Abstract
TRU-016 is a SMIPTM (monospecific protein therapeutic) molecule against the tetraspanin transmembrane family protein CD37 that is currently in Phase 2 trials in Chronic Lymphocytic Leukemia (CLL) and Non-Hodgkin Lymphoma (NHL). In an attempt to enhance the ADCC function of SMIP-016, the chimeric version of TRU-016, SMIP-016GV was engineered with a modification in a glycosylation site in the Fc domain. The wild-type and glycovariant SMIP proteins mediate comparable Type I antibody-like direct cytotoxicity in the presence of anti-human Fc crosslinker and show a similar tyrosine phosphorylation pattern post-treatment. However, NK cells stimulated with the SMIP-016GV exhibit enhanced activation and release 3-fold more interferon-γ compared with SMIP-016. SMIP-016GV shows enhanced ADCC function against cells expressing CD37 with NK cell effectors derived from both normal and CLL-affected individuals. Enhanced ADCC is observed against CLL cells and is sustained at concentrations of SMIP-016GV as low at 5E−6 µg/mL on cells expressing minimal CD37 antigen. In support of the biological relevance of this, SMIP-016GV mediates effective ADCC against primary acute lymphoblastic leukemia (ALL) cells with low surface expression of CD37. Collectively, these data suggest potential use of the novel therapeutic agent SMIP-016GV with enhanced effector function for B cell malignancies, including CLL and ALL therapy.
Keywords: :
Introduction
CD37 is a tetraspanin transmembrane family protein that is expressed on the surface of mature, immunoglobulin-producing B cellsCitation1 but not in CD10+, CD34+ and CD34- B cell precursors found in the bone marrow. Surface CD37 expression becomes strong in CD10- mature B-lymphocytes and its expression further increases as the B-lymphocytes continue to mature and move into the lymph nodes and peripheral blood. Finally, surface CD37 expression is lost in terminally differentiated plasma B cells.Citation2,Citation3 CD37 is also highly expressed on the surface of transformed mature B cell leukemia and lymphoma cells but not on myeloma cells.Citation3 CD37 is dimly expressed on T cells, monocytes and granulocytes and is not expressed on the surface of natural killer (NK) cells, platelet and erythrocytes.Citation1,Citation2 This limited expression makes it an ideal therapeutic target in B cell malignanciesCitation2 such as chronic lymphocytic leukemia (CLL) and acute lymphoblastic leukemia (ALL).
CD37 was first examined as a potential therapeutic target in the late 1980s. Radio-labeled mouse monoclonal antibodies against CD37 were studied in B cell lymphoma patients and were shown to produce anti-tumor responses.Citation4-Citation6 However, due to the perceived targeting potential of CD20, CD37 as a therapeutic target was not further developed until recently with an engineered monoclonal antibody mAb 37.1 that has been shown to be effective in preclinical models of B cell malignancies.Citation7 Furthermore, our laboratory has shown that a novel protein therapeutic directed against CD37, SMIP-016 induces more apoptosis in CLL B cells than rituximabCitation8 in vitro, when it is used alongside an anti-human Fc crosslinking antibody. Its mechanism of action is through a caspase independent pathway, which suggests it can be used in combination therapy with other caspase activation-dependent cytotoxic antibody therapies or chemotherapeutic agents, such as fludarabine. The direct cytotoxic effect of SMIP-016 on CLL B cells is proportional to the amount of CD37 present on the cell surface, making it a highly selective therapy toward malignant B cells. Furthermore, SMIP-016 showed potent anti-lymphoma activity in a Raji/SCID xenograft mouse model. TRU-016, a humanized anti-CD37 SMIP molecule derived from SMIP-016, is currently in Phase 2 clinical trials and showing single agent activity in CLL.Citation9
In addition to direct killing, a major potential mechanism involved in TRU-016 tumor elimination is ADCC. SMIP-016 induced NK cells mediated antibody-dependent cellular cytotoxicity (ADCC) both in vitro and in vivo.Citation8 Monoclonal antibodies with bisected, complex, non-fucosylated oligosaccharides attached to the asparagine 297 residue in the CH2 region, bind with increased affinity to FcγRIIIa.Citation10 This glycoform engineering has been shown to enhance ADCCCitation11 through cells bearing FcγRIIIa, an important component in how monoclonal antibodies are clinically effective.Citation12 For example, afucosylated anti-CD20 antibodies show higher B cell depletion than their fucosylated counterpart by reaching saturated ADCC levels at lower concentrations and through improved FcγRIIIa binding.Citation13 In addition, it has been reported that antibodies lacking the core fucose in Fc oligosaccharides elicit high ADCC responses by two mechanisms.Citation14 On the effector cell side, afucosylated anti-CD20 antibodies were less inhibited by human plasma IgG. On the target cells, cells treated with non-fucosylated anti-CD20 antibodies showed markedly stronger binding to NK cells than fucosylated anti-CD20.Citation14
Due to the success of the parent compound SMIP-016, we sought to determine if modifying the Fc oligosaccharides of a SMIP protein would enhance its activity. Herein, we describe a second generation anti-CD37 SMIP molecule, SMIP-016GV, with an afucosylated Fc receptor binding region designed for enhanced effector function. Our data demonstrates SMIP-016GV has enhanced effector function with NK cells and monocyte derived macrophages (MDM), making it an exciting novel CD37-targeted peptide therapeutic for B cell malignancies.
