
Abstract
Aims: Pro-inflammatory cytokines and chemokines, in particular IL-1β, IFNγ, and CXCL10, contribute to β-cell failure and loss in DM via IL-1R, IFNγR, and TLR4 signaling. IL-1 signaling deficiency reduces diabetes incidence, islet IL-1β secretion, and hyperglycemia in animal models of diabetes. Further, IL-1R antagonism improves normoglycemia and β-cell function in type 2 diabetic patients. Inhibition of lysine deacetylases (KDACi) counteracts β-cell toxicity induced by the combination of IL-1 and IFNγ and reduces diabetes incidence in non-obese diabetic (NOD) mice. We hypothesized that KDACi breaks an autoinflammatory circuit by differentially preventing β-cell expression of the β-cell toxic inflammatory molecules IL-1β and CXCL10 induced by single cytokines.
Results: CXCL10 did not induce transcription of IL-1β mRNA. IL-1β induced β-cell IL-1β mRNA and both IL-1β and IFNγ individually induced Cxcl10 mRNA transcription. Givinostat inhibited IL-1β-induced IL-1β mRNA expression in INS-1 and rat islets and IL-1β processing in INS-1 cells. Givinostat also reduced IFNγ induced Cxcl10 transcription in INS-1 cells but not in rat islets, while IL-1β induced Cxcl10 transcription was unaffected in both.
Materials and Methods: INS-1 cells and rat islets of Langerhans were exposed to IL-1β, IFNγ or CXCL10 in the presence or absence of KDACi (givinostat). Cytokine and chemokine mRNA expressions were quantified by real-time qPCR, and IL-1β processing by western blotting of cell lysates.
Conclusion/Interpretation: Inhibition of β-cell IL-1β expression and processing and Cxcl10 transcription contributes to the β-cell protective actions of KDACi. In vitro β-cell destructive effects of CXCL10 are not mediated via IL-1β transcription. The differential proinflammatory actions of KDACs may be attractive novel drug targets in DM.
Introduction
Diabetes mellitus is a severe chronic disease characterized by hyperglycemia and risk of late diabetic complications including neuropathy, blindness, and accelerated macroangiopathy. Both major forms of diabetes mellitus, type 1 and type 2 diabetes, are caused by absolute or relative failure, respectively, of the insulin-producing pancreatic β-cells to meet insulin demands. Pro-inflammatory cytokines such as IL-1β and IFNγ are believed to contribute to β-cell malfunction and apoptosis in type 1 diabetes mellitus, and there is clinical proof of evidence that IL-1β mediates progressive β-cell failure in type 2 diabetes.Citation1-Citation5 IL-1β and IFNγ in synergy drive the expression of inflammatory genes via NFκB, MAPK and STAT1 signaling pathways.Citation1,Citation2
The IFN-inducible chemokine CXCL10 is produced by β-cells in recent onset type 1 diabetics,Citation6-Citation9 and also in type 2 diabetic patients.Citation10 CXCR3, the receptor for CXCL10, is expressed on Th1 cells infiltrating CXCL10 expressing islets of type 1 diabetics,Citation8 mediating the migration of Th1 cell to the β-cells. Serum levels of CXCL10 in NOD mice correlate with the level of CXCR3 mRNA in pancreatic lymph nodes.Citation11 However, CXCL10 may contribute to the pathogenesis of diabetes in an auto- and paracrine manner by suppressing β-cell function, viability and proliferation via CXCR3 or Toll-like receptor (TLR) 4 signaling pathways shared with IL-1.Citation9,Citation10,Citation12 In support of this notion, DNA vaccination-induced CXCL10 antibody production prevented diabetes in NOD mice without altering islet inflammation.Citation13
Lysine deacetylases (KDACs) are a family of enzymes that, together with lysine acetyltransferases, determine the acetylation balance of histones, transcription factors and many cytosolic, mitochondrial and nuclear proteinsCitation14 and thereby regulate not only gene transcription but also many other cellular processes. We have recently shown that all 11 classical KDACs are expressed and regulated by cytokines in β-cells.Citation15 KDAC inhibitors (KDACi) prevent cytokine-induced β-cell dysfunction and destruction and cytokine production by inflammatory cells, predominantly by reducing the transcriptional activity of NFκB.Citation16,Citation17 Thus, knock down of HDAC1 and 2 significantly inhibits transcription of the directly β cell-toxic inflammatory molecule IL-1β induced by IL-1β and IFNγ in synergy,Citation17 but it is unknown if KDAC inhibition differentially prevents β-cell transcription of IL-1 induced by single cytokines or processing of IL-1, thereby selectively breaking individual autoinflammatory circuits.
