNA/Ad) expressing P. falciparum circumsporozoite protein (CSP) and apical membrane antigen-1 (AMA1) (NMRC-M3V-D/Ad-PfCA Vaccine) induced 27% protection against controlled human malaria infection (CHMI). To investigate the contribution of DNA priming, we tested the efficacy of adenovirus vaccine alone (NMRC-M3V-Ad-PfCA ) in a Phase 1 clinical trial.
Methodology/Principal Findings: The regimen was a single intramuscular injection with two non-replicating human serotype 5 adenovectors encoding CSP and AMA1, respectively. One x 1010 particle units of each construct were combined prior to administration. The regimen was safe and well-tolerated. Four weeks later, 18 study subjects received P. falciparum CHMI administered by mosquito bite. None were fully protected although one showed delayed onset of parasitemia. Antibody responses were low, with geometric mean CSP ELISA titer of 381 (range < 50–1626) and AMA1 ELISA of 4.95 µg/mL (range 0.2–38). Summed ex vivo IFN-γ ELISpot responses to overlapping peptides were robust, with geometric mean spot forming cells/million peripheral blood mononuclear cells [sfc/m] for CSP of 273 (range 38–2550) and for AMA1 of 1303 (range 435–4594). CD4+ and CD8+ T cell IFN-γ responses to CSP were positive by flow cytometry in 25% and 56% of the research subjects, respectively, and to AMA1 in 94% and 100%, respectively.
Significance: In contrast to DNA/Ad, Ad alone did not protect against CHMI despite inducing broad, cell-mediated immunity, indicating that DNA priming is required for protection by the adenovirus-vectored vaccine. ClinicalTrials.gov Identifier: NCT00392015.
Introduction
The malaria parasite has a complex life cycle initiated by the bite of an infected mosquito injecting sporozoites into the skin of the human host. These motile forms enter the blood stream, travel to the liver and quickly invade hepatocytes, emerging 5–6 d later to start a cycle of erythrocyte invasion and lysis that leads to clinical disease and enables transmission back to the vector. A vaccine preventing the development of pre-erythrocytic stages would therefore block both clinical disease and transmission. Immunological protection against malaria sporozoites and liver stages is thought to depend primarily on anti-sporozoite antibodies and CD8+ T cells, respectively. While the exact mechanisms are unclear, studies in animal models demonstrate the critical role of cytotoxic CD8+ T cells in clearing liver stage parasites; these effector cells release cytotoxins that directly lyse the malaria-infected hepatocyte, or interferon-gamma (IFN-γ) that induces expression of nitric oxide within the host cell, thereby killing the resident parasite.Citation1,Citation2
While protein-based subunit vaccines such as RTS,S induce protective antibodies targeting the sporozoite,Citation3 gene-based technologies are a major focus of vaccine research aiming to induce cytotoxic CD8+ T cells targeting the infected liver cell. There are many gene-based vaccines licensed for use in veterinary medicine, but only the live-attenuated yellow fever-Japanese encephalitis chimeric vaccine (IMOJEV) has so far been licensed for human use (Australia).Citation4 Vaccine developers, however, are intensively engaged in the development of optimal platforms, formulations and delivery methods for gene-based vaccines in humans. In one approach, microbial genes are inserted into a DNA plasmid taken up passively by the host cell following injection. In another, the genes may be inserted into a viral vector, which more efficiently transports the DNA into the host cell and nucleus. With either plasmid or viral vectors, parasite proteins are expressed within the cytoplasm rather than supplied exogenously as in the case of protein-based malaria vaccines such as RTS,S. This leads to activation of the MHC class I (endogenous) antigen presentation pathway, generating cell-mediated immunity (CMI) including cytotoxic CD8+ T cells.
We have evaluated the gene-based approach, and have successfully protected mice, non-human primates and humans against malaria with DNA plasmid- and/or virally-vectored vaccines; like others, we have found that heterologous prime-boost regimens are especially effective.Citation5-Citation14 For example, a pentavalent DNA plasmid vaccine encoding P. falciparum CSP and four additional pre-erythrocytic stage antigens (MuStDO5 Vaccine) administered in three monthly doses failed to protect research subjects against controlled human malaria infection (CHMI),Citation15 while a mixture of just two plasmids encoding CSP and apical membrane antigen-1 (AMA1) administered in three monthly doses as the prime, followed by two recombinant adenovectors (human serotype 5) administered as a single heterologous boost, protected 27% of research subjects.Citation16 In another earlier trial, without CHMI, these two adenovectors alone had induced robust CD4+ and CD8+ T cell responses,Citation5 suggesting that they might be protective without the DNA prime. Thus we determined it was essential to test the adenovirus-vectored vaccine alone in a clinical trial without the DNA priming component to determine whether this simple, one-dose regimen might induce protection. A single injection was selected because in a prior study, a boost at eight weeks did not further improve CMI responses.Citation17
The recombinant Ad vaccine (NMRC-M3V-Ad-PfCA, ) was administered to 20 Ad5 seronegative malaria-naïve adult subjects to evaluate safety, tolerability and immunogenicity. Eighteen of the 20 were challenged (). As in the earlier trials, the vaccine proved to be safe, well tolerated and immunogenic, but no research subjects were sterilely protected, and the onset of parasitemia was delayed relative to controls in only one. We concluded that indeed DNA priming is essential for the induction of protective immunity.
Figure 1. Schematic of Adenovirus CSP and AMA1 vaccines. Each panel presents the native protein (top of each panel) and the protein expressed by the Ad construct (bottom of each panel) for the CSP (A) and AMA1 (B) vaccine antigens. N = N-terminus; C = carboxy terminus; TM = transmembrane domain. Identical colors indicate identical sequences.
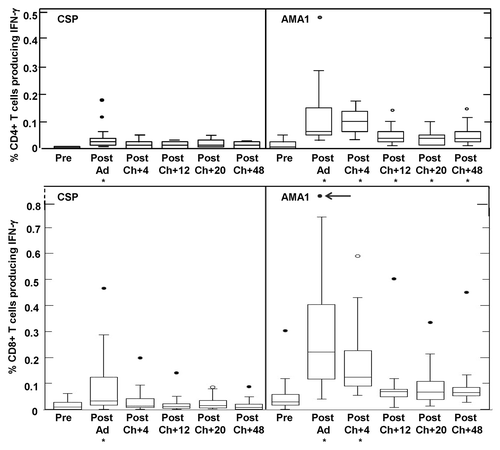
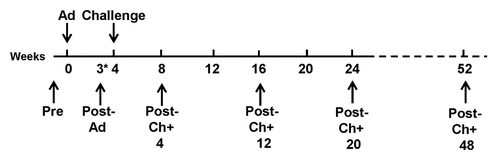
Figure 2. Trial design. Subjects were immunized week 0 and challenged week 4. Samples for measuring cell-mediated immunity (ELISpot assay and flow cytometry) and antibody levels (ELISA and IFA) were collected at six time points (black arrows): Pre (pre-immunization), Post-Ad (*22–23 d after immunization), Post-Ch+4 (four weeks after challenge), Post-Ch+12 (12 weeks after challenge), Post-Ch+20 (20 weeks after challenge), and Post-Ch+48 (48 weeks after challenge).
Results
Participant flow
Ninety-four volunteers were assessed for eligibility. Fifty-four did not meet inclusion criteria (). Forty volunteers met all eligibility criteria, of whom 14 withdrew consent or did not respond when re-contacted. The remaining 26 volunteers, who were all Ad-5 seronegative (neutralizing antibody titer < 1/500Citation18), were enrolled in the immunization group (n = 20) or as infectivity controls (n = 6). Their demographics are shown in . All volunteers in the immunization group completed the single scheduled vaccination, and all were included in the safety analysis. Eighteen immunized volunteers and the six non-immunized infectivity controls underwent CHMI; two immunized volunteers were not challenged, one due to family reasons, and one at the discretion of the study team due to poor compliance during post-immunization safety follow-up visits. One volunteer who was immunized and challenged was withdrawn six months after the final immunization due to deployment; he then returned to the US and participated in annual follow-ups. One infectivity control died during the second year of follow-up for reasons unrelated to participation in the clinical trial. Therefore, safety and tolerability were determined using 20 immunized research subjects, efficacy using 18 immunized and challenged research subjects, and immunogenicity using 18 immunized and challenged research subjects with some not included at every time point as indicated.