Results
Engineering and characterization of glycovariant SMIP protein
SMIP-016GV was generated by treating a DG44 CHO cell line transfected with SMIP-016 cDNA with castanospermine (CS), a potent plant alkaloid inhibitor of glucosidases involved in N-linked processing of glycoproteins such as glucosidase I.Citation15 LC/MS glycoprofiling analysis of the products demonstrated that the observed mass species in both CS treated (SMIP-016GV) and untreated (SMIP-016) preparations were consistent with the expected amino acid sequence with a typical, heterogeneous mammalian glycosylation pattern. A slight difference in amino acid composition was noted between the two preparations with a greater proportion of the SMIP-016GV monomers found to lack the C-terminal lysine compared with the SMIP-016 monomers (89% vs 72%). With respect to the N-linked glycoforms, there was a near total reversal in the percentage of fucosylated and non-fucosylated glycan species between the two preparations (). For SMIP-016, the fucosylated complex glycoforms (94% of total) were composed of 80% G0F and 20% G1F species, with the remaining 6% of glycans being represented by non-fucosylated high hexose forms of (Man)3 + (Man)3(GlcNAc)2 composition. In the case of SMIP-016GV, only 5% of the glycans were of fucosylated complex form (all G0F), while the remaining (95%) were of non-fucosylated high hexose form comprised mostly of (Hex)7 + (Man)3(GlcNAc)2 glycans with the remainder of (Man)5 + (Man)3(GlcNAc)2 composition.
Table 1. Comparison of the N-linked Glycan Composition of SMIP-016 vs. SMIP-016GV
SMIP-016GV and SMIP-016 have equal binding affinity for CD37
To test whether the glycovariant form of SMIP-016 had similar antigen binding affinity as the parent compound, both SMIP proteins were bound to Daudi cells, a human B-lymphoblastoid cell line derived from Burkitt lymphoma that expresses high levels of CD37. A FITC-labeled anti-huIgG antibody was added to bind to the SMIP proteins and the bound fluorescence was measured by flow cytometry. As shown in the , SMIP-016 and its CS generated glycovariant SMIP-016GV yielded virtually identical binding profiles on CD37+ Daudi cells, demonstrating that alteration of the N-linked glycosylation of SMIP-016GV had no effect on target antigen binding of the protein.
Figure 1. Characterization of SMIP-016GV molecule. (A) Binding affinity of SMIP-016 and SMIP-016GV to target antigen CD37. (B) Enhanced binding of SMIP-016 GV with both high and low affinity soluble FcγRIII.
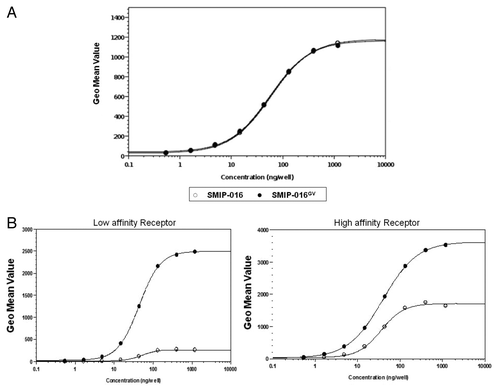
The Fc portion of SMIP-016GV has enhanced affinity for low and high affinity soluble FcγRIII
The single nucleotide polymorphisms (SNP) in FcγRIIIa resulting in either a valine (V) or phenylalanine (F) at position 158 results in low or high affinity receptors in humans, and this has been implicated to be indicative of response to rituximab immunotherapy.Citation16-Citation18 This SNP, however, has shown no correlation to response in CLL.Citation19,Citation20 To test the binding affinities to these receptors, SMIP-016 and SMIP-016GV were bound to CD37+ Daudi cells and their ability to bind to soluble versions of low and high affinity FcγRIIIa was tested by flow cytometry. SMIP-016GV demonstrated a 9.3-fold increase in binding over that observed with SMIP-016 for the FcγRIII low affinity receptor and a 2.1-fold increase in affinity for the high affinity FcγRIII receptor, respectively ().