Results
Givinostat induces hyperacetylation of histone H4
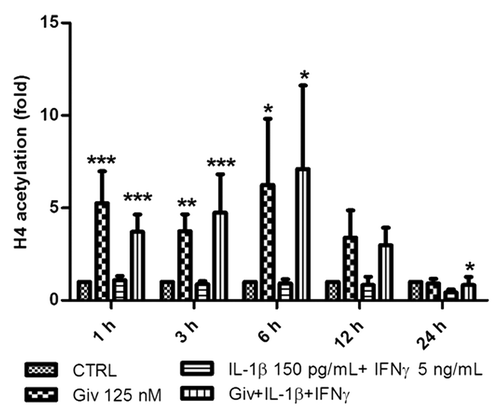
We first investigated the hyperacetylating activity of givinostat on histone H4 in INS-1 cells (). Givinostat in a concentration of 125 nM, shown earlier to reduce IL-1+IFNγ-induced INS-1-cell NO production and apoptosis, robustly hyperacetylated histone H4 after 1–6 h. IL-1+IFNγ did not affect H4 acetylation. After 24 h, IL-1+IFNγ in combination with givinostat slightly but significantly reduced H4 acetylation compared with control.
Figure 1. Givinostat induces hyperacetylation of histone 4. 5 × 105 INS-1 cells were cultured for 1, 3, 6, 12 and 24 h in the presence or absence of IL-1β (150 pg/ml) + IFNγ (5 ng/ml). Givinostat (Giv) was added (125 nM) 1 h prior to cytokine exposure or as a control without cytokine exposure. Cells were lysed and total protein was isolated and subjected to SDS-PAGE and western blot analysis with anti-H4 specific antibody and normalized to β-actin. Data from four independent experiments are presented as fold change compared with controls. Results are shown as means +SEM *p < 0.05, **p < 0.01 and ***p < 0.001 vs. Control (ANOVA with Tukey’s correction for multiple comparisons).
Lysine deacetylase inhibition reduces IL-1β-induced Il1b expression and IL-1β processing
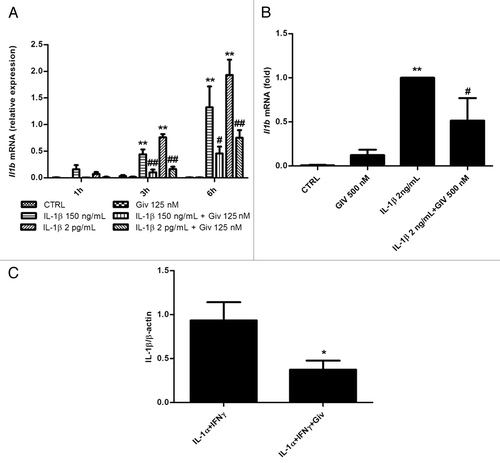
We next exposed INS-1 cells to IL-1β alone for 1, 3 or 6 h, in the absence or presence of givinostat. As seen in , givinostat significantly reduced IL-1β-induced transcription of Il1b in a time-dependent manner, indicating that KDACs regulate IL-1β-induced Il1b transcription. To confirm the biological significance in primary cells of this finding, we also exposed rat islets to IL-1β in the presence or absence of givinostat, and observed a significant reduction in IL-1β induced Il1b mRNA by givinostat (). IL-1α and IL-1β bind and signal through the same receptor although with different potencies.Citation18 Therefore, to examine if givinostat reduced IL-1β processing we exposed INS-1 to IFNγ and IL-1α followed by exposure to ATP to activate the inflammasome for 15 min in the presence or absence of givinostat. Givinostat significantly reduced IL-1β processing compared with control determined as the amount of mature 17.4 kDa IL-1β protein normalized to actin in INS-1 cell lysates ().
