Abstract
Monoclonal antibodies (mAb) have become a mainstay in tumor therapy. Clinical responses to mAb therapy, however, are far from optimal, with many patients presenting native or acquired resistance or suboptimal responses to a mAb therapy. MAbs exert antitumor activity through different mechanisms of action and we propose here a classification of these mechanisms. In many cases mAbs need to interact with immune cells to exert antitumor activity. We summarize evidence showing that interactions between mAbs and immune cells may be inadequate for optimal antitumor activity. This may be due to insufficient tumor accumulation of mAbs or immune cells, or to low-affinity interactions between these components. The possibilities to improve tumor accumulation of mAbs and immune cells, and to improve the affinity of the interactions between these components are reviewed. We also discuss future directions of research that might further improve the therapeutic efficacy of antitumor mAbs.
Introduction: Monoclonal Antibodies, A Growing Class of Antitumor Drugs
Monoclonal antibodies (MAbs) have become an increasingly important class of antitumor drugs since the first regulatory approval of the anti-CD20 antibody rituximab in 1997.Citation1 Since then, seven other antitumor mAbs (only full-length mAbs are herein considered; antibody fragments or immunoconjugates are excluded) have been registered worldwide, and some have become blockbuster drugs with yearly sales exceeding 1 billion USD.Citation2 It does therefore not come as a surprise that a substantial number of antitumor mAbs are at now in active clinical development.Citation2 Clinical results obtained so far suggest, however, that there is still much room for improvement in the therapeutic efficacy of antitumor mAbs. In fact, many patients do not respond or respond suboptimally to the mAb that they are administered, while most responding patients become resistant over time.Citation3,Citation4 In this review we propose a classification of antitumor mAbs on the basis of their mechanism(s) of action, and discuss opportunities to improve tumor accumulation and interactions between antitumor mAbs and cells of the innate or adaptive immune system (hereafter referred to as immune cells) which, in some instances, are crucially involved in the antitumor activity of mAbs.Citation5 Approaches of this kind hold promise to improve the therapeutic efficacy of antitumor mAbs.
Mechanisms of Action of Antitumor mAbs
Antitumor mAbs can act through different mechanisms of action (). In the following we propose a classification of these mechanisms of action that expands and integrates similar classifications that have been proposed in the past.Citation6,Citation7 Beforehand, however, it is worth noting that while individual antitumor mAbs may have more than one mechanism of action as determined in different in vitro or in vivo models, it appears more difficult to identify the mechanism(s) mainly responsible for the antitumor activity in the clinical setting. Additional complexity derives from the possibility that different mechanisms of action may predominate in different malignancies.
Table 1. Mechanisms of action of antitumor mAbs
Direct induction of cell death
MAbs that induce direct cell death can be divided into two subclasses. We refer to this mode of action as direct induction, because cell death is the result of activation of a death program that is the direct consequence of antibody binding. This distinguishes these antibodies from those that induce cell death indirectly, due to deprivation of growth or survival signals (e.g., intracellular signals induced by growth factors), or recruitment of immune cells.
The first subclass of these antibodies induces a form of non-apoptotic programmed cell death that is based on the permeabilization of lysosome membranes, and does not imply any involvement of immune cells.Citation8 The mechanism of action of these antibodies has been most accurately investigated for anti-CD20 antibodies, but appears to apply also to antibodies against other antigens, such as HLA-DR, CD47, CD74 and CD99.Citation9,Citation10 Anti-CD20 antibodies of this subclass do not redistribute CD20 into lipid rafts upon binding, are weak activators of complement, efficient activators of Fcγ receptor (FcγR)-positive immune cells and potent inducers of direct cell death. The form of cell death that these antibodies induce is non-apoptotic, dependent on mAb-induced actin cytoskeletal reorganization, lysosome membrane permeabilization and, eventually, production of reactive oxygen species mediated by nicotinamide adenine dinucleotide phosphate oxidase.Citation10-Citation12
Tositumomab is the only anti-CD20 mAb of this subclass that is US Food and Drug Administration (FDA)-approved; it is administered as part of the BEXXAR radioimmunotherapy regimen.Citation13 In this case, however, the main effector mechanism appears to be radiation-induced cell death, and it is difficult to estimate whether antibody-induced cell death contributes to the overall therapeutic efficacy. Another mAb of this subclass that has reached phase II/III clinical trials is obinutuzumab (GA101). However, also in this case the mechanism of action is not limited to direct cell killing, as this antibody has a glycoengineered Fc region that endows the antibody with improved capacity to engage immune cells and trigger antibody-dependent cellular cytotoxicity (ADCC).Citation14 Results with this antibody in rituximab-resistant lymphoma patients appear very promising.Citation15
The second subclass of antibodies induces apoptotic cell death and includes antibodies against tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-receptors/death receptors (TRAIL-R/DR).Citation16 The ligand, TRAIL, induces apoptosis of tumor cells and has antitumor activity in vivo. Five TRAIL-Rs have been identified, two of which, DR4 (TRAIL-R1) and DR5 (TRAIL-R2), are capable of transducing the apoptosis signal, whereas the others serve as decoy receptors. Expression of DR4 or DR5 is frequently detected in human cancers with low or no expression in normal tissues.Citation16
Administration of recombinant TRAIL in animals induces tumor regression without systemic toxicity. Agonistic anti-DR mAbs induce apoptotic cell death similarly to TRAIL and may be preferable because they can be selected against individual DRs and have a much longer half-life than TRAIL. Several agonistic anti-DR mAbs have demonstrated efficacy in preclinical tumor models, and improved efficacy was observed in combination with chemotherapy. Several forms of resistance to DR-mediated apoptosis have been described.Citation16
Until recently, anti-DR mAbs were thought to induce apoptosis independently of any involvement of effector cells. A recent article, however, has shown that cell death induced by anti-DR4 and anti-DR5 mAbs rests on the expression of FcγRs on tumor-associated immune cells.Citation17 While other antibodies require activatory FcγRs for their antitumor activity (e.g., rituximab), either activatory or inhibitory FcγRs can support induction of tumor cell death by anti-DR4 or -DR5 antibodies.Citation17 This observation implies that FcγR-positive immune cells do not actively contribute to mAb-induced cell death, but merely serve as a platform for multivalent antibody display and mAb-mediated clustering of the TRAIL-R to drive apoptotic signaling. For this reason, we consider cell death induced by anti-DR mAbs as a form of direct cell death that justifies their inclusion in this antibody class.
Several anti-DR4 and -DR5 mAbs are or have been in clinical development: two anti-DR4 antibodies, mapatumumabCitation18 and lexatumumab,Citation19,Citation20 two anti-DR5 antibodies, drozitumabCitation21 and conatumumab,Citation22 and the tetrameric anti-DR5 nanobody (single-chain, camelid antibody) TAS266.Citation23 Some minor responses and several disease stabilizations have been observed in early-stage clinical studies with these antibodies.
Inhibition of tumor-promoting growth or survival signals
Antitumor mAbs can inhibit tumor growth or survival signals in two ways: first, through neutralization of ligands that induce such signals upon binding to cell surface receptors; second, through binding to cell surface receptors or co-receptors.
Neutralization of tumor-promoting ligands
The only mAb of this class that has gained regulatory approval so far (bevacizumab) is directed against vascular endothelial growth factor (VEGF), which supports growth and differentiation of endothelial cells involved in the formation of the tumor vasculature. The first indication that has received approval for bevacizumab therapy is metastatic colorectal cancer (CRC) in combination with chemotherapeutics.Citation24 Bevacizumab is thought to temporarily “normalize” tumor blood vessels, with an overall decrease of tumor perfusion, vascular volume, microvascular density, interstitial fluid pressure (IFP) and an increase of the fraction of vessels with pericyte coverage.Citation25 This normalization of the tumor vasculature is thought to promote uptake of co-administered antitumor drugs.Citation26 In a few instances, bevacizumab has been shown to have antitumor activity on its own, but only at relatively high doses and on highly vascularized tumors, like renal cell cancer.Citation27
There are several other antibodies or antibody-related products against angiogenic growth factors in developmentCitation28-Citation30 but, surprisingly, very few antibodies against other tumor growth-promoting ligands. Antibodies against hepatocyte growth factor,Citation31 and insulin-like growth factorCitation32 are two exceptions. There are different possible reasons for this neglect: (1) factors that promote tumor cell growth and dissemination are probably highly redundant and inhibition of one single factor may lead to the induction of largely overlapping effects by other factors; (2) deprivation of these factors may lead to states of quiescence or autophagy that are particularly conducive to drug resistance;Citation33 (3) neutralization of these factors may require deep tumor penetration to be effective, and this is difficult to achieve, as discussed below.
Binding to cell surface receptors or co-receptors
Engagement of cell surface receptors or co-receptors by antibodies may inhibit signal transmission through multiple modes: inhibition of binding of the native ligand, inhibition of receptor homo- or hetero-dimerization, or increased endocytotic destruction of the receptor.Citation34 The clearest, albeit indirect evidence of the contribution of this mechanism to the clinical efficacy of a mAb comes from the use of anti-EGFR mAbs cetuximab and panitumumab in patients with CRC.Citation35,Citation36 Activating mutations of KRAS, a downstream mediator of EGFR signaling, have been shown to render anti-EGFR antibodies ineffective in the treatment of CRC because of ligand-independent, downstream activation of the EGFR. This has led regulatory authorities to mandate testing for these mutations before using anti-EGFR antibodies in CRC. It should be noted however, that while the tumor-promoting effect of KRAS mutations have been firmly established for CRC, this does not appear to apply to all solid tumors. Thus, KRAS mutations do not seem to identify anti-EGFR nonresponding patients in non-small cell lung cancer.Citation37
Recruitment of FcγR-positive immune cells
As a mechanism of action, recruitment of FcγR-positive immune cells is of particular relevance because a large body of evidence suggests that this is the main, if not exclusive, mechanism of action of rituximab in the treatment of B-cell non-Hodgkin lymphoma (B-NHL).Citation12 FcγRs are expressed on several immune cells that mediate diverse functions like ADCC or phagocytosis. While engagement of FcγR-positive cells appears mandatory for the antitumor activity of rituximab, this does not indicate which FcγR-positive cells exert tumoricidal activity. Thus, natural killer (NK) cells express FcγRIIIA and mediate ADCC upon binding of the antibody Fc portion. Macrophages, on the other hand, express FcγRIIA and mediate phagocytosis upon antibody binding.