SMIP-016 and SMIP-016GV show comparable levels of cytotoxicity through similar mechanisms
Direct cytotoxicity is one of the mechanisms of killing mediated by SMIP-016. To test whether the changes in the glycosylation pattern on this SMIP protein influenced its potential for direct cytotoxicity, we tested SMIP-016 and SMIP016GV on isolated primary CLL cells. SMIP-016GV mediates direct cytotoxicity in primary CLL B cells with goat anti-human Fc crosslinker in comparable levels to those seen with the parent (unmodified) protein (). When cells are drugged for 48 h along with anti-human Fc crosslinker, SMIP-016GV decreased cell viability by about 60%, which was significantly higher than rituximab (p < 0.0001) but comparable to SMIP-016 (p = 0.708; ). Therefore, both SMIP-016 and its glycovariant SMIP-016GV show similar levels of direct cytotoxicity at the same concentration and time points, suggesting they have the similar potential for signaling of direct cytotoxicity in cells expressing CD37 on their surface. Furthermore, the SMIP proteins initiate apoptosis in a Type I antibody-like manner () compared with a Type II anti-CD20 antibody.
Figure 2. SMIP-016 and SMIP-016GV show similar methods of direct cytotoxicity. (A) SMIP-016 and SMIP-016GV show comparable levels of direct cytotoxicity in primary CLL B cells (n = 11) (p = 0.0708). (B) SMIP-016 and SMIP-016GV kills cells in a Type I antibody-like manner. Microscopy images of CLL B cells (i) untreated (ii) Type II antibody (iii) SMIP-016 or (iv) SMIP-016GV for 16 h (n = 3). (C) Both SMIP-016 and SMIP-016GV induced a similar pattern of tyrosine phosphorylation of cellular proteins at 65 kDa. CLL cells were treated with 5μg/mL of SMIP-016 or SMIP-016GV with anti-Fc cross-linking antibody in PBS for 10 min and phospho tyrosine proteins were detected by western blot analysis using anti-phosphotyrosine antibody 4G10 (n = 6). (D) SMIP-016 and SMIP-016GV do not initiate complement dependent cytotoxicity in CLL B cells (n = 6).
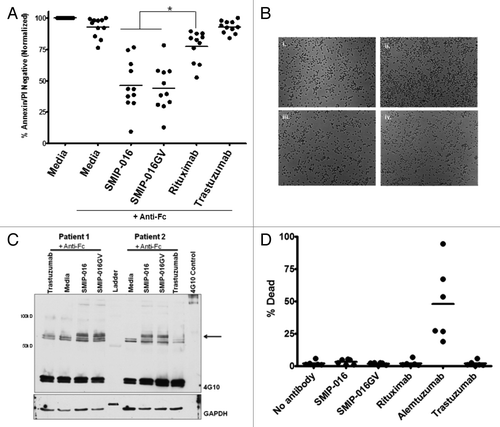
Zhao et al. previously reported that SMIP-016 works in a novel caspase-independent pathway and that in vitro stimulation of CLL B cells with SMIP-016 along with an anti-Fc crosslinker showed activation of protein tyrosine phosphorylation.Citation8 This was subsequently shown to involve SHP1 and a complex array of signaling via the ITIM motif of CD37.Citation21 To test whether the SMIP-016GV was signaling within the cells via the same molecular pathways, we stimulated CLL cells with SMIP-016 or SMIP-016GV along with anti-human Fc crosslinker. Both SMIP-016 and SMIP-016GV show similar patterns of increased tyrosine phosphorylation after 10 min of in vitro stimulation of CLL cells (). In particular, western blot analysis of cellular lysates with an anti-phosphotyrosine antibody revealed a predominant protein at ~65 kD in cells treated with SMIP-016 or SMIP-016GV, an observation previously described by our groupCitation8,Citation21 and determined to be SHP-1 and Lyn phosphorylation. The identical binding affinity to the target antigen CD37 along with the equivalent amount of cytotoxicity against CLL cells via identical phosphorylation of downstream proteins collectively suggests that the parent compound SMIP-016 and the glycovariant SMIP-016GV are working through similar molecular pathways in CLL cells to cause direct cytotoxicity, as previously described.Citation21
Next, we tested if SMIP-016GV had any complement initiating capabilities. We have shown that the SMIP-016 is unable to mediate complement-dependent cytotoxicity.Citation8 This is also seen with the SMIP-016GV (). The alteration of the Fc glycosylation pattern does not affect the SMIP’s inability to initiate complement.