Figure 2.Il1b mRNA is induced by IL-1β exposure and abrogated by givinostat in INS-1 cells and rat islets, and IL-1β processing is inhibited by givinostat in INS-1 cells. (A) Il1b mRNA expression. INS-1 cells (2.5 × 106 per well) were preincubated with 125 nM givinostat for 1 h and then exposed to 150 pg/mL or 2 ng/mL IL-1β. Total RNA was isolated and cDNA generated by reverse transcription, followed by real time PCR quantification. Il1b expression was normalized to Hprt1. Data are presented as relative expression. n = 6, means +SEM. Kruskal-Wallis ANOVA on ranks followed by Student-Newman-Keuls multiple comparison test. Significance levels: **p < 0.01 (cytokines vs. control); ##p < 0.01 (cytokines vs. cytokines + givinostat). (B) Il1b mRNA expression. Rat islets (150 per well) were preincubated with 500 nM givinostat for 1 h and then exposed to 2 ng/mL IL-1β for 3 h. Total RNA was isolated and cDNA generated by reverse transcription, followed by real time PCR quantification. Il1b expression was normalized to Hprt1. Data are presented as relative expression. n = 6, means +SEM. Kruskal-Wallis ANOVA on ranks followed by Student-Newman-Keuls multiple comparison test **p < 0.01 (cytokines vs. control), #p < 0.05 (cytokines vs. cytokines + givinostat). (C) IL-1β processing. INS-1 cells (2.5 x 106 per well) were preincubated with or without 125 nM givinostat for 1 h before exposure to 1500 pg/mL IL-1α + 0.1 ng/mL IFNγ for 6 h and exposed to 1 mM ATP for 15minutes. Mature 17.4 kDa IL-1β protein was quantified, normalized to β-actin. n = 4, means + SEM. Student’s two-way t-test. #p < 0.05 (cytokines vs. cytokines + givinostat)
We next exposed INS-1 cells to IFNγ alone or CXCL10 (0.1 ng/mL) alone for 1, 3 or 6 h in the absence or presence of givinostat to examine if these inflammatory molecules individually could induce Il1b transcription and if so, whether KDACs mediate such a regulation. Il1b was undetectable after IFNγ or CXCL10 exposure, indicating that the role of IFNγ and CXCL10 in β-cell decay is unrelated to β-cell Il1b expression (data not shown).
Lysine deacetylase inhibition differentially regulates IL-1β- or IFNγ- induced Cxcl10 mRNA in INS-1 and rat islets
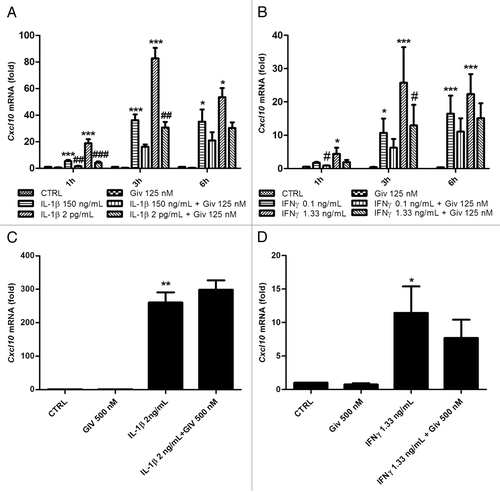
Finally, we wished to investigate the regulation of Cxcl10 by IFNγ or IL-1β alone and the possible effect of givinostat thereupon. In INS-1 cells IL-1β or IFNγ individually induced Cxcl10 transcription in a dose-dependent manner within 6 h (), and givinostat significantly inhibited Cxcl10 induction by IL-1β or IFNγ (). However, when we exposed rat islets to these cytokines, givinostat did not significantly inhibit Cxcl10 induction ().