As to solid tumors, several potential mechanisms of action have been attributed to anti-human epidermal growth factor receptor (HER)2 trastuzumab.Citation34 Clinical studies, however, found a correlation between the level of ADCC activity and clinical response to trastuzumab therapy,Citation38 suggesting that ADCC plays an important role also in the tumoricidal activity of trastuzumab. Evidence for the contribution of FcγR-positive immune cells to the mechanism of action has been brought also for cetuximab. Thus, similar to trastuzumab, combined FcγRIIA/FcγRIIIA polymorphisms have been found to be prognostic factors for disease progression in metastatic CRC patients treated with cetuximab plus irinotecan.Citation39 Favorable genotypes were associated with longer progression-free survival (PFS). These polymorphisms were also clinically relevant in KRAS-mutated, metastatic CRC. In fact, KRAS mutations and FcγR combined status were independent prognostic factors for PFS.
Complement activation
Complement activation plays an important role in the antitumor activity of anti-CD52 alemtuzumab. This antibody has activity as a monotherapy and in combination with fludarabine in patients with relapsed/refractory chronic lymphocytic leukemia (CLL).Citation40 Alemtuzumab lysed cells from CLL samples through complement-dependent cytotoxicity (CDC) much more efficiently than rituximab, presumably because of the higher expression of CD52 than CD20.Citation41 Lack of direct cytotoxiciy, but significant CDC on fresh CLL cells, was confirmed in another study, although a subpopulation of CLL cells were intrinsically resistant.Citation42
Complement activation is crucial also for the antitumor activity of anti-CD20 ofatumumab, which targets a membrane-proximal epitope that is different from the more membrane-distal epitope targeted by rituximab.Citation43 This is the likely reason why, in spite of rituximab and ofatumumab having comparable binding affinities for CD20, ofatumumab induces stronger CDC at lower CD20 densities than rituximab.Citation43 This mechanism forms the basis for the therapeutic activity of ofatumumab in CLL, where the cell surface density of CD20 is lower than in B-NHL. There is evidence, however, that CDC is not the only mechanism of action of ofatumumab. In fact, it induces also ADCC in vivo,Citation43 and this effect may integrate CDC in the overall antitumor activity, with one or the other activity predominating depending on the disease that is treated (e.g., CDC in CLL and ADCC in B-NHL). So far, ofatumumab has gained FDA approval for the treatment of patients with CLL refractory to fludarabine and alemtuzumab.Citation44
On the other hand, considerable evidence suggests that complement activation by antitumor mAbs may have detrimental consequences. Thus, complement activation plays a key role in the toxicity related to infusion of mAbs.Citation45,Citation46 More recently, Wang et al. showed that complement activation inhibits rituximab-induced ADCC and NK-cell activation.Citation47 Components of the complement cascade have also been shown to possess direct or indirect tumor growth-promoting activities.Citation48
Promotion of an adaptive antitumor immune response
Antibodies of this class can be divided into two subclasses: (1) antibodies that upregulate an ongoing antitumor immune response through inhibition of immune suppressive pathways; and (2) antibodies that, through engagement of accessory cells, induce an effect that resembles active antitumor immunization. While several antibodies of the first subclass are in active development, and one (ipilimumab) was recently approved, several antibodies of the second subclass (rituximab, trastuzumab) have been marketed for over a decade. Induction of active antitumor immunity plays a still ill-defined, but possibly important role in their overall antitumor effect.
Antibodies of the first subclass inhibit cell-cell interactions that downregulate immune responses, including antitumor immune responses. The anti-cytotoxic T-lymphocyte antigen (CTLA)-4 mAb ipilimumab received FDA approval for the treatment of metastatic melanoma.Citation49,Citation50 The main characteristics of the antitumor effects of ipilimumab are the following: (1) ipilimumab significantly prolongs overall survival in metastatic melanoma patients both as monotherapy and in combination with a chemotherapeutic; (2) treatment is accompanied in 10–15% of the patients by immune-related (“autoimmune-like”) adverse events, which can be severe, long-lasting, but reversible in most cases; (3) patients that are affected by these adverse events are often those that respond best to ipilimumab. Several other antibodies against the same target or other targets are at different stages of clinical development.Citation51,Citation52
An example of antibodies of the second subclass is rituximab, which has been shown to induce an idiotype-specific T-cell response in follicular lymphoma patients.Citation53 This effect has been suggested to underlie the durable remissions experienced by some patients in response to rituximab.Citation54 Moreover, it has been shown that an anti-CD20 mAb induces protection against a CD20-positive murine lymphoma in a mouse model through a T cell-mediated immune response.Citation55 Induction of an adaptive immune response may be the consequence of rituximab-coated lymphoma cells facilitating the cross-priming of tumor antigens by human dendritic cells and subsequent elicitation of an immune response mediated by CD8-positive T cells.Citation56
Trastuzumab is another antibody that has been shown to promote an adaptive immune response. Indeed, in a murine model the mechanism of tumor regression induced by this antibody required an adaptive immune response.Citation57 The addition of chemotherapeutic drugs enhanced the reduction of tumor burden, but abrogated the antibody-initiated immunity leading to decreased resistance to rechallenge or earlier relapse.Citation57 Evidence for trastuzumab-induced antitumor immunity comes also from human studies. Thus, trastuzumab enhanced lysis of HER2-positive tumor cells by cytotoxic T-lymphocytes recognizing a HER2 peptide, but not cytotoxic T-lymphocytes specific for an unrelated peptide.Citation58 Moreover, trastuzumab increased immune cell infiltration into breast tumors,Citation59 and patients treated with trastuzumab develop anti-HER2 IgGλ antibody responses.Citation60 In this study, antibody responses were observed more frequently in patients with objective responses to trastuzumab, and HER2-specific CD4-positive T-cell responses were generated in response to trastuzumab and chemotherapy.
Obstacles to the Accumulation and Interaction of mAbs and Immune Cells in Tumors
Obstacles to the accumulation of mAbs in tumors
In addition to the mechanism(s) of action, other factors can influence the antitumor efficacy of mAbs. One of these factors is related to the location of the targeted antigen. In solid tumors these antigens can be located on tumor endothelial cells (e.g., VEGF receptors) or on cells of the extravascular compartment (mainly tumor cells, but also immune cells or other cells of the tumor stroma).Citation61,Citation62
Regarding antigens expressed in the extravascular compartment, mAbs encounter, similarly to other antitumor drugs, considerable difficulties in accumulating and penetrating solid tumor tissues.Citation26 A mechanistic model has been proposed to predict the interplay between molecular size, in vivo half-life, affinity and tumor uptake of tumor-targeting agents.Citation63 In the typical size range for proteins, the model uncovers a complex trend in which intermediate-sized targeting agents (~25,000 Da) have the lowest tumor uptake, whereas higher tumor uptake levels are achieved by smaller or larger agents. Small peptides accumulate rapidly in the tumor but are cleared faster and require high affinity to be retained. In contrast, large proteins have slower diffusion rates in tissues, but have longer circulating half-lives, owing to the size and after penetration can achieve similar retention even with > 100-fold weaker binding.Citation63 The poor tumor accumulation of intermediate-sized molecules can be obviated by taking advantage of constant perfusion rather than bolus adminstration.Citation64 Similar approaches are being pursued also for the administration of chemotherapeutics.Citation65
MAbs against antigens expressed on tumor endothelial cells, on the other hand, do not require tumor penetration. Nevertheless, increased resistance to blood flow and impairment of blood supply to the tumor can represent obstacles for the delivery of these antibodies.Citation26
Another case to consider is that of antibodies that target antigens expressed on both tumor endothelial cells and tumor cells.Citation66 While dedicated studies on the tumor penetration of these antibodies are lacking, intuitively one is led to say that they have considerable therapeutic potential. In fact, an antibody with cytotoxic effects against tumor endothelial cells is expected to lead to the breakdown of the tumor endothelial cell barrier which represents the most challenging obstacle for antibody uptake in tumors.Citation67 This would facilitate tumor accumulation and penetration of the antibody, which could then target antigen-positive tumor cells.
Obstacles to the accumulation of immune cells in tumors
As discussed previously, the antitumor activity of mAbs often requires the accessory function of immune cells. To exert their functions, these cells need to extravasate and infiltrate tumors in a manner similar to antibodies. While the impact of these cells on tumor growth and dissemination is being intensively studied,Citation68 the consequences of tumor uptake and infiltration of these cells on the antitumor efficacy of mAbs is a neglected area of research. This is surprising because any investigation on the tumor accumulation of mAbs may be meaningless if the therapeutic effect of an antibody depends on the presence of a threshold number of immune cells, and such a threshold number is not attained in a given tumor.
The obstacles that affect tumor accumulation of mAbs and other antitumor drugs are also likely to affect immune cells. Immune cells, however, may suffer from additional impediments related to the physiological pathways of immune cell extravasation which depends on a multistep cascade of events involving tethering, rolling, firm adhesion and migration. These steps are mediated by distinct adhesion molecules and activation pathways,Citation69 but adhesion molecules are often downregulated on tumor endothelial cells, a phenomenon defined as endothelial cell anergy.Citation70 This impairs the extravasation of immune cells into tumor sites.Citation71-Citation73 Impaired accumulation of immune cells in tumor tissues, on the other hand, has been found in several studies to represent an independent, negative prognostic marker in clinical studies.Citation74
These considerations support the possibility that a sizeable fraction of tumors may not have a sufficient number of immune cells to support the tumoricidal activity of those mAbs that depend on their contribution. This possibility has found strong support in studies with trastuzumab. Indeed, Gennari et al.Citation75 found that patients showing complete or partial remission after treatment with trastuzumab alone were found to have higher ADCC and higher in situ infiltration of cytotoxic lymphocytes, including NK cells.
Obstacles to the interaction between mAbs and immune cells in tumors
A low concentration of mAbs or a low number of immune cells in tumor tissues represent, per se, an obstacle to their interaction and to the generation of tumoricidal activities (ADCC or phagocytosis). In addition, low-affinity interactions between antitumor mAbs and immune cells may adversely affect efficient antitumor activity. This was first documented with rituximab. Thus, clinical studies in B-NHL patients showed that rituximab-treated patients who carry genetic polymorphisms of FcγRIIIA with a high affinity for IgG (158 valine/valine) have much better clinical and molecular responses than patients with low-affinity polymorphisms (158 phenylalanine/phenylalanine or 158 valine/phenylalanine).Citation76 Similar results have been reported by others.Citation77,Citation78 Weng and LevyCitation77 reported that polymorphisms of FcγRIIA can predict outcome after rituximab treatment. Similar results were reported later for trastuzumab in breast cancer patients.Citation79 These authors showed a positive correlation between high affinity allelic variants of FcγRIIIA and FcγRIIA and clinical response to trastuzumab therapy. The combination of the two favorable genotypes was independently associated with better prognosis compared with the other combinations, and FcγRIIIA seemed to play the predominant role in the outcome.