SMIP-016GV shows enhanced phagocytosis by monocyte-derived macrophages
We reported previously that NK cells, and not monocytes, were the major effector population mediating cytotoxicity with SMIP-016,Citation8 but the importance of the role of monocytes and macrophages in the function of therapeutic antibodies is emerging,Citation22-Citation25 especially in anti-CD20 therapy. Similar to what we had seen previously, we saw low ADCC function with SMIP-016 and SMIP-016GV in monocytes against CLL B cells (data not shown).
In addition, plate bound SMIP proteins were not efficient at inducing cytokines in peripheral blood monocytes. TNF production by monocytes stimulated with SMIP-016 and SMIP-016GV for 24 h was decreased compared with non-specific IgG or rituximab (). Furthermore, monocyte-derived macrophages (MDM) stimulated similarly for 24 h also had lower TNF production (data not shown)
Figure 3. SMIP-016GV can mediate cytotoxicity through effector cells. (A) Intact antibodies, F(ab)’2 or SMIP bound to plates induced TNF from peripheral blood monocytes (n = 4). (B) SMIP-016GV mediates enhanced antibody-dependent cellular phagocytosis (ADCP) by monocyte-derived macrophages (MDM) of primary CLL cells compared with SMIP-016, as measured by flow cytometry (n = 3) (p < 0.05). (C) Induction of CD107a on the surface of CD56+ NK cells by SMIP-016GV, as measured by flow cytometry (n = 6). (D) Enhanced induction of interferon gamma from NK cells by SMIP-016GV compared with SMIP-016 (p = 0.009) (n = 4). (E, F) ADCC with normal donor NK cell effectors and Raji (E) or 697 (F) targets. SMIP-016GV shows enhanced ADCC function in the CD37 expressing Raji cells compared with SMIP-016 (p = 0.0013 of average of all E:T ratio > 0), but no activity in the 697 cells (n = 3).
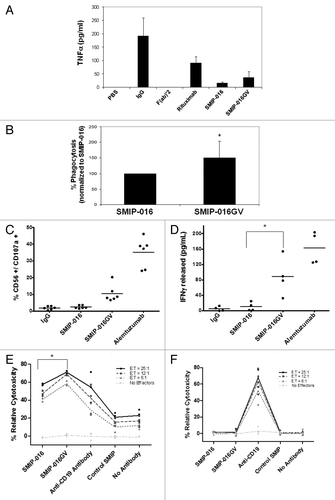
To study if SMIP-016GV could elicit a cytotoxic response from MDM, which express FcγRIIIa,Citation22,Citation26 we tested it in antibody-dependent cellular phagocytosis (ADCP) assays using MDM from normal donors. Primary CLL samples were coated with either SMIP-016 or SMIP-016GV and subsequently co-incubated with MDM. The glycovariant SMIP protein mediates significantly more ADCP than the parent SMIP protein (p < 0.05; ).
SMIP-016GV enhances NK cell activation and ADCC toward CD37-expressing targets
Previous studies have shown that changing the glycosylation pattern of the Fc portion of antibodies can influence their ability to elicit effector function by enhancing their binding affinity to FcγRIIIa on human immune cells.Citation14,Citation27 We saw similar enhanced affinity to FcγRIIIa with SMIP-016GV (). In addition, compared with SMIP-016, SMIP-016GV mediated enhanced activation of NK cells as revealed by the increased expression of surface CD107a following stimulation with the SMIP proteins or IgG control antibodies by 8-fold (p = 0.012) () and by the 16-fold increase in interferon (IFN)-γ production (p = 0.009, ).