Figure 3. Flow diagram of immunized and control volunteers. Thirty-seven volunteers met all eligibility criteria of whom 11 withdrew consent, and 26 volunteers were allocated to the immunization group (n = 20) and infectivity controls (n = 6) respectively. Two immunized volunteers were not challenged due to family reasons and poor compliance, respectively. Approximately two years after challenge, an infectivity control died of causes unrelated to the vaccine.
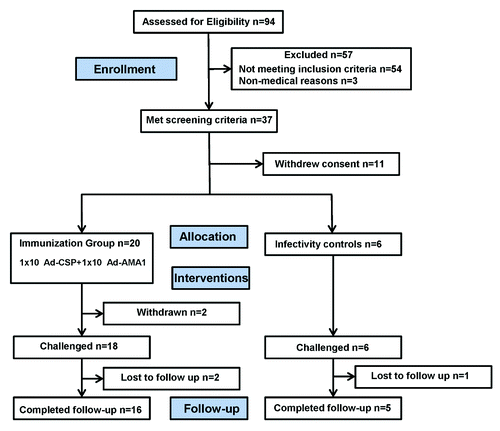
Table 1. Study subjects demographics
Local and systemic adverse events
The vaccine was safe and well tolerated. During the 14 d following immunization, 36 AE’s local to the site of injection were recorded as definitely, probably or possibly related to immunization (). Of these, 29 (81%) were mild (Grade 1, no intervention required) and seven (19%) were moderate (Grade 2, intervention required but able to perform daily activities). The most common local side effects were tenderness (44% of total local AE’s) and pain (42%) at the injection site. Less frequent local side effects were warmth (6%), redness (3%) and induration (3%) at the injection site, and pruritus (3%) that occurred (rather than locally at the injection site) on subject’s right lower extremity and on the back 8 h after immunization, lasting 30 min and resolving spontaneously. No swelling was observed. Unlike the previous trial with NMRC-M3V-Ad-PfCA administered alone,Citation5 no volunteers reported axillary adenopathy or effects on arm motion.
Table 2. Number of volunteers experiencing solicited local and systematic adverse signs and symptoms (days 0–14 post immunization)
Twenty-eight solicited systemic AE’s were recorded during the 14 d after immunization (), of which 25 (89%) were mild (Grade 1) and three (12%) were moderate (Grade 2). The most common systemic symptoms were fatigue (21%), headache (14%), malaise (14%), myalgia (11%), fever (7%), and chills (7%); joint pain, cough, dizziness, pharyngitis, nausea, vomiting, and rash were reported each contributing 4% of AE’s. The following solicited systemic AE’s were not reported: conjunctivitis, eye pain-irritation, nasal congestion, coryza, diarrhea, dysuria, frequent urination or hives.
Laboratory adverse events
Transient neutropenia and/or lymphopenia occurred during the first seven days post immunization in 45% and 30% of research subjects, respectively. All were Grade 1 or 2 except for one Grade 3 neutropenia (ANC: 957 cells/microliter) on day 2 post immunization that improved to Grade 1 the following day (). These drops in neutrophils and lymphocytes were expected based on prior clinical experience with the vaccine, likely representing transient margination (adhesion of leukocytes to blood vessel walls) in response to immunization with adenovectors, and exhibited kinetics similar to those reported earlier, with counts returning to baseline within 7 d in most cases.Citation5,Citation17 Other laboratory abnormalities recorded after immunization did not appear related to the vaccine. Protein was detected in the urine of six research subjects on days 2 or 14, ranging from trace to 100 mg/dl, but in five of these cases either proteinuria or other abnormalities were recorded in the same individuals prior to receiving the vaccine, and therefore these findings were deemed unlikely related to immunization. One research subject had normal urinalyses prior to immunization and was recorded with two-plus protein (30 mg/dl) on day 14 but had normal creatinine and normal protein to creatinine ratio and increased specific gravity suggesting proteinuria may have been related to mild dehydration. Another volunteer (male) was noted to have Grade 3 hematuria 2 d post immunization which had resolved by day 14 post immunization; this volunteer was noted to have Grade 2 hematuria on the day of immunization and thus this finding, like the proteinurias, was deemed unlikely related to immunization. There were also mild elevations of AST, ALT and creatinine, and mild decreases in platelets and hemoglobin, on various study days in several research subjects, all of which resolved spontaneously and were classified as unlikely related to immunization.
Table 3. Laboratory Adverse Events Occurring 1 to 28 Days Post Immunization
Unsolicited adverse events
Volunteers were questioned in open-ended fashion (“do you have any other symptoms?”) for unsolicited symptoms for 28 d after vaccination () and Grade 1 adverse events were documented for three volunteers. One volunteer experienced subjective fever in the evening after immunization (classified as definitely related to vaccination); another volunteer had subjective fever and sweats that began on the day after immunization and resolved by the following morning (classified as probably related to vaccination); and a third volunteer had pruritus of his back and left leg occurring the evening of immunization (classified as possibly related to immunization, same event as mentioned above).
Table 4. Number of volunteers experiencing unsolicited adverse events during 28 d following each immunization
Efficacy
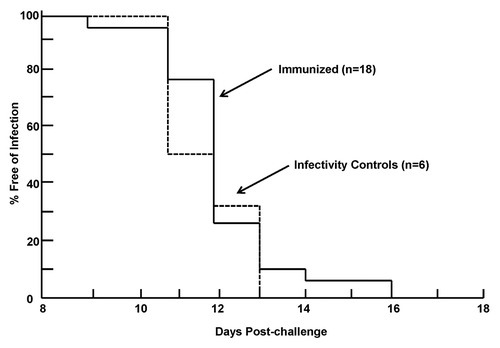
All six infectivity controls and 18 immunized research subjects developed parasitemia, indicating an absence of sterile protection (). The geometric mean time to parasitemia in the infectivity controls (11.8 d, range day 11–13, standard deviation 1.0 d) was similar to that previously reportedCitation16,Citation17 and to that of immunized research subjects (12.1 d, range day 9–16, standard deviation 1.4 d), indicating absence of partial protection (p = 0.489). When research subjects were examined individually, one (v194) who became positive on day 16 exceeded the geometric mean plus two standard deviations of the infectivity controls (11.8 + [2 × 1.0] = 13.8 d), suggesting a significant delay in time to parasitemia for this individual.Citation19
Figure 4. Development of parasitemia in the immunized and infectivity volunteers. Parasitemia-free survival curves (Kaplan-Meier) for immunized volunteers and infectivity controls based on microscopic examination of peripheral blood smears.
Immunogenicity
Antibody responses of all 18 immunized and challenged research subjects were tested using sera collected prior to and after Ad immunization and four, 12 and 20 weeks after challenge. Individual fresh ELISpot assays and batched frozen ICS/flow cytometry assays were tested on PBMC collected 22–23 d after Ad administration, and four, 12, 20, and 48 weeks after challenge. The numbers of research subjects for whom cells were available at these five time points were 18, 16, 15, 15, and 14 for ELISpot assay, and 16, 17, 15, 15, and 13 for flow cytometry, respectively.
Antibody responses
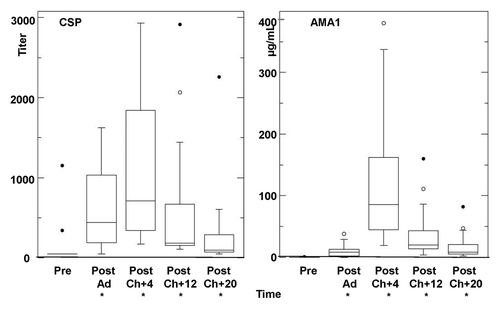
Antibody responses by ELISA titers to CSP significantly rose after Ad immunization in all 18 immunized and challenged volunteers () but were modest (geometric mean titer 381, range < 50–1626) and were similar to those reported in the earlier trial of the NMRC-M3V-Ad-PfCA vaccine.Citation5 At four weeks following challenge, anti-CSP responses rose further in 11/18 volunteers and declined in 7/18 volunteers (geometric mean titer 686, range 172–2933) and overall remained significantly higher than pre-immunization. At 12 weeks and 20 weeks after challenge, anti-CSP titers dropped below those after Ad immunization (12 weeks: geometric mean titer 311, range 109–2914; 20 weeks: geometric mean titer 151, range 62–2259) but remained significantly higher than pre-immunization (). Anti-AMA1 responses also rose in all 18 volunteers after Ad immunization (4.95 µg/mL, range 0.2–38 µg/mL), rose further after challenge (geometric mean 84 µg/ml, range 19–380 µg/ml), and at 12 and 20 weeks after challenge remained elevated above levels after Ad immunization (12 weeks: geometric mean 25 µg/mL, range 4–160 µg/mL; 20 weeks: geometric mean 11 µg/mL, range 4–82 µg/mL), and at all time points were significantly higher than pre-immunization levels ().