Figure 3. IFNγ and IL-1β both induce Cxcl10 mRNA in INS-1 cells and rat islets, and KDAC inhibition decreases Cxcl10 mRNA production in INS-1 cells. (A) INS-1 cells (2.5 × 106 per well) were preincubated with 125 nM givinostat or vehicle for 1 h and then exposed to 150 pg/mL or 2 ng/mL IL-1 β for 1–6 h, (B) INS-1 cells (2.5 × 106 /mL per well) were preincubated with 125 nM givinostat or vehicle for 1 h and then exposed to 0.1 ng/mL or 1.33 ng/mL IFNγ for 1–6 h, (C) Rat islets (150 per well) were preincubated with 500 nM givinostat for 1 h and then exposed to 2 ng/mL IL-1β for 3 h (D) Rat islets (150 per well) were preincubated with 500 nM givinostat for 1 h and then exposed to 1.33 ng/mL IFNγ for 3 h. Total RNA was isolated and cDNA generated by reverse transcription, followed by real time PCR quantification using the 2−ΔΔct method. n = 6, means +SEM. ANOVA with Tukey’s post hoc correction (A and B) Kruskal-Wallis ANOVA on ranks followed by Student-Newman-Keuls multiple comparison test (C and D). Significance levels: *p < 0.05; **p < 0.01; ***p < 0.001 (cytokines vs. control); #p < 0.05; ##p < 0.01; ###p < 0.001 (cytokines vs. cytokines + givinostat).
Discussion
Here we show that KDACs regulate cytokine-induced β-cell transcription of Il1b and Cxcl10, and that neither IFNγ nor CXCL10 induce Il1b mRNA transcription. We find that KDACs regulate Il1b transcription in the β-cell line INS-1 and in isolated rat islets of Langerhans and IL-1β processing in INS-1 cells.
While givinostat, as expected, induced acetylation of histone H4 in INS-1 cells, the acetylation levels were unaffected by cytokines. Thus, induction of IL-1β and CXCL10 mRNA by IL-1β and IFNγ exposure as well as the inhibitory effects by givinostat are not likely to be explained by effects on histone acetylation. Since we have recently shown that knockdown of HDAC3 reduces NFκB transcriptional activity by reducing p65 DNA binding in β-cells,Citation17 this could explain the KDACi mediated reduction in cytokine induced IL-1β mRNA expression in β-cells. A similar mechanism of action may explain the inhibitory effect of givinostat on IL-1β processing, since the expression of the NLPR3 inflammasome is also controlled by NFκB.Citation19
Previous studies of the protective roles of KDAC inhibition in vitro and in vivo have been conducted using a combination of IL-1 and IFN.Citation16,Citation17 Here we show that inhibition of KDACs affects the signaling of the cytokines IL-1 and IFNγ individually in the INS-1 cell. Considering the synergistic effects of the combination of these cytokines in the β-cell, this finding illustrates that the protective effects of KDAC inhibition is independent on converging IL-1 and IFNγ signaling.
We show here that CXCL10 mediated β-cell toxicity is not conveyed through induction of Il1b. Other groups have shown that TLR4 signaling is dispensable for CXCL10 actions in β-cells,Citation20 which may then signal via CXCR3. From this evidence, we propose that Il1b expression is dispensable for the β-cell proapoptotic action of CXCL10. The fact that KDAC inhibition had no significant effect on cytokine induced Cxcl10 mRNA production in rat islets as compared with INS-1 cells, while KDAC inhibition is known to protect β-cells against cytokine mediated demise, further strengthens this conclusion.