To determine whether the afucosylation of glycoproteins on the SMIP-016GV engineered using TRU-ADhanCeTM, a glycovariant optimization technology, influence the molecule’s ability to elicit NK cell ADCC, we performed ADCC assays with NK cells from healthy donors against CD37 positive (Raji) and negative (697) malignant B cell lines targets. As shown in , SMIP-016GV mediated significantly more (p = 0.0013 in all E:T ratios > 0) ADCC against Raji cells than SMIP-016. In contrast, using 697 cells, a human B cell precursor leukemia cell line that does not express CD37, SMIP-016 and SMIP-016GV did not mediate any ADCC effects (), validating the antigen specificity of the two SMIP proteins.
NK cells from CLL patients show enhanced ADCC with SMIP-016GV
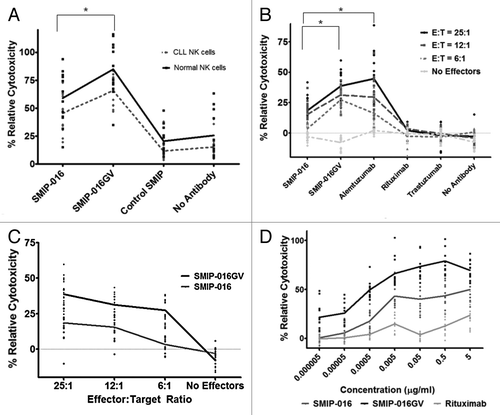
Ziegler et al. showed in a landmark paper that the NK cells from CLL patients are deficient in activity. The NK cell activity was not detectable in patients with advanced disease and six times lower than control in patients with early disease.Citation28 This finding, however, has been challenged by others.Citation29 To test whether CLL NK cells would be capable of mediating an effective response with SMIP-016GV, we used NK cells from patients with early stage disease as effectors in ADCC assays against Raji cells targets. Their response was compared with the response from NK cells isolated from healthy donors. The SMIP-016GV showed increased levels of cytotoxicity than SMIP-016 with normal and CLL NK cells (p < 0.0001 for both) (). The trend in response to the SMIP-016GV from primary NK cells from CLL patients as in NK cells from healthy donors was comparable (p = 0.0154). Another statistical test showed that the difference between the healthy vs. CLL NK cells and the SMIP-016GV is not significant (p = 0.86), indicating that the superiority of SMIP-016GV over SMIP-016 is consistent for CLL and normal NK cells. The enhanced cytotoxicity achieved by SMIP-016GV with CLL NK cells validates our hypothesis that the glycovariant SMIP-016 may be an excellent therapeutic in CLL patients.
Figure 4. SMIP-016GV is effective against CLL B cells. (A) SMIP-016GV shows comparable enhanced cytotoxicity trends in ADCC assays with primary normal donor or CLL NK cells used as effectors against Raji cell targets at an 25:1 E:T ratio (n = 12) (p = 0.01). The levels of cytotoxicity achieved with the CLL NK cells were significantly lower than what is seen with comparable treatments in the normal donor NK cells. (p = 0.0154 for SMIP-16GV and p = 0.0104 for SMIP-016). (B) ADCC with normal donor NK cells against primary CLL B cell targets shows enhanced ADCC function with SMIP-016GV compared with SMIP-016 (p < 0.0001) . Each patient at each E:T ratio is represented by a dot (n = 15). (C) The enhanced ADCC function seen in (B) is sustained over all effector to target ratios tested (average SMIP-016GV vs. SMIP-016 at E:T ratio > 0, p < 0.0001). (D) Dose dependent ADCC function of SMIP-016GV against CLL B cells. SMIP-016GV shows significantly enhanced ADCC compared with SMIP-016 (p < 0.0001) or rituximab (p < 0.0001) even at the lowest concentration (n = 12). The trend showed significantly increased ADCC with SMIP-016GV when compared with SMIP-016 (p < 0.0001) or rituximab (p < 0.0001). The non-specific Control SMIP showed less than 5% cytotoxicity at all concentrations tested (data not shown).
SMIP-016GV shows enhanced ADCC against primary CLL B cells
Given the documented clinical activity of TRU-016, the clinical version of SMIP-016, in CLL, we next compared the ability of SMIP-016 with the SMIP-016GV to elicit effector function against primary CLL B cells. CLL B cells have a wide range of CD37 on their surface.Citation8 SMIP-016GV was able to induce greater cytotoxicity in CLL cell targets compared with SMIP-016 (p < 0.0001; ). SMIP-016GV mediates significantly greater ADCC than rituximab (p < 0.0001) and is comparable to alemtuzumab (p = 0.691); however, SMIP-016GV is more specific to malignant B cells than the anti-CD52 antibody. The superiority of SMIP-016GV over SMIP-016 for ADCC was observed for all of the effector:target ratios tested (average across E:T ratios > 0 vs. E:t = 0 for SMIP-016GV vs. SMIP-016, p < 0.0001) ().