Figure 5. Antibody responses by ELISA to CSP and AMA1. Group geomean CSP and AMA1 ELISA activities for the 18 recipients were significantly higher than baseline (*) CSP: Post-Ad (CSP p = < 0.0001), Post-Ch+4 CSP (p = < 0.0001), Post-Ch+12 (CSP p = < 0.0001) and Post-Ch+20 (CSP p = 0.0003); AMA1: Post-Ad (p = < 0.0001), Post-Ch+4 CSP (p = < 0.0001), Post-Ch+12 (p = < 0.0001) and Post-Ch+20 (p = < 0.0001). For explanation of box plots (including outliers) see statistics section at the end of Methods below.
IFA titers to sporozoites () rose (defined as a 4-fold change) in 15/18 immunized and challenged volunteers after Ad immunization and the group geometric mean titer (283, range < 10–2560) was significantly higher than the pre-immunization titer and similar to that in the earlier trial of the NMRC-M3V-Ad-PfCA Vaccine.Citation5 Following challenge, anti-sporozoite responses rose in 4/18 volunteers, declined in 4/18 volunteers and remained unchanged in 10/18 volunteers (geometric mean 285, range 160–640). At 12 weeks and 20 weeks after challenge, anti-sporozoite titers dropped below those after Ad immunization. Titers at each time point after challenge were significantly higher than pre-immunization titers (12 weeks: geometric mean titer 38, range < 10–320; 20 weeks: geometric mean titer 38, range < 10–320).
Figure 6. Antibody responses by IFA to P. falciparum sporozoites and asexual blood stages. Group geomean IFA activities were significantly higher than baseline (*): Sporozoites: Post-Ad (p = 0. < 0001), Post-Ch+4 (p = 0. < 0001), Post-Ch+12 (p = 0.0015), Post-Ch+20 (p = 0.0002); red blood stages: Post-Ad (p = < 0.0001), Post-Ch+4 (p = < 0.0001), and Post-Ch+12 (p = < 0.0001) and Post-Ch+20 (p = < 0.0001) (mixed linear model). For explanation of box plots (including outliers) see statistics section at the end of Methods below.
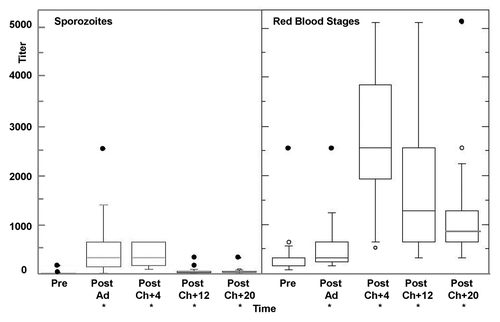
IFA titers to blood stages parasites rose significantly in only 1/18 immunized and challenged volunteers after Ad immunization (defined as a 4-fold rise) but as a group () were significantly higher than pre-immunization (geometric mean 403, range 160–2560) and were similar to the earlier trial.Citation5 After challenge, anti-blood stage parasite responses rose in 14/18 volunteers (geometric mean 2463, range 640–5120), and at 12 weeks and 20 weeks after challenge, titers dropped below those after Ad immunization but activities at each time point after challenge remained significantly higher than pre-immunization titers (12 weeks: geometric mean titer 1159, range 320–2560; 20 weeks: geometric mean titer 1076, range 320–5120).
Ex vivo T cell IFN-γ activities by ELISpot for CSP
ELISpot CSP IFN-γ responses () rose significantly at 22/23 d after immunization (geometric mean response summed across peptide pools for each volunteer, 273 sfc/m, range 38–2550 sfc/m), declined at four weeks after challenge (136 sfc/m, range 24–1327 sfc/m) and continued to decline at 12 weeks (geometric mean 69 sfc/m, range 6–398 sfc/m), and then remained level at 20 weeks (71 sfc/m, range 4–229 sfc/m) and 48 weeks (79 sfc/m, range 18–272 sfc/m) after challenge, with activities of all these time points significantly higher than pre-immunization (). Responses differed among volunteers, and using the criteria to define individual positive responses (see Methods), 13 of the 18 volunteers (72%) were positive after immunization and prior to challenge, with this number declining at four weeks after challenge to 7/16 (44%), and remaining approximately level at 12 weeks (2/15, 13%), 20 weeks (4/15, 27%) and 48 weeks (4/14, 29%) after challenge.
Figure 7. Ex vivo T cell IFN-γ activities by ELISpot Assay for CSP and AMA1. Group geomean IFA activities were significantly higher than baseline (*): CSP: Post-Ad (p = < 0.0001), Post-Ch+4 (p = < 0.0001), Post-Ch+12 (p = 0.0083), Post-Ch+20 (p = 0.0009) Post Ch+48 (p = < 0.0001; AMA1 Post-Ad (p = < 0.0001), Post-Ch+4 (p = < 0.0001), Post-Ch+12 (p = 0.0056), Post-Ch+20 (p = 0.0019), Post Ch+48 (p = < 0.0013). For explanation of box plots (including outliers) see statistics section at the end of Methods below.
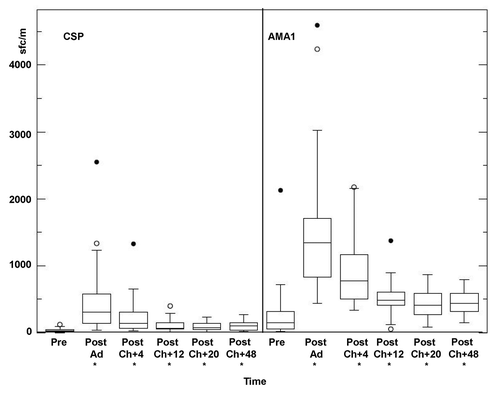
Ex vivo T cell IFN-γ activities by ELISpot for AMA1
As with CSP, ELISpot AMA1 IFN-γ responses () also significantly rose at 22/23 d after immunization (geometric mean 1303 sfc/m, range 435–4594 sfc/m), and declined at four weeks after challenge (803 sfc/m, range 415–2175 sfc/m) and continued to decline at 12 weeks after challenge (447 sfc/m, range 52–1373 sfc/m) and then remained approximately level at 20 weeks (382 sfc/m, range 81–866 sfc/m) and 48 weeks (407 sfc/m, range 150–789 sfc/m) after challenge, with activities at all these time points significantly higher than pre-immunization (). In contrast to CSP, responses to AMA1 were positive in all 18 volunteers (100%) after immunization, and remained positive in 16/16 (100%) volunteers at 4 weeks, 14/15 volunteers (93%) at 12 weeks, 13/15 (87%) volunteers at 20 weeks and 11/14 volunteers (78%) at 48 weeks after challenge.
Total IFN-γ T cell responses by flow cytometry/intracellular cytokine staining (ICS) for CSP
Total CD4+ and CD8+ T cell IFN-γ responses were measured and these included IFN-γ+IL2+TNF+, IFN-γ+IL2-TNF+, IFN-γ+IL2+TNF-, and IFN-γ+IL2-TNF- containing cells ().
Figure 8. CD4+ and CD8+ T cell IFN-γ activities by flow cytometry for CSP and AMA1. The box plots represent IFN-γ-producing CD4+ or CD8+ T cell frequencies as percentage of gated CD4+ or CD8+ T cells, measured by flow cytometry assays after stimulation with a single CSP or AMA1 megapool containing all individual peptide pools for each antigen. Arrow denotes v156 who had very high CD8+ T cell activity to AMA1 (2.08%) at Post-Ad. IFN-γ-producing CD4+ T cell activities for CSP were only significantly higher than baseline (*) at Post-Ad (p = 0.0017); CD4+ T cell activities for AMA1 were significantly higher than baseline (*) Post-Ad (p = 0.0002), Post-Ch+4 (p = 0.0017), Post-Ch+20 (p = 0.0054) and Post-Ch+48 (p = 0.0035). IFN-γ-producing CD8+ T cell activities for CSP were only marginally higher than baseline (*) at Post Ad (p = 0.07); CD8+ T cell activities for AMA1 were significantly higher than baseline (*) at Post-Ad (p = 0.0021) and Post-Ch+4 (p = 0.017). For explanation of box plots (including outliers) see statistics section at the end of Methods below.