KDAC inhibition significantly reduced Cxcl10 induced by INS-1 cell exposure to IFNγ or IL-1β, consistent with the role of KDACs in Cxcl10 transcription upon IFNγ exposure in HeLa cells.Citation21 Interestingly, while TLR4 activation leads to induction of expression of inflammatory cytokines through MyD88 recruitment, a MyD88-independent pathway also exists that signals through IRF3, and STAT1 activation induces a subset of TLR4 dependent genes, among which is Cxcl10.Citation22 Indeed, the KDAC inhibitor SAHA inhibits LPS induced STAT1 phosphorylation and Cxcl10 expression in graft-vs.-host disease.Citation23 Acetylated STAT1 acts as a suppressor of NFκB subunit p65 activity,Citation24 enabling a shift from the default synergistic action of NFκB and JAK/STAT signaling in the β-cell to an inhibitory crosstalk between these signaling pathways. This would provide an explanation for the inhibitory effect of KDACi on NFκB activity Similarly, activating phosphorylation of STAT1 as well as nuclear translocation of STAT1 after IFNα stimulation are inhibited by STAT1 acetylation.Citation25
Thus, it is possible that KDAC regulation of Cxcl10 in INS-1 cells can be attributed to non-MyD88 dependent TLR4 signaling, but this remains to be shown.
In conclusion, although Cxcl10 was upregulated by IL-1β or IFNγ in INS-1 cells, neither IFNγ nor CXCL10 induced Il1b production alone. We thus suggest that the deleterious effects of CXCL10 on β-cells are not secondary to IL-1β production. We found that KDACi inhibited cytokine induced β-cell transcription and processing of IL-1β. The protective effects of KDACi on β-cell dysfunction and loss in vivo and in vitro may thus be due to inhibition of a deleterious feed forward loop of cytokine production. Our findings further strengthen the rationale for considering trials of KDAC inhibitors in T1 and T2DM.
Materials and Methods
Cytokines and KDACi
Recombinant rat (rr) interferon (IFN) γ was from R&D Systems (#585-IF), recombinant mouse (rm) IL-1β from BD PharMingen (554577), rmIL-1α was from Abcam (ab9725) rmCXCL10 from R&D Systems (266IP/CF) and the KDAC inhibitor givinostat (ITF2357) a gift from Italfarmaco.
Cells
INS-1 cells were a gift from C. Wollheim, Department of Cell Physiology and Metabolism, University Medical Center, Geneva, Switzerland. Cells were maintained in RPMI 1640 medium with glutamax (GIBCO, 153732), supplemented with 10% (heat-inactivated) fetal calf serum (GIBCO, 26140-079), 100 U/mL penicillin, 100 μg/mL streptomycin (GIBCO, 15140-122) and 50 μM β-mercaptoethanol (Sigma, M7522) and cultured at 37°C in a humidified atmosphere containing 5% CO2. Once weekly cells were passaged and precultured for 2 d before experiments. Experiments were initiated with change of medium and addition of givinostat or vehicle 1 h prior to cytokine exposure.
RT qPCR
INS-1 cells (2.5 × 106 per condition) were seeded in 6-cm Petri dishes 48 h prior to experiments and precultured for 1 h in the presence or absence of givinostat. Primary neonatal rat islets were isolated from 3- to 6-d-old outbred Wistar Rats (Taconic) as previously describedCitation26 and cultured as previously reported.Citation15 Cells and islets were exposed to rmIL-1β, rrIFNγ or rmCXCL10 in concentrations and for the time periods indicated in the figure legends. Total RNA was harvested and extracted using the Nucleo-Spin kit (Macheray-Nagel, 740933.250) according to the manufacturer’s instructions. Quality and quantity of the extracted RNA was assessed using a NanoDrop 1000 (Thermo Scientific, ND-1000). RNA was converted to cDNA with the iScript™ cDNA Synthesis Kit (BioRad, 170-8891). Real-time qPCR was performed using TaqMan Master Mix (Applied Biosystems, 4369016) TaqMan probes (Il1b: Rn99999009_m1; Cxcl10: Rn00594648_m1; Hprt1: Rn1428093_m1; iNos: Rn00561646_m1, all from Applied Biosystems) on an Applied Biosystems 7900HT Real-Time PCR System (Applied Biosystems, 7900HT) and data analyzed using SDS 2.3 (Applied Biosystems). PCR products were quantified using the 2−ΔΔct methodCitation27 compared with the housekeeping gene hypoxanthine-guanine phosphoribosyltransferase (Hprt1), which we have previously validated to be unaffected by the experimental conditions in our system.Citation15 A standard dilution curve was used as control for amplification efficiency; when efficiency failed to reach 90% or exceeded 110% experiments were discarded. Expression relative to a standard curve, was reported when mRNA expression in control conditions was below detection threshold, as the 2−ΔΔct-method is invalid for quantification when there is no expression of the gene of interest in the control condition.