SMIP-016GV at low concentrations is effective in enhancing NK cell ADCC
Our initial ADCC experiments were done using 5 µg/mL of both SMIP proteins, which was the optimal dose found to be effective for direct cytotoxicity in CLL B cells.Citation8 Given the enhanced ADCC by SMIP-016GV, we hypothesized that this may maintain the enhanced ADCC at lower SMIP protein concentrations. It has been suggested that serum plasma and IgG can affect the concentrations of therapeutic monoclonal antibodies within the body,Citation12 and, therefore, efficacy at low concentrations is desirable for therapy. SMIP-016, SMIP-016GV and rituximab were used at decreasing log concentrations in an ADCC assay using normal donor NK cells as effectors and CLL B cells as targets. The SMIP-016GV showed enhanced ADCC compared with the SMIP-016 across a wide range of concentrations, even as low as 5 x 10−6 µg/mL (p < 0.0001 compared with both SMIP-016 and rituximab, ). The cytotoxicity with SMIP-016 reaches a plateau after the 0.005 µg/mL concentration while SMIP-016GV continues to increase. The trend in cytotoxicity for SMIP-016GV was significantly steeper than for SMIP-016 (p < 0.0001) and for rituximab (p < 0.0001).
SMIP-016GV enhances ADCC in cells expressing low levels of surface CD37
SMIP-016-mediated direct cytotoxicity has been shown to be dependent on antigen density.Citation8,Citation21 We wanted to test whether antigen density played a role in ADCC with the SMIP-016 and whether the glycovariant could overcome this. To test this, we created a model system by retrovirally transducing 697 cells (CD37 negative cell line) with a pBABE-CD37 vector construct and isolated clones with differing levels of surface CD37 by limiting dilution cloning. We validated that the clones were producing CD37 mRNA by RT-PCR (data not shown) and quantified the amount of surface CD37 protein by flow cytometry (). These clones expressed between 5000 and 80000 molecules of CD37 on their surface, substantially less than Raji cells, which have hundreds of thousands of CD37 molecules. To test a range of CD37 antigen levels, low, medium and high CD37 antigen density clones were chosen for our assays.
Figure 5. SMIP-016GV mediates enhanced ADCC in cells expressing a range of surface CD37. (A) Quantification of surface CD37 on parental 697 (697), 697-CD37 clones V1, V2 and V3 by flow cytometry. (B) ADCC assay with normal donor NK cell effectors against the different 697 cell clones as targets at a 25:1 E:T ratio. SMIP-016 and SMIP-016GV were used in decreasing concentrations (n = 4 NK donors). Increasing surface CD37 levels enhanced ADCC with both the SMIP-016GV (p = 0.002) and SMIP-016 (p = 0.0005).
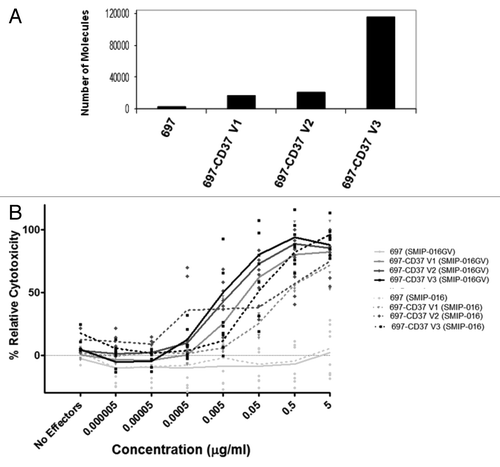
Our experimental system allowed us to determine if antigen density played a role in the level of NK cell-mediated ADCC by SMIP-016GV. Using normal donor NK cells as effectors and the CD37-expressing 697 clones as targets, ADCC assays were performed to test varied amounts of antigen on the target cells against decreasing SMIP protein concentrations (). As shown previously (), the parent 697 cells were not susceptible to ADCC by the CD37 specific SMIP protein, but despite a relatively low level of CD37 expression in the 697-CD37 V1 clone (under 20,000 molecules/cell), SMIP-016GV elicited excellent NK-mediated ADCC in these cells, up to ~80% relative cytotoxicity (). In addition, the results showed that with SMIP-016GV, cytotoxicity increased with increasing CD37 surface expression (p = 0.002). Enhanced ADCC with increasing CD37 antigen was also seen in SMIP-016 treated cells (p = 0.0005).