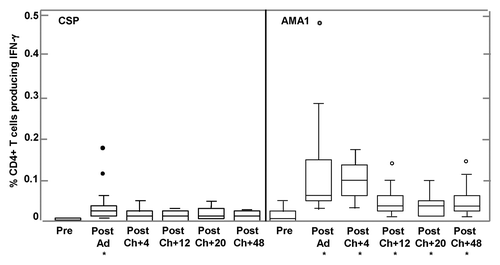
As previously reported for Ad vaccinesCitation16,Citation17,Citation20 both CD4+ and CD8+ T cell IFN-γ responses to CSP were induced (), with geometric mean responses rising significantly after Ad-CA immunization for both CD4 (p = 0.0017) and CD8 (p = 0.0002) T cell responses. However, mean responses remained below the 0.03% cut off for positivity at each time point for both cell populations (see Methods). When individual CD4 T cell IFN- γ responses were examined, applying positivity criteria (see Methods), 4/16 volunteers (25%) were positive after immunization, declining at 4, 12, 20 and 48 weeks after challenge to 1/17 (6%), 0/15 (0%), 2/15 (13%), and 0/13 (0%), respectively. For CD8 T cell IFN- γ responses, numbers positive were 9/16 volunteers (56%) after immunization, declining at 4, 12, 20 and 48 weeks after challenge to 5/17 (29%), 3/15 (20%), 3/15 (20%), and 1/13 (8%). Overall CD8+ T cell responses were higher than CD4+ T cell responses, but the difference was not statistically significant (p = 0.51).
The largest component of the CD8+ T cell IFN-γ responses of the 9 positive volunteers to CSP after Ad immunization (geometric mean 0.093%, range 0.03–0.47%) were single IFN-γ-secreting cells (IFN-γ+IL2-TNF-α-; geometric mean 0.086%, range 0.03–0.41%) that represented 92% (range 87–99%) of total CD8+ IFN-γ responses.
Total IFN-γ T cell responses by flow cytometry / intracellular cytokine staining (ICS) for AMA1
As with CSP above, both CD4+ and CD8+ T cell IFN-γ responses to AMA1 were induced (). In contrast to CSP, group geomean CD4+ and CD8+ T cell responses were all positive at each post-immunization time point (above the 0.03% cut off). After Ad immunization, CD4+ T cell responses significantly rose (p = 0.0002) and were approximately five times higher than those to CSP (geometric mean 0.090%, range 0.027–0.478). CD4+ responses remained level at four weeks after challenge (0.091%, range 0.036–0.176%) and then declined and remained level at 12 weeks (0.038%, range 0.01–0.143%), 20 weeks (0.032%, range 0.009–0.098%) and 48 weeks (0.036%, range 0.009–0.14%). CD4+ T cell responses at each time point after challenge were significantly higher than pre-immunization activities (). After Ad immunization 15/16 (94%) and four weeks after challenge 17/17 (100%) volunteers were positive, declining at 12 weeks 9/15 (60%), 20 weeks 9/15 (60%) volunteers, and 48 weeks 8/13 (62%) after challenge.
CD8+ T cell responses to AMA1 also significantly rose (p = 0.0021) after immunization and were higher than CD4+ responses (0.216%, range 0.04–2.08%). CD8+ responses declined at four weeks after challenge (0.126%, range 0.019–0.59%) but remained significantly higher than pre-immunization activities (p = 0.017), then declined further and remained approximately level at 12 weeks (0.065%, range 0.008–0.51%), 20 weeks (0.038%, range 0.0001–0.335%), and 48 weeks (0.065%, range 0.026–0.451%) after challenge but were not significantly higher than pre-immunization activities ().
The numbers of volunteers with positive CD8+ positive responses to AMA1 were more persistent than for CD4+ responses; after Ad-CA immunization 16/16 (100%) volunteers, at four weeks after challenge 16/17 (94%) volunteers and 12 weeks after challenge 14/15 (93%) volunteers were positive. This declined slightly at 20 weeks to 13/15 (87%) volunteers and at 48 weeks to 11/13 (85%) volunteers. Overall CD8+ T cell responses to AMA1 were significantly higher than CD4+ T cell responses (p = 0.001).
The largest component of the CD8+ T cell IFN-γ responses to AMA1 of the 16 positive volunteers after Ad immunization (geometric mean 0.216%, range 0.04–2.08) were single IFN-γ -secreting cells (IFN-γ+ IL2- TNF-α-; geometric mean 0.183%, range 0.03–1.76) that represented 84% (range 52–100%) of total CD8 IFN-γ.
Pre-immunization activity
We observed four volunteers with measurable pre-immunization activities in more than one assay: v118 (ELISA AMA1 0.59 µg/mL; CD8+ AMA1 0.0326%); v119 (ELISA CSP 341; ELISpot CSP 121 sfc/m); v127 (ELISpot AMA1 2127 sfc/m; CD8+ AMA1 0.0776%); v173 (ELISA AMA1 0.57 µg/mL; IFA red blood stages 2560; CD4+ AMA1 0.0437%; CD8+ AMA1 0.0511%). In addition, v156 had a high pre-immunization ELISA CSP titer (1152). In the previous trial, we also observed a high pre-immunization ELISA and IFA sporozoite activity in one volunteer with CSP (ELISA titer 838; IFA titer 5120). It is unclear whether these activities represent cross-reactive antigensCitation21 including superantigens,Citation22 or whether they reflect undisclosed prior exposure to malaria or participation in malaria drug or vaccine trials. All volunteers were carefully screened and denied prior exposure to malaria or participation in malaria drug or vaccine trials. These values were all included in the analyses except for the pre-immunization ELISpot assay result of 2127 sfc/m for AMA1 in v127.
Discussion
Interpretation
Safety
The first primary objective of this study was to assess, in healthy, malaria-naïve, Ad5 seronegative adults, the safety and tolerability of the NMRC-M3V-Ad-PfCA Vaccine, a mixture of two recombinant adenovirus constructs expressing CSP and AMA1, respectively. The same vaccine has previously been tested in a dose escalation trial in two groups of six volunteers, with the dose used here, 2 × 1010 pu, identical to the lower dose used in the earlier trial.Citation5 The lower dose was selected from the earlier study for further testing because it was better tolerated and had statistically significantly stronger ELISpot responses than the 5 fold higher dose (1 × 1011 pu).
Adverse event profiles were similar between the prior and current trials. In the current study, there were 36 local AE’s recorded in 20 research subjects (1.8/injection), compared with 1.3/injection in the six research subjects receiving the same dose in the earlier trial, and there were no Grade 3 AE’s recorded in either. Pain and tenderness at the injection site were the most frequent AE’s in both trials. Lymphadenopathy and limited arm motion, observed in the first trial in a few research subjects, did not occur in the current trial, but that could be a chance effect. One episode of pruritus that occurred in the current trial did not occur in the prior trial. There were 28 systemic AE’s in the current trial (1.4/injection) compared with 1.5/injection in the earlier trial. Headache was the most frequent systemic AE in both studies, followed by myalgia, nausea/vomiting and malaise, and frequencies were similar. Two Grade 1 episodes of objective fever and two grade 1 episodes of subjective fever occurred in this trial in 20 research subjects, compared with one episode of subjective fever in six research subjects in the earlier trial. Fatigue was noted only in the earlier trial. Overall there were 64 local and systemic AE’s in the current trial (3.2/injection) compared with 3.0/injection in the earlier trial.
Neutropenia was the only apparent vaccine-related laboratory abnormality. In the earlier trial, there were one Grade 1 and two Grade 2 decreases among six research subjects receiving the 2x1010 pu dose, whereas in the current trial, there were five Grade 1, three Grade 2 and one Grade 3 neutropenias among 20 research subjects—thus rates were similar. Neutropenia was transient and appeared to entail no clinical risk. It likely represented margination of neutrophils in response to the injection of the adenoviral particles. Overall, we concluded that the vaccine is safe and well tolerated.