Overall, these results imply that clinical responses depend on the affinity of the interaction between an antitumor mAb (e.g., rituximab, trastuzumab) and FcγR-positive immune cells, with higher affinity interactions allowing for better antitumor responses. There are, however, some observations that run against this knowledge. Thus, it was found that M2 macrophages expressing the high-affinity variant of FcγRIIIA release immunosuppressive and tumor-promoting cytokines upon coculture with EGFR-positive tumor cells loaded with low concentrations of cetuximab.Citation80 This observation can explain the results of a previous clinical trial in metastatic CRC patients, where the addition of cetuximab to bevacizumab plus chemotherapy was tested.Citation81 In this study, patients expressing the high-affinity allele for FcγRIIIA had a shorter PFS, but only when cetuximab was added.
These observations introduce another condition that needs to be satisfied to achieve productive interactions between antitumor mAbs and immune cells: the mAb needs to interact with the right immune cell subpopulation to give rise to tumoricidal activity. Indeed, a high affinity interaction between a mAb and M2 macrophages in tumor tissues may downregulate effector functions.Citation80 Regarding adaptive antitumor immune responses, it has been recognized that a high frequency of immunosuppressive regulatory T cells (Treg) in tumor tissues is of negative prognostic significance.Citation82,Citation83 Intuitively, but not yet demonstrated, the same phenomenon might also inhibit mAb-induced adaptive antitumor immune responses.
Approaches to Improve Tumor Accumulation and Interactions Between mAbs and Immune Cells
Inadequate antitumor activity that may be the consequence of insufficient tumor accumulation of mAbs or immune cells, an inadequate qualitative profile of infiltrating immune cells, or low-affinity interactions between these components, has been discussed; in this section we will discuss approaches to bypass these obstacles, thereby improving the antitumor activity of mAbs. In accordance with the nomenclature that we proposed in a previous article,Citation26 we will refer to those drugs that improve accumulation of mAbs or immune cells as promoter drugs.
Approaches to improve tumor accumulation of mAbs
As already discussed in previous articles,Citation26,Citation67 promoter drugs that enhance tumor uptake and penetration of antitumor drugs are likely to be effective also for mAbs. In several instances this has been directly demonstrated and we discuss some of these cases here ().
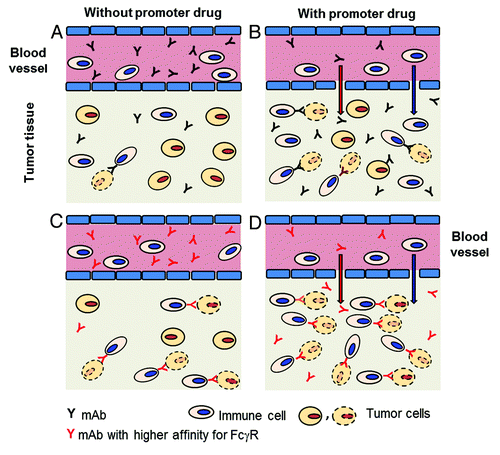
Figure 1. Improving accumulation and interactions between mAbs and immune cells in tumors. (A) In tumor tissues levels of mAbs or immune cells may be too low to interact and kill tumor cells by ADCC or phagocytosis. (B) In the presence of promoter drug(s) tumor accumulation of mAbs or immune cells may achieve levels sufficient to interact and kill some tumor cells (tumor cell with dotted contour). (C) In the presence of mAbs binding with higher affinity to FcγR-positive immune cells, interactions may take place and kill some tumor cells. (D) In the presence of promoter drug(s) and mAbs binding with higher affinity to FcγR-positive immune cells, a higher number of interactions may take place than with either approach alone, and a large number of tumor cells may be killed.
Modulation of tumor blood flow is one approach that has been investigated. Thus, Netti et al.Citation84 studied the effect of periodic and continuous angiotensin II administration on tumor uptake of the anti-TAG-72 mAb CC49 in LS174T CRC xenografts. Both periodic and chronic infusion of angiotensin II resulted in 40% enhancement of specific antibody (CC49) uptake at 4 h after injection, whereas the tumor uptake of a nonspecific antibody was unaffected.
Several other classes of promoter drugs modify the barrier function of tumor vessels. AG-01376 is a VEGF receptor tyrosine kinase inhibitor that acts as promoter drug by normalizing tumor vessels, similarly to other VEGF inhibitors and anti-VEGF mAbs. AG-01376 improves transport of antibodies from the surviving “normalized” blood vessels.Citation85 Antibodies were found to accumulate in the sleeves of the basement membrane left behind by the regressing vessels, which were proposed to serve as the preferential routes of distribution in tumors.
Promoter drugs can act also by enhancing the permeability of tumor vessels. NHS76/PEP2, for example, is a fusion protein between mAb NHS76, which targets necrosis and cellular degeneration commonly found in solid tumors, and a 37-amino-acid linear sequence of interleukin (IL)-2, designated vasopermeability-enhancing peptide (PEP), which contains the vasopermeability activity of IL-2. Using a 2-h pretreatment, an optimal dose of NHS76/PEP2 (30 μg) induced greatly increased and specific tumor accumulation of the mAb B72.3.Citation86 This approach increased also tumor accumulation and therapeutic efficacy of the chemotherapeutic drugs etoposide, doxorubicin, paclitaxel (Taxol®), docetaxel (Taxotere®), 5-fluorouracil, or vinblastine. The same group had previously shown that conjugation of tumor necrosis factor (TNF)-α to the tumor necrosis-targeting mAb TNT-1 caused a three-fold increase of the tumor uptake of the F(ab’)2 fragment of the same antibody. In this regard, the TNFα-antibody conjugate was more effective in improving the uptake compared with other vasoactive agents used in the study, including IL-1, leukotriene B4, histamine, bradykinin and physalaemin. Only an IL-2-TNT-1 conjugate induced a greater increase in the tumor uptake of the antibody fragment.Citation87
Another molecule that acts through a related mechanism of action is the tumor-penetrating peptide “internalizing arginine-glycine-aspartic acid” (iRGD), which increases vascular and tissue permeability in a tumor-specific and neuropilin-1-dependent manner.Citation88 Systemic injection of this peptide improved the therapeutic index of drugs of various compositions including a small molecule (doxorubicin), nanoparticles (nab-paclitaxel and doxorubicin liposomes), and a mAb (trastuzumab). Co-injection of iRGD and trastuzumab resulted in a 40-fold enhanced accumulation of trastuzumab in the tumor. At a clinical dose (9 mg/kg) of trastuzumab, the combination eradicated all tumors in mice, whereas the equivalent dose of trastuzumab alone only slowed tumor growth.Citation89
A recent new class of promoter drugs acts by loosening intercellular junctions between tumor cells. Thus, Beyer et al.Citation90 reported on the protein, junction opener 1 (JO-1), which binds to the epithelial junction protein desmoglein 2. JO-1 mediates cleavage of desmoglein 2 dimers and activates intracellular signaling pathways that reduce E-cadherin expression in tight junctions. These effects allowed for increased intratumoral penetration of trastuzumab. This effect translated directly into increased therapeutic efficacy of trastuzumab in mouse xenograft tumor models. JO-1 improved also the antitumor efficacy of cetuximab. This article also showed that the combination of relaxin expression and JO-1 treatment stopped tumor growth. Tumors did not recur when treatment was terminated, in contrast to groups that received either relaxin+trastuzmab or JO-1+trastuzumab therapy. This is an important result because it demonstrates that the combined use of promoter drugs with different mechanisms of action may achieve, as it has been suggested,Citation26 additive or synergistic promoter effects.
Several promoter drugs have been shown to improve the accumulation of antitumor mAbs by acting on the tumor stroma. Relaxin, for example, is a peptide hormone that degrades the tumor extracellular matrix. It does so by decreasing the synthesis and secretion of interstitial collagens and upregulating matrix metalloproteinase and collagenase expression. Beyer et al.Citation91 showed that hematopoietic stem cell-mediated intratumoral relaxin expression in breast cancer-bearing mice resulted in a decrease of extracellular matrix proteins and enabled control of tumor growth.
Several other approaches that improve the penetration of antitumor agents through remodeling of the tumor stroma have been described. Although not yet demonstrated, these approaches are expected to also improve tumor accumulation of mAbs. The proteoglycan decorin has been used to enhance spreading and tumor tissue penetration of an oncolytic virus.Citation92 The tumoricidal effects of the viruses were lessened, as the binding affinity to collagen was decreased. Also stromal cells are potential targets. Tumor-associated fibroblasts are the primary source of collagen type I, which contributes to decreased drug penetration in tumors. Mice were treated with an oral DNA vaccine targeting fibroblast activation protein, which is specifically overexpressed by fibroblasts in the tumor stroma.Citation62 This vaccine decreased collagen type I expression and led to an up to 70% greater uptake of chemotherapeutic drugs. Vaccinated mice treated with chemotherapy showed a three-fold prolongation in lifespan and marked suppression of tumor growth, with 50% of the animals completely rejecting a tumor cell challenge.
In accordance with the hypothesis that these approaches may improve tumor accumulation of mAbs, is the observation that collagenase overcomes stromal barriers and enhances tumor uptake and penetration of antibodies. In a study using osteosarcoma xenografts, collagenase treatment decreased both IFP and microvascular pressure with differing kinetics such that it transiently increased the transcapillary pressure gradient. This was associated with a ~2-fold increase in the uptake of TP-3, a mAb specific for osteosarcoma.Citation97 In comparison with a nonspecific mAb, TP-3 also exhibited increased penetration and diffused farther away from blood vessels in response to collagenase treatment.
Hyaluronic acid is another important component of the extracellular matrix. Periodic intratumoral injection of hyaluronidase has been shown to greatly increase the tumor-specific uptake of a mAb (TP-3) that targets an osteosarcoma-associated antigen.Citation98
Specific and nonspecific inhibitors of platelet-derived growth factor receptors reduce tumor IFP and enhance uptake of a marker molecule.Citation93 Inhibitory platelet-derived growth factor aptamers and STI571 were shown to enhance the accumulation and antitumor efficacy of the effector drugs paclitaxel (Taxol®), 5-fluoruracil and epothilone B.Citation94,Citation95 Co-administration of the radiolabeled antitumor antibody B72.3 and STI571 yielded long-lasting growth arrest of the human colorectal adenocarcinoma LS174T grown as xenografts in mice.Citation96 Increased antibody accumulation was accompanied by reduction of tumor IFP (by > 50%). The attenuation of IFP increased also the homogeneity of antibody distribution. The increased uptake of radioimmunotherapy into the tumor resulted in > 400% increase in the tumor-absorbed radiation doses and led to growth arrest in mice treated with STI571 and the radiolabeled antibody.
In conclusion, tumor uptake and accumulation of mAbs can be enhanced at different levels of the tumor architecture.