Interestingly, the expression of CD37 amplifies the response difference between SMIP-016GV and SMIP-016 in the low and medium CD37-expressing 697 cell clones, 697-CD37 V1 and V2. In 697-CD37 V3, the trend difference between the SMIP-016 and the glycovariant was not significant. This result may be due to the high amount of cytotoxicity already obtained with SMIP-016. The SMIP-016GV mediates greater killing but reaches 100%, thus reaching the maximal limit of the assay. Therefore, the SMIP-016GV could be beneficial for B cell malignancies with a low level of surface CD37.
SMIP-016GV shows enhanced ADCC function against acute lymphoblastic leukemia cells
Acute lymphoblastic leukemia (ALL) cells have been reported as having low surface expression levels of CD37.Citation3 From our results in , we hypothesized that the SMIP-016GV would be capable of enhanced NK cell mediated ADCC against these leukemic cells. First, we quantified the levels of CD37 on the surface of primary ALL bone marrow cells, CLL cells and normal B cells (). With an average of ~12,000 molecules of surface CD37, the surface levels of CD37 on ALL cells are 3-fold lower than CLL cells (p = 0.0051), which have an average of ~36,000 molecules, and 5-fold lower than normal peripheral B cells (p < 0.0001), which have an average of about 63,000 molecules on their surface. As shown previously,Citation8 CLL cells have a wide range of CD37 expression.
Figure 6. SMIP-016GV mediates effective NK cell ADCC against primary Acute Lymphoblastic Leukemia cell. (A) Quantification of surface CD37 by flow cytometry shows ALL cells (n = 9) have significantly lower levels of surface CD37 than CLL B cells (n = 20) (p = 0.0051) and normal B cells (n = 5) (p < 0.0001). (B) SMIP-016 and SMIP-016GV show comparable levels of modest direct cytotoxicity against primary ALL bone marrow samples (n = 4) (p = 0.74) (C) ADCC using ALL bone marrow samples as targets and normal donor NK cell effectors shows enhanced ADCC with SMIP-016GV compared with SMIP-016. (n = 8 ALL samples × 3 NK cell effectors each) (p < 0.0036 for all E:T ratios tested.
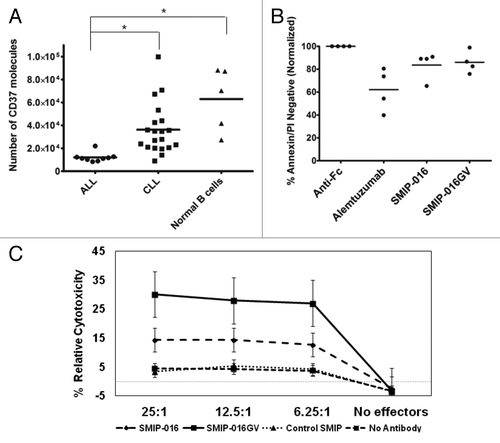
Direct cytotoxicity was tested with SMIP-016, SMIP-016GV and alemtuzumab in primary ALL samples. Viability analysis by flow cytometry at 24 h revealed that both SMIP proteins exhibited comparable cytotoxicity of 20% () (p = 0.7425), while alemtuzumab mediated about 40% cytotoxicity in these cells.
Finally, the primary ALL samples were used as targets in ADCC experiments with normal donor NK cell effectors. We saw a minimal response with SMIP-016 compared with the control SMIP protein. The SMIP-016GV was able to elicit an enhanced ADCC response against ALL samples, despite the low levels of surface CD37 (). This is significantly enhanced cytotoxicity compared with SMIP-016 (p < 0.0036 for all effector to target ratios tested). Collectively, this suggests SMIP-016GV might serve as a potential therapeutic for ALL.