Efficacy
A second primary objective was to determine whether the vaccine provided protection against experimental P. falciparum sporozoite challenge (CHMI), which had not been assessed in the earlier trial.Citation5 No volunteer was protected, although one of 18 showed a delay to patency according to an accepted measure.Citation19 When the CSP-expressing component of the vaccine had been tested by itself in a second earlier clinical trial, when given as two separate doses eight weeks apart, there was likewise no sterile protection, although two of 11 immunized volunteers showed significant delays to patency.Citation17 In both these CHMI trials of Ad given by itself (CSP alone and the current trial with both CSP and AMA1), the antibody and T cell responses of the volunteers showing a delay to patency were similar to the geometric means of each assay for the comparable group as a whole, suggesting no association between delay and the measured immunological outcomes.
In contrast to these Ad-alone studies, the first clinical assessment of a DNA prime/Ad boost regimen using CSP and AMA1Citation16 induced sterile immunity in four of 15 research subjects (27% protection overall, 40% protection in the ten Ad5 seronegative research subjects) that was statistically significantly associated with IFN-γ ELISpot and CD8+ T cell responses to one of the vaccine antigens, AMA1, and showed a trend for CSP. In contrast, antibody responses showed no association with protection. Comparing those results to the current trial, where there was no protection and no indication of immunological correlates in the one research subject with delayed onset of parasitemia, we conclude that DNA priming is critical to the induction of protective responses by the Ad vaccine. Although not specifically assessed using CSP and AMA1-encoding plasmids, we do not believe that DNA alone could protect, given that a mixture of DNA plasmids encoding CSP and four other antigens failed to protect in an earlier study.Citation15
Immunogenicity
A secondary objective was to assess the immunogenicity of the NMRC-M3V-Ad-PfCA Vaccine. As in prior studies,Citation5,Citation17 antibody responses measured by ELISA and IFA were low. Malaria challenge boosted antibody responses to AMA1, likely because it is expressed in the blood stages as well as in the sporozoite and liver stages, but not to CSP, which is not expressed by blood stages. Antibody responses to CSP, AMA1, sporozoites and blood stage parasites were all short-lived and declined to near pre-immunization levels by 20 weeks after challenge (24 weeks after immunization).
In contrast, IFN-γ ELISpot responses summed over peptide pools were robust, also seen in prior studies. They were greater to AMA1 than to CSP as previously reported,Citation5 at least partially reflecting the larger size of AMA1 (622 amino acids vs. 333 for CSP). While ELISpot responses to CSP and AMA1 were not boosted by challenge, they were relatively persistent, especially for AMA1, also a finding from earlier studies. An interesting observation was that CMI appeared to be stronger overall in this non-protective trial, compared with the protective DNA/Ad trial. For example, geometric mean ELISpot IFN-γ responses to CSP and AMA1 recorded prior to challenge in this trial were 273 and 1303 sfc/m respectively, while in the DNA/Ad trial, they were 86 and 348 sfc/m respectively. We considered that this might be partially explained by the inclusion of five research subjects with neutralizing antibody titers to Ad5 greater than 500 in the DNA/Ad trial, but even if those five are excluded from the calculation of the group mean, the magnitudes of the IFN-γ responses are about the same – 118 and 382 sfc/m for CSP and AMA1, respectively. Moreover, Ad5 seropositivity was not statistically associated with lower IFN-γ responses in the DNA/Ad trial, although a trend was observed for both CSP and AMA1. Five of 18 and 11/18 volunteers in the current study developed higher ELISpot responses to CSP or AMA1, respectively, than the protected volunteers in the DNA/Ad trial, and 6/15 and 4/15 volunteers developed higher CD8+ T cell responses to CSP and AMA1, respectively, yet were not protected. Moreover, despite their lack of greater magnitude, the IFN-γ responses in DNA/Ad were statistically significantly associated with sterile protection (AMA1) or showed a strong trend (CSP). This indicates that there must be attributes of the effector cell populations that are not being measured by standard IFN-γ ELISpot assays or flow cytometry assays for CD4+ T cells, CD8+ T cells or the cytokine profiles assessed in the two trials.
A potential insight into the differing qualities of the responses in the two trials is that in three of the four protected research subjects in the DNA/Ad trial, responses to single peptide pools appeared to predominate, indicating that perhaps DNA priming focused the immune response following Ad boost on particular peptides or epitopes, whereas in the current trial, responses were very broadly distributed among the peptide pools (data not shown). These broad responses may include counter-protective T cell populations, such as regulatory T cells, or dysfunctional transgene-specific CD8+ T cells that are impaired in cytokine production or effector functions.Citation23 Alternatively, differences between this trial and the DNA /Ad trial may reflect differences in memory cell phenotypes. DNA/Ad regimens for preventing HIV infection have been reported to induce terminally differentiated effector memory CD8+ T cells that are cytotoxic,Citation24 possibly by induction of activated CD4+ T cells.Citation25 Studies in mice with rodent malaria have suggested that persistent effector memory T cells maintained in the liver, blood and spleen protected against sporozoite challenge.Citation26,Citation27 We are currently determining central and effector memory responses in both trials. However, since we can only measure peripheral responses that may not reflect activities at the site of infection in the liver, (although it has been suggested that in mice antigen-specific T cells circulate between the liver and peripheryCitation26), it may prove difficult to discern a correlate of protection in our human studies.
As in earlier trials, we searched for multifunctional T cells. IFN-γ responses to CSP and AMA1, however, were largely derived from monosecreting CD4+ or CD8+ T cells (IFN-γ+ IL2- TNF-), a similar finding to the DNA/Ad trialCitation16 and the two prior Ad-alone trials.Citation5,Citation17 Multifunctional T cell responses, T cells in which two or more cytokines are secreted, have been proposed as mediators of protection in some diseases.Citation28 Recent evidence from mouse malaria vaccine studies suggests that multifunctionality correlated with protection in miceCitation10; however, the prime-boost vaccine regimen used in that trial (modified vaccinia virus Ankara and adenovirus vectors) differs from our trial, and it is possible that a multifunctional response is not a requirement for protection against malaria in humans.
Generalizability
This trial adds to the compendium of safety and tolerability data on adenovirus vectors in humans. It also demonstrates that the protective immunity induced by the DNA/Ad prime-boost regimen is dependent on priming with the DNA components, at least for the CSP and AMA1 antigen combination.
Limitations
Our findings may not apply to other malaria antigens, to Ad5 seropositive populations, or to other adenovectors currently under development as malaria vaccines.
Overall Evidence
Human adenovirus serotype 5 is a potent platform for inducing broad CD4+ and CD8+ T cell responses to malaria antigens in humans. However, these responses may not necessarily be protective, and may require DNA priming to focus responses on protective epitopes.
Methods
Objectives
The primary objectives of this study were to assess the safety, tolerability and efficacy of the recombinant adenovirus vaccine expressing CSP and AMA1 in healthy malaria-naïve adults. Secondary objectives were to assess the immunogenicity of humoral responses measured by enzyme linked immunosorbent assay (ELISA) and immunofluorescence assay (IFA), and cellular responses measured by enzyme linked immunospot assay (ELISpot) and flow cytometry / intracellular cytokine staining (ICS).
Ethics
The protocol for this clinical trial was approved by the National Naval Medical Center (NNMC, now the Walter Reed National Military Medical Center), Naval Medical Research Center (NMRC) and Walter Reed Army Institute of Research Institutional Review Boards, in compliance with all applicable federal regulations governing the protection of human subjects. All study subjects gave written informed consent. This study was conducted according to all Federal Regulations regarding the protection of human participants in research including The Nuremberg Code, The Belmont Report, 32 CFR 219 (The Common Rule) and all pertinent standards for the responsible conduct of research set by the Department of Defense, the Department of the Navy, the Department of the Army, the Navy Bureau of Medicine and Surgery, the NNMC, the NMRC and the US Army Medical Research and Materiel Command. NMRC holds a Department of Defense/Department of the Navy Federal Wide Assurance for human subject protections, and a Federal Wide Assurance from the Office for Human Research Protections for cooperation with the Department of Health and Human Services (FWA 0152). All NMRC personnel were certified as having completed mandatory human research ethics education curricula and training under the direction of the NMRC Office of Research Administration and Human Subjects Protections Program. The trial was performed under United States Food and Drug Investigational New Drug Application BB-IND-13003.