Approaches to improve tumor accumulation of immune cells
As already mentioned, Gennari et al.Citation75 demonstrated that patients undergoing complete or partial remission in response to trastuzumab had higher ADCC and higher in situ infiltration of cytotoxic lymphocytes, including NK cells. This result suggested that, in non-responding patients, the number of tumor-infiltrating leukocytes may not have been sufficient to support effective tumoricidal activity, and that mAb-induced antitumor activity might be potentiated by improving tumor accumulation of immune cells ().
It is important to note that mAbs are endowed with the capacity to induce recruitment of immune cells to support effector functions. Thus, patients treated with trastuzumab have significantly increased numbers of tumor-associated NK cells and increased lymphocyte expression of cytolytic markers compared with controls.Citation59 It is likely that this recruitment is the result of the release of chemokines from FcγR-positive immune cells upon binding of the mAb.Citation99 Thus, the very fact that an antibody binds to an immune cell induces a positive feedback loop that recruits further immune cells able to support antibody-induced effector functions. Nevertheless, the results of Gennari et al.Citation75 suggest that this mechanism may not be sufficient to support effective antitumor activity, and it may be useful to improve immune cell accumulation in tumor tissues.
One strategy to improve immune cell accumulation in tumors involves co-administration of an antitumor mAb with cytokines or immunomodulatory drugs. A clinical trial was performed to test whether IL-2, by increasing FcγRIII-positive NK cell numbers and cytolytic function in vivo, when added to a trastuzumab treatment regimen, could increase antitumor efficacy.Citation100 In vitro assays showed NK cell expansion and trastuzumab-mediated increase of ADCC against breast cancer targets in a HER2-specific manner, but these effects did not correlate with clinical responses. Similar results were reported in a previous clinical trial with trastuzumab and IL-2.Citation101 The combination of IL-2 with rituximab has also been investigated. Two parallel Phase 1 studies evaluated the combined use of rituximab and IL-2 in relapsed or refractory B cell non-Hodgkin lymphoma (NHL).Citation102 A thrice-weekly IL-2 dosing regimen resulted in NK cell expansion that correlated with clinical response.
Granulocyte-macrophage colony-stimulating factor (GM-CSF) is another cytokine that has been used to improve accumulation and activation of immune cells in tumors. In a recent clinical trial, patients with high-risk neuroblastoma received multiple treatment cycles of anti-disialoganglioside GD2 antibody 3F8 plus subcutaneous GM-CSF. GM-CSF–induced granulocyte activation in these patients was associated with improved clinical outcome.Citation103 It was inferred that these activated granulocytes supported 3F8-mediated ADCC of neuroblastoma cells.
One clinical trial investigated the combined use of IL-2 and GM-CSF with the antitumor ch14.18, a chimeric anti-GD2 mAb. Ch14.18 was administered to high-risk neuroblastoma patients in combination with IL-2, GM-CSF and isotretinoin in a randomized Phase 3 study.Citation104 Control patients were given standard therapy alone (isotretinoin). The results showed that immunotherapy with ch14.18, GM-CSF, and IL-2 was associated with a significantly improved outcome compared with standard therapy in this patient population.
Another class of drugs that has been reported to improve the antitumor activity of mAbs by inducing expansion and tumor accumulation of immune cells are the immunomodulatory drugs. Two thalidomide derivatives were tested in combination with rituximab in lymphoma models in mice.Citation105 Synergistic antitumor activity was observed with these drugs and rituximab compared with rituximab monotherapy. The enhancement of rituximab activity by these drugs was abrogated by in vivo depletion of NK cells.
An adenovirus expressing the LIGHT gene was used in a preclinical tumor model.Citation57 Intratumoral expression of LIGHT can attract various immune cells, including substantial numbers of FcγR-positive cells, dendritic cells and T cells.Citation106 Park et al.Citation57 treated mice bearing established tumors with a suboptimal dose of anti-HER2/neu antibody in close combination with intratumoral injection of Ad-LIGHT. Neither suboptimal anti-HER2/neu antibody, nor Ad-LIGHT alone was sufficient to control tumors. In contrast, combination treatment of anti-HER2/neu antibody and Ad-LIGHT resulted in rejection of all transplanted tumors, and no relapse was detected up to 8 weeks after completion of treatment. Notably, this combination treatment generated a specific immune response that was sufficient to specifically protect mice from a lethal tumor rechallenge. These results strongly suggest that improving tumor accumulation of immune cells significantly improves short-term tumoricidal activity of antitumor mAbs, and longer-term adaptive immune responses.
Moschetta et al. have reported on F8-IL2, an immunocytokine that encompasses an antibody derivative specific for the extra domain A of fibronectin and IL-2.Citation107 Curative effects were obtained with paclitaxel administered before the immunocytokine. Administration of paclitaxel increased the uptake of F8 in xenografted melanomas, enhancing tumor perfusion and permeability.Citation107 Paclitaxel also boosted the recruitment of F8-IL2-induced NK cells to the tumor, suggesting a host response as part of the observed therapeutic benefit. In support of this, NK cell depletion impaired the antitumor effect of paclitaxel plus F8-IL2. These results suggest that paclitaxel can promote the uptake of both the antibody moiety (here, an immunocytokine), as well as immune cells. It remains to be established whether similar results can be obtained with other promoter drugs.
Several other approaches have reported on the favorable consequences of promoting constitutive or vaccine-induced antitumor immune responses and on the adoptive transfer of immune cells. In some cases it has also been possible to induce the preferential accumulation of immune cell subpopulations that have antitumor activities, at the expense of subpopulations that have immunosuppressive and tumor-promoting activities. In the following, we will briefly summarize some recent approaches in view of the possibility of testing them in combination with antitumor mAbs.
NGR-TNF is a fusion protein encompassing the inflammatory cytokine TNF and a tumor-vasculature homing peptide that contains the NGR sequence, a ligand of a CD13 isoform expressed by neo-angiogenic vessels.Citation108,Citation109 Systemic administration of picogram doses of NGR-TNF caused rapid and transient modification of the tumor microenvironment, which in turn, enhanced tumor accumulation of either fully-activated endogenous or adoptively transferred T cells.Citation110 As a consequence, NGR-TNF increased the therapeutic efficacy of tumor vaccines, adoptive immunotherapy, and the combination between immunotherapy and chemotherapy.Citation110 A transient reduction of the tumor endothelial barrier function and the selective activation of endothelial cells enhanced T-cell extravasation and infiltration of the tumor tissues.Citation111 A similar compound, RGR-TNF, has been shown to enhance active immunotherapy in experimental pancreatic neuroendocrine tumors.Citation112 RGR-TNF enhanced CD8-positive T-cell infiltration by stabilizing the vascular network and improving vessel perfusion. Vascular remodeling in tumors was mediated, at least in part, by resident macrophages reprogrammed to secrete immune and angiogenic modulators. These recent observations make vessel-targeted TNF a very attractive tool for enhancing tumor infiltration by T-cells after treatment with antibodies, particularly with antibodies whose mechanism of action involves activation of an adaptive immune response.
Both angiogenesis-promoting, and angiogenesis-inhibiting molecules have been shown to enhance accumulation of immune cells. Thus, high levels of angiopoietin-2 were shown to induce leaky vasculatures that facilitate the extravasation of lymphocytes and promote the adhesion of rolling leukocytes to blood vessels.Citation113 Moreover, tumors with overexpressed angiopoietin-2 are more vascularized and have higher numbers of monocytes expressing the angiopoietin receptor Tie-2.Citation114 On the other hand, lymphocyte infiltration in tumors can also be promoted by antiangiogenic compounds, such as anti-VEGF mAbsCitation115 that inhibit formation of new blood vessels and promote vascular normalization.Citation116
Treatment with a combination of drugs that improve tumor accumulation of immune cells yielding a synergistic antitumor effect has been described recently. Thus, combined treatment of mice with anti-CTLA-4 (a T-cell co-inhibitory receptor) and anti-4–1BB (a co-stimulatory receptor) mAbs in the context of a melanoma vaccine promoted synergistic levels of tumor rejection.Citation117 4-1BB activation elicited strong accumulation of CD8-positive T-cells in the tumor and drove the proliferation of these cells, while CTLA-4 blockade did the same for CD4-positive effector T-cells. Anti-4-1BB also depressed tumor accumulation of immunosuppressive Tregs. 4-1BB activation strongly stimulated inflammatory cytokine production in the vaccine and tumor draining lymph nodes and in the tumor itself.
Hyperthermia is an interesting, non-pharmacological approach that has been shown to enhance tumor accumulation of immune cells. Thus, systemic thermal therapy (core temperature elevated to 39.5°C ± 0.5°C for 6 h) activated an IL-6 trans-signaling program in tumor blood that promoted E/P-selectin- and intercellular adhesion molecule-1-dependent extravasation of cytotoxic CD8-positive T cells in tumors.Citation118 A concomitant decrease in tumor accumulation of Tregs was observed.
Approaches to improve the affinity of interaction between antitumor mAbs and immune cells
These approaches rest on the glyco- or protein-engineering of antibody molecules that endow their Fc region with higher affinity for FcγR-positive immune cells. It is the most advanced approach of those described so far, and several engineered antibodies of this kind are currently being investigated in clinical trials ().
A first glyco-engineering approach is based on the expression of antibody molecules lacking fucose.Citation119 This can be achieved by expressing antibodies in fucosyltransferase-negative cell lines.Citation120 Defucosylated antibodies show greatly improved affinity for FcγRIIIA and, as a result, greatly enhanced ADCC.Citation121,Citation122 Importantly, improved binding and enhanced ADCC is observed for FcγRIIIA isoforms that bind IgG molecules with either low or high affinity, with the improvement being greater for low-affinity isoforms. A similar effect has been observed for other engineering approaches that will be discussed. Defucosylated versions of mAbs against several molecular targets are currently in clinical trials.Citation15,Citation123
A second glyco-engineering approach foresees the expression of antibodies in cell lines encoding β(1,4)-N-acetylglucosaminyltransferase III, a glycosyltransferase catalyzing formation of bisected oligosaccharides that endow antibodies with enhanced ADCC.Citation124 A third approach rests on the use of yeast cells in which the genes that are responsible for yeast-specific glycosylation have been knocked out and reconstituted with 14 genes that replicate the sequential steps of human glycosylation.Citation125,Citation126
Glycoengineering approaches have provided anti-CD20 variants with increased affinity for FcγRIIIA and enhanced ADCC. These variants show superior therapeutic efficacy in preclinical lymphoma models. Several glycoengineered anti-CD20 antibodies with enhanced ADCC activity are now in clinical development.Citation12
Protein engineering has also been used to obtain antibody variants with enhanced effector functions. By using computational design algorithms and high-throughput screening, Fc variants with optimized FcγR affinity and specificity were engineered.Citation127 The designed variants displayed > 2 orders of magnitude enhancement of in vitro effector function, in particular ADCC, enabled efficacy against cells expressing low levels of target antigen, and resulted in increased cytotoxicity in an in vivo preclinical model. Recent articles have extensively reviewed both glyco- and protein engineering approaches to improve the affinity of IgG mAbs for FcγRs.Citation128,Citation129
Conclusions
We have proposed here a classification of mAbs according to their mechanism of action, and we reviewed current knowledge on the tumor accumulation of mAbs, immune cells and the interactions that need to take place to generate efficient antitumor activity. We also discussed possible approaches to improve tumor accumulation and interactions between mAbs and immune cells. For the future, there are several points to be considered to achieve further progress in this area.