Immunoblotting
Histone acetylation: 5 × 105 INS-1 cells were seeded in 12-well plates in complete medium. Mouse IL-1β and rat IFNγ were added for the indicated time periods. Cells were lysed, protein content measured by the Bradford method, and lysates adjusted for protein concentration and prepared for gel electrophoresis as previously described.Citation28 A minimum of 8 μg protein was loaded and separated by gel electrophoresis. Fluorescence based Q-Dot technology (Invitrogen) was used for detection of anti-acetylated histone H4 antibody (Millipore, 06-866). Light emission was captured digitally with the Flourchem Q BioImager System (Kem-En-Tec). Q-dot 605 and 705 were detected using 605 ± 30 nm and 705 ± 30 nm band width filters (Kem-En-Tec).
IL-1β processing: 2.5 × 106/mL cells per condition were seeded in 6 cm Petri dishes (Nunc) 48 h prior to experiments and precultured for one hour with/without givinostat and exposed to IL-1β and IFNγ or vehicle for 6 h, washed and exposed to Na2ATP (Sigma-Aldrich, A3377) for the time periods indicated before cells were lysed. A minimum of 15 μg of protein was loaded. Primary antibodies were from eBioscience against IL-1β (16-7012) and Santa Cruz against β-actin (sc-5274). Only mature 17.4 kDa IL-1β was quantified and normalized to actin. Secondary antibodies: biotin goat anti-Armenian hamster was from Abcam (ab5744) and biotin-XX goat anti-mouse was from Invitrogen (B-2763).
Statistical analysis
Comparisons between groups were by ANOVA followed by paired two-way t-test with Tukey’s correction using SAS® 9.1.3 for normally distributed data, and by Kruskal-Wallis ANOVA on ranks followed by Student-Newman-Keuls multiple comparison test for non-normally distributed data using SigmaPlot 11.0® (Systat Software). Comparisons between single conditions were by Student’s two-way t-test.
| Abbreviations: | ||
| CXCL10 | = | C-X-C motif chemokine ligand 10 |
| CXCR3 | = | CXCR3 chemokine (C-X-C motif) receptor 3 |
| IFNγ | = | interferon-gamma |
| IL-1β | = | interleukin-1 β |
| INS-1 | = | insulinoma-1 cell line |
| JAK | = | janus kinase |
| KDAC | = | lysine deacetylase |
| LPS | = | lipopolysaccharide |
| MAPK | = | mitogen-activated protein kinase |
| MyD88 | = | myeloid differentiation primary response gene (88) |
| NFκB | = | nuclear factor κ B |
| NOD | = | non-obese diabetic |
| qPCR | = | quantitative polymerase chain reaction |
| STAT1 | = | signal transducers and activators of transcription 1 |
| T1/T2 DM | = | type 1/2 diabetes mellitus |
| TLR4 | = | toll-like receptor 4 |
Acknowledgments
G.K. Singh is thanked for excellent technical assistance. This work was supported by grants from JDRF (26-2008-893), the EFSD/JDRF/Novo Nordisk European Programme in Type 1 Diabetes Research, the University of Copenhagen and the Novo Nordisk Foundation.
Disclosure of Potential Conflicts of Interest
Paolo Mascagni is employed by Italfarmaco. The other authors disclose no conflicting interests.