Study design
This study was an open-label, Phase 1 trial, with CHMI by mosquito bite to assess protection. Volunteers received a single dose of the NMRC-M3V-D-PfCA vaccine and were monitored for adverse signs and symptoms, laboratory abnormalities, and humoral and cellular immune responses (). Eighteen immunized volunteers, plus six unimmunized infectivity controls, were challenged with P. falciparum (strain 3D7) via five infectious mosquito bites at day 28. Blood was collected and Giemsa-stained malaria smears were read by certified microscopists on days 6 through 21 post-challenge, then every other day through day 28 in volunteers remaining smear negative. Positive volunteers were treated with 1500 mg chloroquine base over three days and followed daily until three consecutive negative smears had been documented.
Study subjects and eligibility
Enrollment and screening began in July 2010 and were completed in October 2010. All potential study subjects provided written, informed consent before screening and enrollment and had to pass an assessment of understanding as previously described.Citation5 Enrollment was limited to healthy malaria-naïve civilian and military adult men and women, age 18–50 y who passed screening by medical history, physical examination, electrocardiogram and laboratory testing (criteria in the supplement). Cardiac risk screening was conducted to identify and exclude individuals at moderate to high risk of developing symptomatic coronary artery disease during the next 5 y, based on gender, blood pressure, body mass index, smoking history and presence or absence of diabetes.Citation29 This was done to avoid the physiologic stress of malaria infection in individuals with occult coronary artery disease. Recruitment was according to a protocol approved by the National Naval Medical Center (NNMC) Institutional Review Board and the Army Surgeon General’s Human Subjects Research Review Board. The study was conducted at the NMRC Clinical Trials Center in Bethesda, Maryland. Enrollment was restricted to Ad5 seronegative or low titer (NAb neutralizing antibody titer < 500Citation18) volunteers as other studies have suggested pre-existing NAb may reduce T cell responsesCitation30 and this had been observed, to a limited degree, in the protective DNA/Ad Vaccine trial.Citation16
Vaccine
The vaccine, two non-replicating adenovirus-derived constructs expressing P. falciparum strain 3D7 CSP and AMA1, respectively, is called NMRC-M3V-Ad-PfCA (NMRC - Multi-antigen Multi-stage, Malaria Vaccine - Adenovectored - P. falciparum CSP and AMA1 antigens). It has been previously describedCitation5 and is shown in . Briefly, the CSP gene from 3D7 strain was modified by deletion of 16 of the central repeat sequences (64 amino acids) and by adding a 23 amino acid segment from the transcriptional terminator of bovine growth hormone at the C terminus (appears to increase expression when the glycosylphosphatidylinositol anchor is still present, as it is in this construct), while the AMA1 gene from 3D7 was full length, including the transmembrane and cytoplasmic domains in addition to the ectodomain. The CSP and AMA1 3D7 genes were codon-optimized for expression in mammalian cells and were inserted into the E1 region of the adenovirus vector under the transcriptional control of a modified human cytomegalovirus promoter (CMV-IE). The serotype 5 Ad vector was derived from GV11D (GenVec, Inc.) and was missing the E1 and E4 regions required for replication as well as part of the E3 region. The two constructs were vialed separately and were mixed prior to intramuscular administration by needle as a single 1 mL deltoid injection at 2 × 1010 particle units (pu) per dose (1 × 1010 pu each construct). Immunization was initiated in July 2010 using 20 volunteers.
Safety and tolerability
After each immunization, subjects were monitored for at least 30 min before being discharged from the clinic. To evaluate safety, tolerability and reactogenicity, solicited local and systemic adverse events (AEs) were recorded during a 14 d follow-up period while unsolicited events were collected over a 28 d follow-up period. Solicited local AE’s were: tenderness, pain, warmth, redness, induration, pruritus and swelling at injection site; axillary adenopathy; and limited range of arm motion. Solicited systemic AE’s were: fatigue, headache, malaise, myalgia, objective fever, chills, joint pain, cough, dizziness, pharyngitis, nausea, rash, vomiting, diarrhea, hives, coryza, nasal congestion, eye pain/irritation, conjunctivitis, dysuria and frequent urination. Serious AEs were collected throughout the duration of the study for each group. Safety assessments were recorded by direct observation and questioning (daily from day 0, 1, 2, 3, 5, 7, 14, and 28 (days 0 to 7, for self-recording of oral temperature and symptoms). Safety laboratory assays, including complete blood count and a chemistry panel, were performed on samples collected at the in-person visits on days 0, 1, 2, 3, 5, 7, 14, and 28 (CBC with differential leukocyte count) and on days 0, 2, 7, 14, and 28 (biochemistry, consisting of creatinine, AST and ALT). In addition, urinalysis was performed on days 0 and 14. All clinical laboratory tests were performed at the NNMC clinical laboratory. Only those laboratory abnormalities that were gradable by protocol or FDA/CBER guidelines are reported here. The in-person follow up period was approximately one year. Local, systemic, and laboratory AEs were graded using severity scales previously described.Citation5
Sample size
The sample size was powered to detect a two day mean delay in patency in the immunized group compared with the infectivity controls (80% power, α = 0.05, one-sided).Citation31
Immunological endpoints
Antibody and T cell responses were measured at 7 d before immunization (Pre), 22/23 d after immunization (Post-Ad), four weeks after challenge (Post-Ch+4), 12 weeks after challenge (Post-Ch+12), 20 weeks after challenge (Post-Ch+20) and 48 weeks after challenge (Post-Ch+48). Activities of the 18 challenged subjects are reported herein, although not all volunteers were measured at each time point as indicated above (See immunogenicity results).
Antibody responses
Anti-CSP and AMA1 antibodies were measured using enzyme-linked immunosorbent assay (ELISA) using the P. falciparum peptide (NANP)6 or recombinant ectodomain of AMA1 as the capture antigens. Titer was defined as the serum dilution yielding an optical density reading of 1.0, or µg/mL were use comparing test to a standardized immune serum. P. falciparum sporozoite-specific antibodies were assayed by immunofluorescent staining of air-dried P. falciparum sporozoites as described previously.Citation5
T cell responses
Ex vivo enzyme-linked immunospot (ELISpot) IFN-γ activity
This was performed as previously described.Citation5 Stimulants were pools of 15 amino acid (aa) synthetic peptides overlapping by 11 aa (Chiron Technologies) covering full length CSP and AMA1 vaccine antigens. These were combined into 9 CSP pools (Cp1-Cp9) containing 3–12 peptides per pool and 12 AMA1 pools (Ap1-Ap12) containing 10–13 peptides per pool, with pools used individually as stimulants for fresh PBMCs. T cell responses were measured using ELISpot IFN-γ assay of fresh peripheral blood mononuclear cells (PBMC) stimulated for 36 h with the peptide pools. Results of each individual peptide pool were summed and activity reported as spot forming cells/million PBMC (sfc/m). ELISpot assay results were defined as positive when there was a statistically significant difference (p = < 0.05) between the average of the sfc in test and negative control wells (Student’s two tailed t-test), plus at least a doubling of sfc in test compared with control wells, plus a difference of at least 10 sfc between test and control wells. A volunteer was considered a responder when positive against at least one of the CSP or AMA1 pools.
Intracellular cytokine staining (ICS)
For each volunteer, matched pre- and post-vaccination samples were tested simultaneously using frozen PBMCs. ICS was performed under a similar protocol as published previously,Citation5 using all 15mer CSP or AMA1 peptides combined into a single CSP megapool or a single AMA1 megapool. Using 7-color flow cytometry, cells were phenotyped as CD4+ or CD8+ T cells and assessed for functionality by staining for IFN-γ. A positive response was defined as a frequency of cytokine-stained CD4+ or CD8+ cells exceeding the geometric mean + 3 standard deviations of the medium stimulated controls (0.03%).
Efficacy
Protective efficacy of 18 vaccinated volunteers was assessed using a homologous 3D7 strain sporozoite challenge 28 d after the NMRC-M3V-Ad-PfCA vaccine immunization. Six unvaccinated volunteers served as infectivity controls. Each volunteer underwent a standardized challenge by bite of 5 P. falciparum-infected mosquitoes as previously described.Citation19 Beginning 7 d after challenge, volunteers were housed at a hotel in Bethesda, MD. Each volunteer was monitored daily for the onset of signs and symptoms of malaria and by Giemsa-stained thick blood films with positive films confirmed by a second reader. The identity of immunized and non-immunized volunteers was known to the clinical trial staff but not to the microscopists reading the malaria smears. Symptomatic, undiagnosed volunteers had additional smears performed at the discretion of the study doctor, not to exceed one smear every 8–12 h. Volunteers who developed malaria were treated with a standard oral dose of chloroquine: total 1500 mg base (2500 mg chloroquine phosphate) given in divided doses: 600 mg initially followed by 300 mg at 6, 24 and 48 h under direct observation.