Permeability of the tumor endothelium is a limiting factor for the uptake of antibodies, and the tumor antibody concentration is always 100–1,000 fold lower than the plasma concentration.Citation67 Thus, to reach tumor cells mAbs need to overcome an important obstacle and it appears of obvious interest to improve extravasation of antitumor mAbs. It is less clear whether improved extravascular accumulation needs to be accompanied also by increased penetration into tumor tissues. In fact, others have proposed a modelCitation130 whereby cytotoxic effector drugs that are administered repeatedly at regular intervals cause ‘peeling’ of increasing numbers of tumor cell layers until tumor regression is observed. We have recently suggested that a similar model can be considered for mAbs that exert tumoricidal activity with or without the contribution of effector cells.Citation131 On the other hand, we have proposed that deep tumor penetration is important to prevent or reverse new or existing drug resistance.Citation131 If this holds true also for mAbs, which suffer from native and acquired resistance similarly to cytotoxic drugs, albeit through different mechanisms, remains to be established.
Another question that needs to be addressed is which patients should be considered for treatment with promoter drugs that improve tumor accumulation of mAbs, immune cells or the affinity between them. Over the short-term, the most likely strategy that will be pursued is to treat patients that are resistant to an individual mAb. In fact, any positive result in these patients will be useful to accelerate the approval of the investigated drug(s). Over the longer term, different options are available. If trials in resistant patients are successful, one possibility is to treat all patients that are candidates for an individual mAb, with a given promoter drug. Of greater interest would be to identify those patients that are most likely to respond successfully to a given promoter drug treatment. Thus, certain patients may require treatment with a promoter drug that improves selectively the uptake and accumulation of a mAb. Other patients may need to be treated to achieve a selective increase of the tumor accumulation of immune cells. Still other patients may have threshold levels of a mAb and immune cells in tumor tissue, but may profit most from a mAb with enhanced affinity for FcγR-positive immune cells. To achieve this goal, predictive biomarkers must be available for each situation. While it appears unrealistic that such a goal will be achieved in the near future, this longer-term opportunity should foster research aimed at identifying predictive biomarkers for the different situations.
Last but not least, the possibility should also be considered to combine approaches that work through different mechanisms of action to verify whether additive or synergistic antitumor effects can be achieved. Thus, it would be interesting to use an antibody with enhanced affinity for FcγRs in combination with a promoter drug that improves its tumor accumulation (). One could also use an antibody with enhanced affinity for FcγRs in combination with a promoter drug that enhances tumor accumulation of immune cells. Given the current state of the art, some of these possibilities are testable in preclinical and clinical studies.
In conclusion, antibody therapy of tumors appears to be sufficiently mature to enter a new stage. So far, antitumor antibodies have achieved encouraging, and in some cases even good therapeutic results. Yet, there is still much room for improvement in enhancing potency, the accumulation of antibodies and immune cells in tumor tissues, and the interaction between these two components. In this review, we have indicated some routes that can be pursued to achieve this goal.
| Abbreviations: | ||
| ADCC | = | antibody-dependent cellular cytotoxicity |
| B-NHL | = | B-cell non-Hodgkin lymphoma |
| CDC | = | complement-dependent cytotoxicity |
| CLL | = | chronic lymphocytic leukemia |
| CRC | = | colorectal cancer |
| CTLA | = | cytotoxic T-lymphocyte antigen |
| DR | = | death receptor |
| EGFR | = | epidermal growth factor |
| FcγR | = | Fcγ receptor |
| FDA | = | Food and Drug Administration |
| GM-CSF | = | granulocyte-macrophage colony-stimulating factor |
| HER | = | human epidermal growth factor receptor |
| IL | = | interleukin |
| IFP | = | interstitial fluid pressure |
| iRGD | = | internalizing arginine-glycine-aspartic acid |
| JO-1 | = | junction opener 1 |
| mAb | = | monoclonal antibody |
| NGR-TNF | = | asparagine-glycine-arginine-TNF |
| NK | = | natural killer |
| PFS | = | progression-free survival |
| TNF | = | tumor necrosis factor |
| TRAIL | = | tumor necrosis factor-related apoptosis-inducing ligand |
| TRAIL-R | = | TRAIL-receptor |
| Treg | = | regulatory T cell |
| VEGF | = | vascular endothelial growth factor |
Acknowledgments
The authors’ work was supported by grants from the Italian Association for Cancer Research (AIRC, Milan, grants numbers 9965, IG11692 and IG9180), the Ministry of Health (Rome), and the Ministry of University and Research (Rome).
Related Research Data
References
- Reichert JM, Rosensweig CJ, Faden LB, Dewitz MC. Monoclonal antibody successes in the clinic. Nat Biotechnol 2005; 23:1073 - 8; http://dx.doi.org/10.1038/nbt0905-1073; PMID: 16151394
- Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer 2012; 12:278 - 87; http://dx.doi.org/10.1038/nrc3236; PMID: 22437872
- Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 2008; 8:592 - 603; http://dx.doi.org/10.1038/nrc2442; PMID: 18650835
- Pohlmann PR, Mayer IA, Mernaugh R. Resistance to trastuzumab in breast cancer. Clin Cancer Res 2009; 15:7479 - 91; http://dx.doi.org/10.1158/1078-0432.CCR-09-0636; PMID: 20008848
- Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med 2000; 6:443 - 6; http://dx.doi.org/10.1038/74704; PMID: 10742152
- Glennie MJ, van de Winkel JGJ. Renaissance of cancer therapeutic antibodies. Drug Discov Today 2003; 8:503 - 10; http://dx.doi.org/10.1016/S1359-6446(03)02714-4; PMID: 12818520
- Weiner GJ. Monoclonal antibody mechanisms of action in cancer. Immunol Res 2007; 39:271 - 8; http://dx.doi.org/10.1007/s12026-007-0073-4; PMID: 17917071
- Chan HTC, Hughes D, French RR, Tutt AL, Walshe CA, Teeling JL, et al. CD20-induced lymphoma cell death is independent of both caspases and its redistribution into triton X-100 insoluble membrane rafts. Cancer Res 2003; 63:5480 - 9; PMID: 14500384
- Ivanov A, Beers SA, Walshe CA, Honeychurch J, Alduaij W, Cox KL, et al. Monoclonal antibodies directed to CD20 and HLA-DR can elicit homotypic adhesion followed by lysosome-mediated cell death in human lymphoma and leukemia cells. J Clin Invest 2009; 119:2143 - 59; PMID: 19620786
- Honeychurch J, Alduaij W, Azizyan M, Cheadle EJ, Pelicano H, Ivanov A, et al. Antibody-induced nonapoptotic cell death in human lymphoma and leukemia cells is mediated through a novel reactive oxygen species-dependent pathway. Blood 2012; 119:3523 - 33; http://dx.doi.org/10.1182/blood-2011-12-395541; PMID: 22354003
- Lim SH, Beers SA, French RR, Johnson PWM, Glennie MJ, Cragg MS. Anti-CD20 monoclonal antibodies: historical and future perspectives. Haematologica 2010; 95:135 - 43; http://dx.doi.org/10.3324/haematol.2008.001628; PMID: 19773256
- Alduaij W, Illidge TM. The future of anti-CD20 monoclonal antibodies: are we making progress?. Blood 2011; 117:2993 - 3001; http://dx.doi.org/10.1182/blood-2010-07-298356; PMID: 21209380
- Link BK, Martin P, Kaminski MS, Goldsmith SJ, Coleman M, Leonard JP. Cyclophosphamide, vincristine, and prednisone followed by tositumomab and iodine-131-tositumomab in patients with untreated low-grade follicular lymphoma: eight-year follow-up of a multicenter phase II study. J Clin Oncol 2010; 28:3035 - 41; http://dx.doi.org/10.1200/JCO.2009.27.8325; PMID: 20458031
- Mössner E, Brünker P, Moser S, Püntener U, Schmidt C, Herter S, et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood 2010; 115:4393 - 402; http://dx.doi.org/10.1182/blood-2009-06-225979; PMID: 20194898
- Salles G, Morschhauser F, Lamy T, Milpied N, Thieblemont C, Tilly H, et al. Phase 1 study results of the type II glycoengineered humanized anti-CD20 monoclonal antibody obinutuzumab (GA101) in B-cell lymphoma patients. Blood 2012; 119:5126 - 32; http://dx.doi.org/10.1182/blood-2012-01-404368; PMID: 22431570
- Buchsbaum DJ, Forero-Torres A, LoBuglio AF. TRAIL-receptor antibodies as a potential cancer treatment. Future Oncol 2007; 3:405 - 9; http://dx.doi.org/10.2217/14796694.3.4.405; PMID: 17661715
- Wilson NS, Yang B, Yang A, Loeser S, Marsters S, Lawrence D, et al. An Fcγ receptor-dependent mechanism drives antibody-mediated target-receptor signaling in cancer cells. Cancer Cell 2011; 19:101 - 13; http://dx.doi.org/10.1016/j.ccr.2010.11.012; PMID: 21251615
- Trarbach T, Moehler M, Heinemann V, Köhne CH, Przyborek M, Schulz C, et al. Phase II trial of mapatumumab, a fully human agonistic monoclonal antibody that targets and activates the tumour necrosis factor apoptosis-inducing ligand receptor-1 (TRAIL-R1), in patients with refractory colorectal cancer. Br J Cancer 2010; 102:506 - 12; http://dx.doi.org/10.1038/sj.bjc.6605507; PMID: 20068564
- Plummer R, Attard G, Pacey S, Li L, Razak A, Perrett R, et al. Phase 1 and pharmacokinetic study of lexatumumab in patients with advanced cancers. Clin Cancer Res 2007; 13:6187 - 94; http://dx.doi.org/10.1158/1078-0432.CCR-07-0950; PMID: 17947486
- Wakelee HA, Patnaik A, Sikic BI, Mita M, Fox NL, Miceli R, et al. Phase I and pharmacokinetic study of lexatumumab (HGS-ETR2) given every 2 weeks in patients with advanced solid tumors. Ann Oncol 2010; 21:376 - 81; http://dx.doi.org/10.1093/annonc/mdp292; PMID: 19633048
- Camidge DR, Herbst RS, Gordon MS, Eckhardt SG, Kurzrock R, Durbin B, et al. A phase I safety and pharmacokinetic study of the death receptor 5 agonistic antibody PRO95780 in patients with advanced malignancies. Clin Cancer Res 2010; 16:1256 - 63; http://dx.doi.org/10.1158/1078-0432.CCR-09-1267; PMID: 20145186
- Herbst RS, Kurzrock R, Hong DS, Valdivieso M, Hsu CP, Goyal L, et al. A first-in-human study of conatumumab in adult patients with advanced solid tumors. Clin Cancer Res 2010; 16:5883 - 91; http://dx.doi.org/10.1158/1078-0432.CCR-10-0631; PMID: 20947515
- Huet H, Schuller A, Li J, Johnson J, Dombrecht B, Meerschaert K, et al. TAS266, a novel tetrameric nanobody agonist targeting death receptor 5 (DR5), elicits superior antitumor efficacy than conventional DR5-targeted approaches. In: Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research; 2012 Mar 31-Apr 4; Chicago, IL. Philadelphia (PA): AACR; Cancer Res 2012; 72(8 Suppl):Abstract nr 3853. doi:1538-7445.AM2012-3853.
- Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004; 350:2335 - 42; http://dx.doi.org/10.1056/NEJMoa032691; PMID: 15175435
- Willett CG, Boucher Y, di Tomaso E, Duda DG, Munn LL, Tong RT, et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med 2004; 10:145 - 7; http://dx.doi.org/10.1038/nm988; PMID: 14745444
- Marcucci F, Corti A. How to improve exposure of tumor cells to drugs: promoter drugs increase tumor uptake and penetration of effector drugs. Adv Drug Deliv Rev 2012; 64:53 - 68; http://dx.doi.org/10.1016/j.addr.2011.09.007; PMID: 21983328
- Yang JC, Haworth L, Sherry RM, Hwu P, Schwartzentruber DJ, Topalian SL, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med 2003; 349:427 - 34; http://dx.doi.org/10.1056/NEJMoa021491; PMID: 12890841
- Fischer C, Jonckx B, Mazzone M, Zacchigna S, Loges S, Pattarini L, et al. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell 2007; 131:463 - 75; http://dx.doi.org/10.1016/j.cell.2007.08.038; PMID: 17981115
- Herbst RS, Hong D, Chap L, Kurzrock R, Jackson E, Silverman JM, et al. Safety, pharmacokinetics, and antitumor activity of AMG 386, a selective angiopoietin inhibitor, in adult patients with advanced solid tumors. J Clin Oncol 2009; 27:3557 - 65; http://dx.doi.org/10.1200/JCO.2008.19.6683; PMID: 19546406
- Huang H, Lai J-Y, Do J, Liu D, Li L, Del Rosario J, et al. Specifically targeting angiopoietin-2 inhibits angiogenesis, Tie2-expressing monocyte infiltration, and tumor growth. Clin Cancer Res 2011; 17:1001 - 11; http://dx.doi.org/10.1158/1078-0432.CCR-10-2317; PMID: 21233403
- Rosen PJ, Sweeney CJ, Park DJ, Beaupre DM, Deng H, Leitch IM, et al. A phase Ib study of AMG 102 in combination with bevacizumab or motesanib in patients with advanced solid tumors. Clin Cancer Res 2010; 16:2677 - 87; http://dx.doi.org/10.1158/1078-0432.CCR-09-2862; PMID: 20406832
- Bagatell R, Herzog CE, Trippett TM, Grippo JF, Cirrincione-Dall G, Fox E, et al. Pharmacokinetically guided phase 1 trial of the IGF-1 receptor antagonist RG1507 in children with recurrent or refractory solid tumors. Clin Cancer Res 2011; 17:611 - 9; http://dx.doi.org/10.1158/1078-0432.CCR-10-1731; PMID: 21127194
- Huang J, Ni J, Liu K, Yu Y, Xie M, Kang R, et al. HMGB1 promotes drug resistance in osteosarcoma. Cancer Res 2012; 72:230 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-11-2001; PMID: 22102692
- Hudis CA. Trastuzumab--mechanism of action and use in clinical practice. N Engl J Med 2007; 357:39 - 51; http://dx.doi.org/10.1056/NEJMra043186; PMID: 17611206
- Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008; 359:1757 - 65; http://dx.doi.org/10.1056/NEJMoa0804385; PMID: 18946061
- Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 2008; 26:1626 - 34; http://dx.doi.org/10.1200/JCO.2007.14.7116; PMID: 18316791
- Roberts PJ, Stinchcombe TE, Der CJ, Socinski MA. Personalized medicine in non-small-cell lung cancer: is KRAS a useful marker in selecting patients for epidermal growth factor receptor-targeted therapy?. J Clin Oncol 2010; 28:4769 - 77; http://dx.doi.org/10.1200/JCO.2009.27.4365; PMID: 20921461
- Varchetta S, Gibelli N, Oliviero B, Nardini E, Gennari R, Gatti G, et al. Elements related to heterogeneity of antibody-dependent cell cytotoxicity in patients under trastuzumab therapy for primary operable breast cancer overexpressing Her2. Cancer Res 2007; 67:11991 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-07-2068; PMID: 18089830
- Bibeau F, Lopez-Crapez E, Di Fiore F, Thezenas S, Ychou M, Blanchard F, et al. Impact of FcγRIIa-FcγRIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J Clin Oncol 2009; 27:1122 - 9; http://dx.doi.org/10.1200/JCO.2008.18.0463; PMID: 19164213
- Elter T, Borchmann P, Schulz H, Reiser M, Trelle S, Schnell R, et al. Fludarabine in combination with alemtuzumab is effective and feasible in patients with relapsed or refractory B-cell chronic lymphocytic leukemia: results of a phase II trial. J Clin Oncol 2005; 23:7024 - 31; http://dx.doi.org/10.1200/JCO.2005.01.9950; PMID: 16145065
- Golay J, Manganini M, Rambaldi A, Introna M. Effect of alemtuzumab on neoplastic B cells. Haematologica 2004; 89:1476 - 83; PMID: 15590398
- Zent CS, Secreto CR, LaPlant BR, Bone ND, Call TG, Shanafelt TD, et al. Direct and complement dependent cytotoxicity in CLL cells from patients with high-risk early-intermediate stage chronic lymphocytic leukemia (CLL) treated with alemtuzumab and rituximab. Leuk Res 2008; 32:1849 - 56; http://dx.doi.org/10.1016/j.leukres.2008.05.014; PMID: 18584865
- Cheson BD. Ofatumumab, a novel anti-CD20 monoclonal antibody for the treatment of B-cell malignancies. J Clin Oncol 2010; 28:3525 - 30; http://dx.doi.org/10.1200/JCO.2010.27.9836; PMID: 20458041
- Lemery SJ, Zhang J, Rothmann MD, Yang J, Earp J, Zhao H, et al. U.S. Food and Drug Administration approval: ofatumumab for the treatment of patients with chronic lymphocytic leukemia refractory to fludarabine and alemtuzumab. Clin Cancer Res 2010; 16:4331 - 8; http://dx.doi.org/10.1158/1078-0432.CCR-10-0570; PMID: 20601446
- Tawara T, Hasegawa K, Sugiura Y, Harada K, Miura T, Hayashi S, et al. Complement activation plays a key role in antibody-induced infusion toxicity in monkeys and rats. J Immunol 2008; 180:2294 - 8; PMID: 18250438
- van der Kolk LE, Grillo-López AJ, Baars JW, Hack CE, van Oers MH. Complement activation plays a key role in the side-effects of rituximab treatment. Br J Haematol 2001; 115:807 - 11; http://dx.doi.org/10.1046/j.1365-2141.2001.03166.x; PMID: 11843813
- Wang SY, Racila E, Taylor RP, Weiner GJ. NK-cell activation and antibody-dependent cellular cytotoxicity induced by rituximab-coated target cells is inhibited by the C3b component of complement. Blood 2008; 111:1456 - 63; http://dx.doi.org/10.1182/blood-2007-02-074716; PMID: 18024795
- Rutkowski MJ, Sughrue ME, Kane AJ, Mills SA, Parsa AT. Cancer and the complement cascade. Mol Cancer Res 2010; 8:1453 - 65; http://dx.doi.org/10.1158/1541-7786.MCR-10-0225; PMID: 20870736
- Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711 - 23; http://dx.doi.org/10.1056/NEJMoa1003466; PMID: 20525992
- Robert C, Thomas L, Bondarenko I, O’Day S, M D JW, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011; 364:2517 - 26; http://dx.doi.org/10.1056/NEJMoa1104621; PMID: 21639810
- Vonderheide RH, LoRusso PM, Khalil M, Gartner EM, Khaira D, Soulieres D, et al. Tremelimumab in combination with exemestane in patients with advanced breast cancer and treatment-associated modulation of inducible costimulator expression on patient T cells. Clin Cancer Res 2010; 16:3485 - 94; http://dx.doi.org/10.1158/1078-0432.CCR-10-0505; PMID: 20479064
- Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol 2010; 28:3167 - 75; http://dx.doi.org/10.1200/JCO.2009.26.7609; PMID: 20516446
- Hilchey SP, Hyrien O, Mosmann TR, Livingstone AM, Friedberg JW, Young F, et al. Rituximab immunotherapy results in the induction of a lymphoma idiotype-specific T-cell response in patients with follicular lymphoma: support for a “vaccinal effect” of rituximab. Blood 2009; 113:3809 - 12; http://dx.doi.org/10.1182/blood-2008-10-185280; PMID: 19196657
- Hainsworth JD, Litchy S, Burris HA 3rd, Scullin DC Jr., Corso SW, Yardley DA, et al. Rituximab as first-line and maintenance therapy for patients with indolent non-hodgkin’s lymphoma. J Clin Oncol 2002; 20:4261 - 7; http://dx.doi.org/10.1200/JCO.2002.08.674; PMID: 12377971
- Abès R, Gélizé E, Fridman WH, Teillaud JL. Long-lasting antitumor protection by anti-CD20 antibody through cellular immune response. Blood 2010; 116:926 - 34; http://dx.doi.org/10.1182/blood-2009-10-248609; PMID: 20439625
- Selenko N, Maidic O, Draxier S, Berer A, Jäger U, Knapp W, et al. CD20 antibody (C2B8)-induced apoptosis of lymphoma cells promotes phagocytosis by dendritic cells and cross-priming of CD8+ cytotoxic T cells. Leukemia 2001; 15:1619 - 26; http://dx.doi.org/10.1038/sj.leu.2402226; PMID: 11587221
- Park SG, Jiang Z, Mortenson ED, Deng L, Radkevich-Brown O, Yang X, et al. The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity. Cancer Cell 2010; 18:160 - 70; http://dx.doi.org/10.1016/j.ccr.2010.06.014; PMID: 20708157
- zum Büschenfelde CM, Hermann C, Schmidt B, Peschel C, Bernhard H. Antihuman epidermal growth factor receptor 2 (HER2) monoclonal antibody trastuzumab enhances cytolytic activity of class I-restricted HER2-specific T lymphocytes against HER2-overexpressing tumor cells. Cancer Res 2002; 62:2244 - 7; PMID: 11956077
- Arnould L, Gelly M, Penault-Llorca F, Benoit L, Bonnetain F, Migeon C, et al. Trastuzumab-based treatment of HER2-positive breast cancer: an antibody-dependent cellular cytotoxicity mechanism?. Br J Cancer 2006; 94:259 - 67; http://dx.doi.org/10.1038/sj.bjc.6602930; PMID: 16404427