References
- Donath MY, Størling J, Berchtold LA, Billestrup N, Mandrup-Poulsen T. Cytokines and beta-cell biology: from concept to clinical translation. Endocr Rev 2008; 29:334 - 50; http://dx.doi.org/10.1210/er.2007-0033; PMID: 18048762
- Eizirik DL, Mandrup-Poulsen T. A choice of death--the signal-transduction of immune-mediated beta-cell apoptosis. Diabetologia 2001; 44:2115 - 33; http://dx.doi.org/10.1007/s001250100021; PMID: 11793013
- Böni-Schnetzler M, Thorne J, Parnaud G, Marselli L, Ehses JA, Kerr-Conte J, et al. Increased interleukin (IL)-1beta messenger ribonucleic acid expression in beta -cells of individuals with type 2 diabetes and regulation of IL-1beta in human islets by glucose and autostimulation. J Clin Endocrinol Metab 2008; 93:4065 - 74; http://dx.doi.org/10.1210/jc.2008-0396; PMID: 18664535
- Dinarello CA, Donath MY, Mandrup-Poulsen T. Role of IL-1beta in type 2 diabetes. Curr Opin Endocrinol Diabetes Obes 2010; 17:314 - 21; PMID: 20588114
- Larsen CM, Faulenbach M, Vaag A, Vølund A, Ehses JA, Seifert B, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 2007; 356:1517 - 26; http://dx.doi.org/10.1056/NEJMoa065213; PMID: 17429083
- Roep BO, Kleijwegt FS, van Halteren AG, Bonato V, Boggi U, Vendrame F, et al. Islet inflammation and CXCL10 in recent-onset type 1 diabetes. Clin Exp Immunol 2010; 159:338 - 43; http://dx.doi.org/10.1111/j.1365-2249.2009.04087.x; PMID: 20059481
- Ribaux P, Ehses JA, Lin-Marq N, Carrozzino F, Böni-Schnetzler M, Hammar E, et al. Induction of CXCL1 by extracellular matrix and autocrine enhancement by interleukin-1 in rat pancreatic beta-cells. Endocrinology 2007; 148:5582 - 90; http://dx.doi.org/10.1210/en.2007-0325; PMID: 17702850
- Uno S, Imagawa A, Saisho K, Okita K, Iwahashi H, Hanafusa T, et al. Expression of chemokines, CXC chemokine ligand 10 (CXCL10) and CXCR3 in the inflamed islets of patients with recent-onset autoimmune type 1 diabetes. Endocr J 2010; 57:991 - 6; http://dx.doi.org/10.1507/endocrj.K10E-076; PMID: 20966598
- Morimoto J, Yoneyama H, Shimada A, Shigihara T, Yamada S, Oikawa Y, et al. CXC chemokine ligand 10 neutralization suppresses the occurrence of diabetes in nonobese diabetic mice through enhanced beta cell proliferation without affecting insulitis. J Immunol 2004; 173:7017 - 24; PMID: 15557199
- Schulthess FT, Paroni F, Sauter NS, Shu L, Ribaux P, Haataja L, et al. CXCL10 impairs beta cell function and viability in diabetes through TLR4 signaling. Cell Metab 2009; 9:125 - 39; http://dx.doi.org/10.1016/j.cmet.2009.01.003; PMID: 19187771
- Shigihara T, Oikawa Y, Kanazawa Y, Okubo Y, Narumi S, Saruta T, et al. Significance of serum CXCL10/IP-10 level in type 1 diabetes. J Autoimmun 2006; 26:66 - 71; http://dx.doi.org/10.1016/j.jaut.2005.09.027; PMID: 16309891
- Shimada A, Oikawa Y, Yamada Y, Okubo Y, Narumi S. The role of the CXCL10/CXCR3 system in type 1 diabetes. Rev Diabet Stud 2009; 6:81 - 4; http://dx.doi.org/10.1900/RDS.2009.6.81; PMID: 19806237
- Shigihara T, Shimada A, Oikawa Y, Yoneyama H, Kanazawa Y, Okubo Y, et al. CXCL10 DNA vaccination prevents spontaneous diabetes through enhanced beta cell proliferation in NOD mice. J Immunol 2005; 175:8401 - 8; PMID: 16339582