Statistics
All antibody (ELISA and IFA) and T cell (ELISpot, flow cytometry) assays were displayed as box plotsCitation32 for the numbers of immunized and challenged research subjects tested (see Immunogenicity). The lower quartile (25th percentile), median and upper quartile (75th percentile) are the base, transecting line and top of each box (defining the interquartile range or “likely range of variation”), and the upper and lower bars represent the maximum and minimum values unless outliers or suspected outliers are present. Suspected outliers and outliers are defined as exceeding 1.5 times and 3.0 times the interquartile range, respectively, above or below the box. When outliers are present, the bar is set at 1.5 times the interquartile range, leaving the outliers or suspected outliers beyond the bar. Suspected outliers and outliers are represented as open and filled dots respectively.
A mixed linear model with compound covariance structure was used to compare geometric mean immunogenicity readings between baseline and post-immunization for antibody, ELISpot and ICS responses, adjusting comparisons between baseline and post-immunization using Dunnett’s method. All antibody responses were log10 transformed. Vaccine efficacy was represented by a Kaplan-Meier plot and evaluated using the log rank test.
| Abbreviation: | ||
| Ad5 | = | human serotype 5 adenovirus vector |
| AE | = | adverse event |
| ALT | = | alanine aminotransferase |
| AMA1 | = | apical membrane antigen-1 |
| ANC | = | absolute neutrophil count |
| AST | = | aspartate aminotransferase |
| CBC | = | complete blood count |
| CBER | = | Center for Biologics Evaluation and Research, US FDA |
| CD4 | = | cluster of differentiation 4 |
| CD8 | = | cluster of differentiation 8 |
| CFR | = | Code of Federal Regulations |
| CHMI | = | controlled human malaria infection |
| CSP | = | circumsporozoite protein |
| DNA | = | deoxyribonucleic acid |
| ELISA | = | enzyme linked immunosorbent assay |
| ELISpot | = | enzyme linked immunospot assay |
| FDA | = | Food and Drug Administration |
| ICS | = | intracellular cytokine staining |
| IFA | = | immunofluorescence assay |
| IFN-γ | = | interferon-gamma |
| IL2 | = | interleukin 2 |
| IMOJEV | = | live-attenuated yellow fever-Japanese encephalitis chimeric vaccine |
| μg | = | microgram |
| mg | = | milligram |
| MHC | = | major histocompatibility complex |
| MIDRP | = | Military Infectious Disease Research Program |
| NAb | = | neutralizing antibody |
| NMRC | = | Naval Medical Research Center |
| NMRC-M3V-Ad-PfCA | = | NMRC - Multi-antigen Multi-stage, Malaria Vaccine - Adenovectored - P. falciparum CSP & AMA1 antigens |
| NMRC-M3V-D/Ad-PfCA | = | NMRC - Multi-antigen Multi-stage, Malaria Vaccine – DNA/Adenovectored - P. falciparum CSP & AMA1 antigens |
| NNMC | = | National Naval Medical Center (now called Walter Reed National Military Medical Center) |
| NVITAL | = | NIAID Vaccine Immune T-cell and Antibody Laboratory |
| PBMC | = | peripheral blood mononuclear cells |
| pu | = | particle unit |
| sfc | = | spot forming cells |
| TNF | = | tumor necrosis factor |
| WRAIR | = | Walter Reed Army Institute of Research |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank all the study volunteers who participated in the trial. We also extend our thanks to Dr. Robert Bailer of NVITAL, Gaithersburg, MD, who performed the adenovirus neutralization assays, and CDR Kyle Peterson of NMRC who was the clinical monitor.
Active duty military personnel at the time they contributed to this work: CT, IC, MK, JM, EM, JEE and TLR. M.S. (Sedegah) and J.K. were US Government employees. The work of these individuals was prepared as part of official government duties. Title 17 U.S.C. §105 provides that ‘Copyright protection under this title is not available for any work of the United States Government.’ Title 17 U.S.C. §101 defines a US Government work as a work prepared by a military service member or employee of the US Government as part of that person’s official duties. The study protocol for the clinical trial presented in this manuscript was approved by the WRAIR and NMRC Institutional Review Boards, in compliance with all applicable Federal Regulations governing protection of human subjects. All volunteers gave written informed consent. The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of the Navy, the Department of the Army, the Department of Defense, or the US Government.
Financial support
This work was supported by (1) USAID “Development of Adenovirus-Vectored Malaria Vaccines” Grant #: GHA-P-00-03-00006-01, Project Number 936–3118; (2) the Congressionally Directed Medical Research Program “Development of Recombinant Adenoviral-based Vaccines against Malaria” Grant #: W81XWH-05-2-0041; and (3) the Military Infectious Disease Research Program (MIDRP) project “Phase 1/2a clinical trials assessing the safety, tolerability, immunogenicity & protective efficacy of Ad5-CA, a two-antigen, adenovirus-vectored Plasmodium falciparum malaria vaccine, in healthy, malaria-naïve adults,” work unit number 62787A 870 F 1432.
Trial Registration:www.clinicaltrials.gov NCT00870987
References
- Overstreet MG, Cockburn IA, Chen YC, Zavala F. Protective CD8 T cells against Plasmodium liver stages: immunobiology of an ‘unnatural’ immune response. Immunol Rev 2008; 225:272 - 83; http://dx.doi.org/10.1111/j.1600-065X.2008.00671.x; PMID: 18837788
- Butler NS, Schmidt NW, Vaughan AM, Aly AS, Kappe SH, Harty JT. Superior antimalarial immunity after vaccination with late liver stage-arresting genetically attenuated parasites. Cell Host Microbe 2011; 9:451 - 62; http://dx.doi.org/10.1016/j.chom.2011.05.008; PMID: 21669394
- Moorthy VS, Ballou WR. Immunological mechanisms underlying protection mediated by RTS,S: a review of the available data. Malar J 2009; 8:312; http://dx.doi.org/10.1186/1475-2875-8-312; PMID: 20042088
- Halstead SB, Thomas SJ. New Japanese encephalitis vaccines: alternatives to production in mouse brain. Expert Rev Vaccines 2011; 10:355 - 64; http://dx.doi.org/10.1586/erv.11.7; PMID: 21434803
- Sedegah M, Tamminga C, McGrath S, House B, Ganeshan H, Lejano J, et al. Adenovirus 5-vectored P. falciparum vaccine expressing CSP and AMA1. Part A: safety and immunogenicity in seronegative adults. PLoS One 2011; 6:e24586; http://dx.doi.org/10.1371/journal.pone.0024586; PMID: 22003383
- Sheehy SH, Duncan CJ, Elias SC, Choudhary P, Biswas S, Halstead FD, Collins KA, Edwards NJ, Douglas AD, Anagnostou NA, et al. ChAd63-MVA-vectored blood-stage malaria vaccines targeting MSP1 and AMA1: assessment of efficacy against mosquito bite challenge in humans. Mol Ther 2012; 20:2355 - 68; http://dx.doi.org/10.1038/mt.2012.223; PMID: 23089736
- Webster DP, Dunachie S, Vuola JM, Berthoud T, Keating S, Laidlaw SM, et al. Enhanced T cell-mediated protection against malaria in human challenges by using the recombinant poxviruses FP9 and modified vaccinia virus Ankara. Proc Natl Acad Sci U S A 2005; 102:4836 - 41; http://dx.doi.org/10.1073/pnas.0406381102; PMID: 15781866