- Taylor C, Hershman D, Shah N, Suciu-Foca N, Petrylak DP, Taub R, et al. Augmented HER-2 specific immunity during treatment with trastuzumab and chemotherapy. Clin Cancer Res 2007; 13:5133 - 43; http://dx.doi.org/10.1158/1078-0432.CCR-07-0507; PMID: 17785568
- Zhou Z, Bolontrade MF, Reddy K, Duan X, Guan H, Yu L, et al. Suppression of Ewing’s sarcoma tumor growth, tumor vessel formation, and vasculogenesis following anti vascular endothelial growth factor receptor-2 therapy. Clin Cancer Res 2007; 13:4867 - 73; http://dx.doi.org/10.1158/1078-0432.CCR-07-0133; PMID: 17699866
- Loeffler M, Krüger JA, Niethammer AG, Reisfeld RA. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Invest 2006; 116:1955 - 62; http://dx.doi.org/10.1172/JCI26532; PMID: 16794736
- Schmidt MM, Wittrup KD. A modeling analysis of the effects of molecular size and binding affinity on tumor targeting. Mol Cancer Ther 2009; 8:2861 - 71; http://dx.doi.org/10.1158/1535-7163.MCT-09-0195; PMID: 19825804
- Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler M, Knop S, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 2008; 321:974 - 7; http://dx.doi.org/10.1126/science.1158545; PMID: 18703743
- O’Dwyer PJ, Manola J, Valone FH, Ryan LM, Hines JD, Wadler S, et al. Fluorouracil modulation in colorectal cancer: lack of improvement with N -phosphonoacetyl- l -aspartic acid or oral leucovorin or interferon, but enhanced therapeutic index with weekly 24-hour infusion schedule--an Eastern Cooperative Oncology Group/Cancer and Leukemia Group B Study. J Clin Oncol 2001; 19:2413 - 21; PMID: 11331320
- Ning S, Tian J, Marshall DJ, Knox SJ. Anti-alphav integrin monoclonal antibody intetumumab enhances the efficacy of radiation therapy and reduces metastasis of human cancer xenografts in nude rats. Cancer Res 2010; 70:7591 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-10-1639; PMID: 20841470
- Thurber GM, Schmidt MM, Wittrup KD. Antibody tumor penetration: transport opposed by systemic and antigen-mediated clearance. Adv Drug Deliv Rev 2008; 60:1421 - 34; http://dx.doi.org/10.1016/j.addr.2008.04.012; PMID: 18541331
- Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature 2008; 454:436 - 44; http://dx.doi.org/10.1038/nature07205; PMID: 18650914
- Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell 1994; 76:301 - 14; http://dx.doi.org/10.1016/0092-8674(94)90337-9; PMID: 7507411
- Piali L, Fichtel A, Terpe H-J, Imhof BA, Gisler RH. Endothelial vascular cell adhesion molecule 1 expression is suppressed by melanoma and carcinoma. J Exp Med 1995; 181:811 - 6; http://dx.doi.org/10.1084/jem.181.2.811; PMID: 7530765
- Wu NZ, Klitzman B, Dodge R, Dewhirst MW. Diminished leukocyte-endothelium interaction in tumor microvessels. Cancer Res 1992; 52:4265 - 8; PMID: 1638539
- Nooijen PTGA, Westphal JR, Eggermont AMM, Schalkwijk C, Max R, de Waal RMW, et al. Endothelial P-selectin expression is reduced in advanced primary melanoma and melanoma metastasis. Am J Pathol 1998; 152:679 - 82; PMID: 9502409
- Zhang H, Issekutz AC. Down-modulation of monocyte transendothelial migration and endothelial adhesion molecule expression by fibroblast growth factor: reversal by the anti-angiogenic agent SU6668. Am J Pathol 2002; 160:2219 - 30; http://dx.doi.org/10.1016/S0002-9440(10)61169-8; PMID: 12057924
- Pagès F, Galon J, Dieu-Nosjean M-C, Tartour E, Sautès-Fridman C, Fridman W-H. Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene 2010; 29:1093 - 102; http://dx.doi.org/10.1038/onc.2009.416; PMID: 19946335
- Gennari R, Menard S, Fagnoni F, Ponchio L, Scelsi M, Tagliabue E, et al. Pilot study of the mechanism of action of preoperative trastuzumab in patients with primary operable breast tumors overexpressing HER2. Clin Cancer Res 2004; 10:5650 - 5; http://dx.doi.org/10.1158/1078-0432.CCR-04-0225; PMID: 15355889
- Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 2002; 99:754 - 8; http://dx.doi.org/10.1182/blood.V99.3.754; PMID: 11806974
- Weng W-K, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol 2003; 21:3940 - 7; http://dx.doi.org/10.1200/JCO.2003.05.013; PMID: 12975461
- Treon SP, Hansen M, Branagan AR, Verselis S, Emmanouilides C, Kimby E, et al. Polymorphisms in FcgammaRIIIA (CD16) receptor expression are associated with clinical response to rituximab in Waldenström’s macroglobulinemia. J Clin Oncol 2005; 23:474 - 81; http://dx.doi.org/10.1200/JCO.2005.06.059; PMID: 15659493
- Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol 2008; 26:1789 - 96; http://dx.doi.org/10.1200/JCO.2007.14.8957; PMID: 18347005
- Pander J, Heusinkveld M, van der Straaten T, Jordanova ES, Baak-Pablo R, Gelderblom H, et al. Activation of tumor-promoting type 2 macrophages by EGFR-targeting antibody cetuximab. Clin Cancer Res 2011; 17:5668 - 73; http://dx.doi.org/10.1158/1078-0432.CCR-11-0239; PMID: 21788356
- Tol J, Koopman M, Cats A, Rodenburg CJ, Creemers GJ, Schrama JG, et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med 2009; 360:563 - 72; http://dx.doi.org/10.1056/NEJMoa0808268; PMID: 19196673
- Bonertz A, Weitz J, Pietsch D-HK, Rahbari NN, Schlude C, Ge Y, et al. Antigen-specific Tregs control T cell responses against a limited repertoire of tumor antigens in patients with colorectal carcinoma. J Clin Invest 2009; 119:3311 - 21; PMID: 19809157
- Liotta F, Gacci M, Frosali F, Querci V, Vittori G, Lapini A, et al. Frequency of regulatory T cells in peripheral blood and in tumour-infiltrating lymphocytes correlates with poor prognosis in renal cell carcinoma. BJU Int 2011; 107:1500 - 6; http://dx.doi.org/10.1111/j.1464-410X.2010.09555.x; PMID: 20735382
- Netti PA, Hamberg LM, Babich JW, Kierstead D, Graham W, Hunter GJ, et al. Enhancement of fluid filtration across tumor vessels: implication for delivery of macromolecules. Proc Natl Acad Sci U S A 1999; 96:3137 - 42; http://dx.doi.org/10.1073/pnas.96.6.3137; PMID: 10077650
- Nakahara T, Norberg SM, Shalinsky DR, Hu-Lowe DD, McDonald DM. Effect of inhibition of vascular endothelial growth factor signaling on distribution of extravasated antibodies in tumors. Cancer Res 2006; 66:1434 - 45; http://dx.doi.org/10.1158/0008-5472.CAN-05-0923; PMID: 16452199
- Khawli LA, Hu P, Epstein AL. NHS76/PEP2, a fully human vasopermeability-enhancing agent to increase the uptake and efficacy of cancer chemotherapy. Clin Cancer Res 2005; 11:3084 - 93; http://dx.doi.org/10.1158/1078-0432.CCR-04-2310; PMID: 15837764
- Khawli LA, Miller GK, Epstein AL. Effect of seven new vasoactive immunoconjugates on the enhancement of monoclonal antibody uptake in tumors. Cancer 1994; 73:Suppl 824 - 31; http://dx.doi.org/10.1002/1097-0142(19940201)73:3+<824::AID-CNCR2820731312>3.0.CO;2-V; PMID: 8306266
- Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Girard OM, et al. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell 2009; 16:510 - 20; http://dx.doi.org/10.1016/j.ccr.2009.10.013; PMID: 19962669
- Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Greenwald DR, et al. Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science 2010; 328:1031 - 5; http://dx.doi.org/10.1126/science.1183057; PMID: 20378772
- Beyer I, van Rensburg R, Strauss R, Li ZY, Wang H, Persson J, et al. Epithelial junction opener JO-1 improves monoclonal antibody therapy of cancer. Cancer Res 2011; 71:7080 - 90; http://dx.doi.org/10.1158/0008-5472.CAN-11-2009; PMID: 21990319
- Beyer I, Li Z, Persson J, Liu Y, van Rensburg R, Yumul R, et al. Controlled extracellular matrix degradation in breast cancer tumors improves therapy by trastuzumab. Mol Ther 2011; 19:479 - 89; http://dx.doi.org/10.1038/mt.2010.256; PMID: 21081901
- Choi IK, Lee YS, Yoo JY, Yoon AR, Kim H, Kim DS, et al. Effect of decorin on overcoming the extracellular matrix barrier for oncolytic virotherapy. Gene Ther 2010; 17:190 - 201; http://dx.doi.org/10.1038/gt.2009.142; PMID: 19907500
- Pietras K, Östman A, Sjöquist M, Buchdunger E, Reed RK, Heldin C-H, et al. Inhibition of platelet-derived growth factor receptors reduces interstitial hypertension and increases transcapillary transport in tumors. Cancer Res 2001; 61:2929 - 34; PMID: 11306470
- Pietras K, Rubin K, Sjöblom T, Buchdunger E, Sjöquist M, Heldin C-H, et al. Inhibition of PDGF receptor signaling in tumor stroma enhances antitumor effect of chemotherapy. Cancer Res 2002; 62:5476 - 84; PMID: 12359756
- Pietras K, Stumm M, Hubert M, Buchdunger E, Rubin K, Heldin C-H, et al. STI571 enhances the therapeutic index of epothilone B by a tumor-selective increase of drug uptake. Clin Cancer Res 2003; 9:3779 - 87; PMID: 14506171