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009; 325:834 - 40; http://dx.doi.org/10.1126/science.1175371; PMID: 19608861
- Lundh M, Christensen DP, Rasmussen DN, Mascagni P, Dinarello CA, Billestrup N, et al. Lysine deacetylases are produced in pancreatic beta cells and are differentially regulated by proinflammatory cytokines. Diabetologia 2010; 53:2569 - 78; http://dx.doi.org/10.1007/s00125-010-1892-8; PMID: 20878317
- Larsen L, Tonnesen M, Ronn SG, Størling J, Jørgensen S, Mascagni P, et al. Inhibition of histone deacetylases prevents cytokine-induced toxicity in beta cells. Diabetologia 2007; 50:779 - 89; http://dx.doi.org/10.1007/s00125-006-0562-3; PMID: 17265033
- Lundh M, Christensen DP, Damgaard Nielsen M, Richardson SJ, Dahllöf MS, Skovgaard T, et al. Histone deacetylases 1 and 3 but not 2 mediate cytokine-induced beta cell apoptosis in INS-1 cells and dispersed primary islets from rats and are differentially regulated in the islets of type 1 diabetic children. Diabetologia 2012; 55:2421 - 31; http://dx.doi.org/10.1007/s00125-012-2615-0; PMID: 22772764
- Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 2009; 27:519 - 50; http://dx.doi.org/10.1146/annurev.immunol.021908.132612; PMID: 19302047
- Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 2009; 183:787 - 91; http://dx.doi.org/10.4049/jimmunol.0901363; PMID: 19570822
- Yamada Y, Okubo Y, Shimada A, Oikawa Y, Yamada S, Narumi S, et al. Role of CXCR3 in the beta Cell Proliferation in Type 1 Diabetes. Diabetes 2011; 60:Supplement 1 A527
- Guo JJ, Li QL, Zhang J, Huang AL. Histone deacetylation is involved in activation of CXCL10 upon IFNgamma stimulation. Mol Cells 2006; 22:163 - 7; PMID: 17085967
- Kawai T, Takeuchi O, Fujita T, Inoue J, Mühlradt PF, Sato S, et al. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J Immunol 2001; 167:5887 - 94; PMID: 11698465
- Leng C, Gries M, Ziegler J, Lokshin A, Mascagni P, Lentzsch S, et al. Reduction of graft-versus-host disease by histone deacetylase inhibitor suberonylanilide hydroxamic acid is associated with modulation of inflammatory cytokine milieu and involves inhibition of STAT1. Exp Hematol 2006; 34:776 - 87; http://dx.doi.org/10.1016/j.exphem.2006.02.014; PMID: 16728283
- Krämer OH, Baus D, Knauer SK, Stein S, Jäger E, Stauber RH, et al. Acetylation of Stat1 modulates NF-kappaB activity. Genes Dev 2006; 20:473 - 85; http://dx.doi.org/10.1101/gad.364306; PMID: 16481475
- Krämer OH, Knauer SK, Greiner G, Jandt E, Reichardt S, Gührs KH, et al. A phosphorylation-acetylation switch regulates STAT1 signaling. Genes Dev 2009; 23:223 - 35; http://dx.doi.org/10.1101/gad.479209; PMID: 19171783
- Brunstedt J. Rapid isolation of functionally intact pancreatic islets from mice and rats by percollTM gradient centrifucation. Diabete Metab 1980; 6:87 - 9; PMID: 6250926
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25:402 - 8; http://dx.doi.org/10.1006/meth.2001.1262; PMID: 11846609
- Larsen L, Størling J, Darville M, Eizirik DL, Bonny C, Billestrup N, et al. Extracellular signal-regulated kinase is essential for interleukin-1-induced and nuclear factor kappaB-mediated gene expression in insulin-producing INS-1E cells. Diabetologia 2005; 48:2582 - 90; http://dx.doi.org/10.1007/s00125-005-0039-9; PMID: 16283237