- Wang R, Epstein J, Baraceros FM, Gorak EJ, Charoenvit Y, Carucci DJ, et al. Induction of CD4(+) T cell-dependent CD8(+) type 1 responses in humans by a malaria DNA vaccine. Proc Natl Acad Sci U S A 2001; 98:10817 - 22; http://dx.doi.org/10.1073/pnas.181123498; PMID: 11526203
- Wang R, Doolan DL, Charoenvit Y, Hedstrom RC, Gardner MJ, Hobart P, et al. Simultaneous induction of multiple antigen-specific cytotoxic T lymphocytes in nonhuman primates by immunization with a mixture of four Plasmodium falciparum DNA plasmids. Infect Immun 1998; 66:4193 - 202; PMID: 9712767
- Reyes-Sandoval A, Berthoud T, Alder N, Siani L, Gilbert SC, Nicosia A, et al. Prime-boost immunization with adenoviral and modified vaccinia virus Ankara vectors enhances the durability and polyfunctionality of protective malaria CD8+ T-cell responses. Infect Immun 2010; 78:145 - 53; http://dx.doi.org/10.1128/IAI.00740-09; PMID: 19858306
- Jiang G, Shi M, Conteh S, Richie N, Banania G, Geneshan H, et al. Sterile protection against Plasmodium knowlesi in rhesus monkeys from a malaria vaccine: comparison of heterologous prime boost strategies. PLoS One 2009; 4:e6559; http://dx.doi.org/10.1371/journal.pone.0006559; PMID: 19668343
- Sedegah M, Hedstrom R, Hobart P, Hoffman SL. Protection against malaria by immunization with plasmid DNA encoding circumsporozoite protein. Proc Natl Acad Sci U S A 1994; 91:9866 - 70; http://dx.doi.org/10.1073/pnas.91.21.9866; PMID: 7937907
- Le TP, Coonan KM, Hedstrom RC, Charoenvit Y, Sedegah M, Epstein JE, et al. Safety, tolerability and humoral immune responses after intramuscular administration of a malaria DNA vaccine to healthy adult volunteers. Vaccine 2000; 18:1893 - 901; http://dx.doi.org/10.1016/S0264-410X(99)00407-7; PMID: 10699338
- Epstein JE, Gorak EJ, Charoenvit Y, Wang R, Freydberg N, Osinowo O, et al. Safety, tolerability, and lack of antibody responses after administration of a PfCSP DNA malaria vaccine via needle or needle-free jet injection, and comparison of intramuscular and combination intramuscular/intradermal routes. Hum Gene Ther 2002; 13:1551 - 60; http://dx.doi.org/10.1089/10430340260201644; PMID: 12228010
- Richie TL, Charoenvit Y, Wang R, Epstein JE, Hedstrom RC, Kumar S, et al. Clinical trial in healthy malaria-naïve adults to evaluate the safety, tolerability, immunogenicity and efficacy of MuStDO5, a five-gene, sporozoite/hepatic stage Plasmodium falciparum DNA vaccine combined with escalating dose human GM-CSF DNA. Hum Vaccin Immunother 2012; 8:1564 - 84; http://dx.doi.org/10.4161/hv.22129; PMID: 23151451
- Chuang I, Sedegah M, Cicatelli S, Spring M, Polhemus M, Tamminga C, et al. DNA prime/adenovirus boost malaria vaccine encoding P. falciparum CSP and AMA1 induces sterile protection associated with cell-mediated immunity. PLoS ONE 2013; 8:e55571; PMID: 23457473
- Tamminga C, Sedegah M, Regis D, Chuang I, Epstein JE, Spring M, et al. Adenovirus-5-vectored P. falciparum vaccine expressing CSP and AMA1. Part B: safety, immunogenicity and protective efficacy of the CSP component. PLoS One 2011; 6:e25868; http://dx.doi.org/10.1371/journal.pone.0025868; PMID: 22003411
- Sprangers MC, Lakhai W, Koudstaal W, Verhoeven M, Koel BF, Vogels R, et al. Quantifying adenovirus-neutralizing antibodies by luciferase transgene detection: addressing preexisting immunity to vaccine and gene therapy vectors. J Clin Microbiol 2003; 41:5046 - 52; http://dx.doi.org/10.1128/JCM.41.11.5046-5052.2003; PMID: 14605137
- Chulay JD, Schneider I, Cosgriff TM, Hoffman SL, Ballou WR, Quakyi IA, et al. Malaria transmitted to humans by mosquitoes infected from cultured Plasmodium falciparum. Am J Trop Med Hyg 1986; 35:66 - 8; PMID: 3511753
- Sheehy SH, Duncan CJ, Elias SC, Biswas S, Collins KA, O’Hara GA, et al. Phase Ia clinical evaluation of the safety and immunogenicity of the Plasmodium falciparum blood-stage antigen AMA1 in ChAd63 and MVA vaccine vectors. PLoS One 2012; 7:e31208; http://dx.doi.org/10.1371/journal.pone.0031208; PMID: 22363582
- Good MF, Quakyi IA, Saul A, Berzofsky JA, Carter R, Miller LH. Human T clones reactive to the sexual stages of Plasmodium falciparum malaria. High frequency of gamete-reactive T cells in peripheral blood from nonexposed donors. J Immunol 1987; 138:306 - 11; PMID: 2431058
- Fell AH, Silins SL, Baumgarth N, Good MF. Plasmodium falciparum-specific T cell clones from non-exposed and exposed donors are highly diverse in TCR beta chain V segment usage. Int Immunol 1996; 8:1877 - 87; http://dx.doi.org/10.1093/intimm/8.12.1877; PMID: 8982772
- Holst PJ, Ørskov C, Thomsen AR, Christensen JP. Quality of the transgene-specific CD8+ T cell response induced by adenoviral vector immunization is critically influenced by virus dose and route of vaccination. J Immunol 2010; 184:4431 - 9; http://dx.doi.org/10.4049/jimmunol.0900537; PMID: 20212099
- De Rosa SC, Thomas EP, Bui J, Huang Y, deCamp A, Morgan C, et al, National Institute of Allergy and Infectious Diseases HIV Vaccine Trials Network. HIV-DNA priming alters T cell responses to HIV-adenovirus vaccine even when responses to DNA are undetectable. J Immunol 2011; 187:3391 - 401; http://dx.doi.org/10.4049/jimmunol.1101421; PMID: 21844392
- Park JY, Jin DH, Lee CM, Jang MJ, Lee SY, Shin HS, et al. CD4+ TH1 cells generated by Ii-PADRE DNA at prime phase are important to induce effectors and memory CD8+ T cells. J Immunother 2010; 33:510 - 22; http://dx.doi.org/10.1097/CJI.0b013e3181d75cef; PMID: 20463596
- Reyes-Sandoval A, Wyllie DH, Bauza K, Milicic A, Forbes EK, Rollier CS, et al. CD8+ T effector memory cells protect against liver-stage malaria. J Immunol 2011; 187:1347 - 57; http://dx.doi.org/10.4049/jimmunol.1100302; PMID: 21715686
- Jobe O, Donofrio G, Sun G, Liepinsh D, Schwenk R, Krzych U. Immunization with radiation-attenuated Plasmodium berghei sporozoites induces liver cCD8alpha+DC that activate CD8+T cells against liver-stage malaria. PLoS One 2009; 4:e5075; http://dx.doi.org/10.1371/journal.pone.0005075; PMID: 19347042
- Seder RA, Darrah PA, Roederer M. T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol 2008; 8:247 - 58; http://dx.doi.org/10.1038/nri2274; PMID: 18323851
- Gaziano TA, Young CR, Fitzmaurice G, Atwood S, Gaziano JM. Laboratory-based versus non-laboratory-based method for assessment of cardiovascular disease risk: the NHANES I Follow-up Study cohort. Lancet 2008; 371:923 - 31; http://dx.doi.org/10.1016/S0140-6736(08)60418-3; PMID: 18342687
- Harro CD, Robertson MN, Lally MA, O’Neill LD, Edupuganti S, Goepfert PA, et al, Merck V520-007/012 Study Teams. Safety and immunogenicity of adenovirus-vectored near-consensus HIV type 1 clade B gag vaccines in healthy adults. AIDS Res Hum Retroviruses 2009; 25:103 - 14; http://dx.doi.org/10.1089/aid.2008.0212; PMID: 19108693
- Stoute JA, Slaoui M, Heppner DG, Momin P, Kester KE, Desmons P, et al. A preliminary evaluation of a recombinant circumsporozoite protein vaccine against Plasmodium falciparum malaria. RTS,S Malaria Vaccine Evaluation Group. N Engl J Med 1997; 336:86 - 91; http://dx.doi.org/10.1056/NEJM199701093360202; PMID: 8988885
- McGill R. Variations of Box Plots. Am Stat 1978; 32:12 - 6