- Baranowska-Kortylewicz J, Abe M, Pietras K, Kortylewicz ZP, Kurizaki T, Nearman J, et al. Effect of platelet-derived growth factor receptor-β inhibition with STI571 on radioimmunotherapy. Cancer Res 2005; 65:7824 - 31; PMID: 16140951
- Eikenes L, Bruland OS, Brekken C, Davies CdeL. Collagenase increases the transcapillary pressure gradient and improves the uptake and distribution of monoclonal antibodies in human osteosarcoma xenografts. Cancer Res 2004; 64:4768 - 73; http://dx.doi.org/10.1158/0008-5472.CAN-03-1472; PMID: 15256445
- Brekken C, Hjelstuen MH, Bruland OS, de Lange Davies C. Hyaluronidase-induced periodic modulation of the interstitial fluid pressure increases selective antibody uptake in human osteosarcoma xenografts. Anticancer Res 2000; 20:5B 3513 - 9; PMID: 11131655
- Roda JM, Parihar R, Magro C, Nuovo GJ, Tridandapani S, Carson WE 3rd. Natural killer cells produce T cell-recruiting chemokines in response to antibody-coated tumor cells. Cancer Res 2006; 66:517 - 26; http://dx.doi.org/10.1158/0008-5472.CAN-05-2429; PMID: 16397268
- Repka T, Chiorean EG, Gay J, Herwig KE, Kohl VK, Yee D, et al. Trastuzumab and interleukin-2 in HER2-positive metastatic breast cancer: a pilot study. Clin Cancer Res 2003; 9:2440 - 6; PMID: 12855616
- Fleming GF, Meropol NJ, Rosner GL, Hollis DR, Carson WE 3rd, Caligiuri M, et al. A phase I trial of escalating doses of trastuzumab combined with daily subcutaneous interleukin 2: report of cancer and leukemia group B 9661. Clin Cancer Res 2002; 8:3718 - 27; PMID: 12473581
- Gluck WL, Hurst D, Yuen A, Levine AM, Dayton MA, Gockerman JP, et al. Phase I studies of interleukin (IL)-2 and rituximab in B-cell non-hodgkin’s lymphoma: IL-2 mediated natural killer cell expansion correlations with clinical response. Clin Cancer Res 2004; 10:2253 - 64; http://dx.doi.org/10.1158/1078-0432.CCR-1087-3; PMID: 15073100
- Cheung IY, Hsu K, Cheung N-KV. Activation of peripheral-blood granulocytes is strongly correlated with patient outcome after immunotherapy with anti-GD2 monoclonal antibody and granulocyte-macrophage colony-stimulating factor. J Clin Oncol 2012; 30:426 - 32; http://dx.doi.org/10.1200/JCO.2011.37.6236; PMID: 22203761
- Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al, Children’s Oncology Group. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med 2010; 363:1324 - 34; http://dx.doi.org/10.1056/NEJMoa0911123; PMID: 20879881
- Hernandez-Ilizaliturri FJ, Reddy N, Holkova B, Ottman E, Czuczman MS. Immunomodulatory drug CC-5013 or CC-4047 and rituximab enhance antitumor activity in a severe combined immunodeficient mouse lymphoma model. Clin Cancer Res 2005; 11:5984 - 92; http://dx.doi.org/10.1158/1078-0432.CCR-05-0577; PMID: 16115943
- Yu P, Lee Y, Liu W, Chin RK, Wang J, Wang Y, et al. Priming of naive T cells inside tumors leads to eradication of established tumors. Nat Immunol 2004; 5:141 - 9; http://dx.doi.org/10.1038/ni1029; PMID: 14704792
- Moschetta M, Pretto F, Berndt A, Galler K, Richter P, Bassi A, et al. Paclitaxel enhances therapeutic efficacy of the F8-IL2 immunocytokine to EDA-fibronectin-positive metastatic human melanoma xenografts. Cancer Res 2012; 72:1814 - 24; http://dx.doi.org/10.1158/0008-5472.CAN-11-1919; PMID: 22392081
- Curnis F, Sacchi A, Borgna L, Magni F, Gasparri A, Corti A. Enhancement of tumor necrosis factor α antitumor immunotherapeutic properties by targeted delivery to aminopeptidase N (CD13). Nat Biotechnol 2000; 18:1185 - 90; http://dx.doi.org/10.1038/81183; PMID: 11062439
- Curnis F, Sacchi A, Corti A. Improving chemotherapeutic drug penetration in tumors by vascular targeting and barrier alteration. J Clin Invest 2002; 110:475 - 82; PMID: 12189241
- Calcinotto A, Grioni M, Jachetti E, Curnis F, Mondino A, Parmiani G, et al. Targeting TNF-α to neoangiogenic vessels enhances lymphocyte infiltration in tumors and increases the therapeutic potential of immunotherapy. J Immunol 2012; 188:2687 - 94; http://dx.doi.org/10.4049/jimmunol.1101877; PMID: 22323546
- Bellone M, Calcinotto A, Corti A. Won’t you come on in? How to favor lymphocyte infiltration in tumors. OncoImmunology 2012; 1:98 - 9; http://dx.doi.org/10.4161/onci.20213
- Johansson A, Hamzah J, Payne CJ, Ganss R. Tumor-targeted TNFα stabilizes tumor vessels and enhances active immunotherapy. Proc Natl Acad Sci U S A 2012; 109:7841 - 6; http://dx.doi.org/10.1073/pnas.1118296109; PMID: 22547817
- Fiedler U, Reiss Y, Scharpfenecker M, Grunow V, Koidl S, Thurston G, et al. Angiopoietin-2 sensitizes endothelial cells to TNF-alpha and has a crucial role in the induction of inflammation. Nat Med 2006; 12:235 - 9; http://dx.doi.org/10.1038/nm1351; PMID: 16462802
- Coffelt SB, Tal AO, Scholz A, De Palma M, Patel S, Urbich C, et al. Angiopoietin-2 regulates gene expression in TIE2-expressing monocytes and augments their inherent proangiogenic functions. Cancer Res 2010; 70:5270 - 80; http://dx.doi.org/10.1158/0008-5472.CAN-10-0012; PMID: 20530679
- Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res 2010; 70:6171 - 80; http://dx.doi.org/10.1158/0008-5472.CAN-10-0153; PMID: 20631075
- Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 2005; 307:58 - 62; http://dx.doi.org/10.1126/science.1104819; PMID: 15637262
- Curran MA, Kim M, Montalvo W, Al-Shamkhani A, Allison JP. Combination CTLA-4 blockade and 4-1BB activation enhances tumor rejection by increasing T-cell infiltration, proliferation, and cytokine production. PLoS One 2011; 6:e19499; http://dx.doi.org/10.1371/journal.pone.0019499; PMID: 21559358
- Fisher DT, Chen Q, Skitzki JJ, Muhitch JB, Zhou L, Appenheimer MM, et al. IL-6 trans-signaling licenses mouse and human tumor microvascular gateways for trafficking of cytotoxic T cells. J Clin Invest 2011; 121:3846 - 59; http://dx.doi.org/10.1172/JCI44952; PMID: 21926464
- Shinkawa T, Nakamura K, Yamane N, Shoji-Hosaka E, Kanda Y, Sakurada M, et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem 2003; 278:3466 - 73; http://dx.doi.org/10.1074/jbc.M210665200; PMID: 12427744
- Yamane-Ohnuki N, Kinoshita S, Inoue-Urakubo M, Kusunoki M, Iida S, Nakano R, et al. Establishment of FUT8 knockout Chinese hamster ovary cells: an ideal host cell line for producing completely defucosylated antibodies with enhanced antibody-dependent cellular cytotoxicity. Biotechnol Bioeng 2004; 87:614 - 22; http://dx.doi.org/10.1002/bit.20151; PMID: 15352059
- Okazaki A, Shoji-Hosaka E, Nakamura K, Wakitani M, Uchida K, Kakita S, et al. Fucose depletion from human IgG1 oligosaccharide enhances binding enthalpy and association rate between IgG1 and FcgammaRIIIa. J Mol Biol 2004; 336:1239 - 49; http://dx.doi.org/10.1016/j.jmb.2004.01.007; PMID: 15037082
- Junttila TT, Parsons K, Olsson C, Lu Y, Xin Y, Theriault J, et al. Superior in vivo efficacy of afucosylated trastuzumab in the treatment of HER2-amplified breast cancer. Cancer Res 2010; 70:4481 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-09-3704; PMID: 20484044
- Ishida T, Joh T, Uike N, Yamamoto K, Utsunomiya A, Yoshida S, et al. Defucosylated anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia-lymphoma: a multicenter phase II study. J Clin Oncol 2012; 30:837 - 42; http://dx.doi.org/10.1200/JCO.2011.37.3472; PMID: 22312108
- Umaña P, Jean-Mairet J, Moudry R, Amstutz H, Bailey JE. Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity. Nat Biotechnol 1999; 17:176 - 80; http://dx.doi.org/10.1038/6179; PMID: 10052355
- Hamilton SR, Davidson RC, Sethuraman N, Nett JH, Jiang Y, Rios S, et al. Humanization of yeast to produce complex terminally sialylated glycoproteins. Science 2006; 313:1441 - 3; http://dx.doi.org/10.1126/science.1130256; PMID: 16960007
- Li H, Sethuraman N, Stadheim TA, Zha D, Prinz B, Ballew N, et al. Optimization of humanized IgGs in glycoengineered Pichia pastoris. Nat Biotechnol 2006; 24:210 - 5; http://dx.doi.org/10.1038/nbt1178; PMID: 16429149
- Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci U S A 2006; 103:4005 - 10; http://dx.doi.org/10.1073/pnas.0508123103; PMID: 16537476
- Desjarlais JR, Lazar GA. Modulation of antibody effector function. Exp Cell Res 2011; 317:1278 - 85; http://dx.doi.org/10.1016/j.yexcr.2011.03.018; PMID: 21459085
- Hogarth PM, Pietersz GA. Fc receptor-targeted therapies for the treatment of inflammation, cancer and beyond. Nat Rev Drug Discov 2012; 11:311 - 31; http://dx.doi.org/10.1038/nrd2909; PMID: 22460124
- Trédan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst 2007; 99:1441 - 54; http://dx.doi.org/10.1093/jnci/djm135; PMID: 17895480
- Marcucci F, Corti A. Improving drug penetration to curb tumor drug resistance. Drug Discov Today 2012; 17:1139 - 46; http://dx.doi.org/10.1016/j.drudis.2012.06.004; PMID: 22706017