Discussion
Herein we report successful modification of the Fc binding region of a SMIP protein to effectively remove fucosylation in a manner that still allows efficient production of the protein for clinical use. Production of SMIP-016 in CHO cultures that contain CS results in a final product that is afucosylated. This modification of the molecule does not affect its binding affinity to CD37 and subsequent direct cytotoxicity, nor does it alter SMIP-016GV from signaling through mechanisms previously described by our group.Citation8,Citation21 From these data, we hypothesized that SMIP-016GV’s direct cytotoxicity is dependent on surface levels of CD37 on CLL cells and is irrespective of prognostic factors such as IgVH mutational status, Rai stage and common cytogenetic abnormalities, as described with SMIP-016.Citation21 SMIP-016GV, however, shows enhanced NK cell-mediated ADCC against primary CLL B cells compared with its parent compound SMIP-016. We demonstrated that NK cell effectors from CLL patients are not as effective in SMIP-016-mediated ADCC as normal NK cell effectors. The level of ADCC obtained with SMIP-016GV and CLL NK cell effectors, however, were similar to that achieved with the parent compound SMIP-016 and normal NK cell effectors, indicating that the SMIP-016GV can overcome these deficiencies. Finally, we demonstrated that SMIP-016GV mediates superior ADCC against both low surface level expressing 697 cell lines and also primary ALL cells, whereas SMIP-016 lacks activity against these. Collectively, these data provide a strong rationale for clinical development of SMIP-016GV in B cell malignancies expressing high, as well as low, amounts of surface CD37.
With the success of antibody therapy, substantial effort has gone into modifying the Fc domain to enhance effector cell recruitment, ability to fix complement and also increased affinity for FcRn to enhance serum half life. These manipulations of function can occur through amino acid engineering via substitutions in the constant region of antibodies and have been extensively described.Citation30-Citation32 An alternative way to modify antibody effector function with enhanced recruitment of FcγRIIIa binding is to reduce Fc region fucosylation through genetic manipulation of the antibody-producing cell line.Citation33,Citation34 This has been described for CD20-directed antibody therapeutics where studies have shown enhanced NK cell ADCC function at lower antibody concentrations.Citation13,Citation35,Citation36 It provides the opportunity for using less therapeutic antibody, thereby diminishing production and ultimately treatment cost.
Modification of Fc glycosylation of clinically-relevant alternative peptide therapies such as SMIP proteins has not been previously described. Afucosylation of the FcγR binding region had potential to affect stability or other properties important for commercial development. In this paper, we demonstrate that the production process yielded functional afucosylated SMIP-016, which suggests that it may also enhance effector cell function of other immunoglycoprotein therapeutics. Given SMIP-016 has already demonstrated single agent activity against CLL and low grade B cell lymphoma,Citation9 further pursuit of SMIP-016GV with enhanced NK cell effector cell function seems worthwhile.
The Phase 1 study of TRU-016 in CLL demonstrated that this agent has significant single agent activity in both symptomatic untreated CLL and also those patients having received 1 or 2 prior therapies.Citation9 Exploration of TRU-016 in ALL was not warranted based upon very modest expression of this antigen in less mature B cells.Citation3 Our in vitro data showing that SMIP-016 lacks ADCC against ALL cells provide further justification for this decision but studies done with low-expressing CD37 transfected 697 cell lines suggest that SMIP-016GV was more effective at mediating ADCC at low copy number of CD37 antigen. Outside of kinase inhibitors targeting the Philidelphia chromosome positive ALL, little therapeutic progress has been made over the past two decades in the treatment of B cell ALL. A small proportion of patients are cured with intensive chemotherapy-based approaches or allogeneic stem cell transplant, but many ultimately die from their disease. This is particularly true for elderly patients who do not tolerate current therapy well. Immunotherapy for adult ALL is being studied in clinical trials; with rituximab and alemtuzumab, very modest benefit has been observed.Citation37-Citation40 This in part relates to low antigen density of CD20 and populations of cells not expressing CD52 in ALL.Citation40-Citation42 Herein, we demonstrate that SMIP-016GV is effective at NK cell-mediated ADCC against primary ALL cells, which have lower CD37 surface expression. This provides support for use of SMIP-016GV as a potential immune therapy of ALL.
In summary, our data suggest the potential use of the SMIP-016GV with enhanced ADCC function as a new alternative for therapy in B cell malignancies, including CLL and ALL. SMIP-016GV mediated cytotoxicity mainly through FcγR mechanisms. The capacity of monoclonal antibodies to interact with FcγRs in humans has been shown to be vital for therapeutic efficacy.Citation11 This same concept seems to apply to SMIP proteins, with SMIP-016GV being highly effective against CLL and ALL cells due to interactions with FcγRs on effector cells.
Materials and Methods
Production and structural analysis of SMIP-016 and SMIP-016GV
SMIP-016GV was generated by culturing SMIP-016, clone 8g5, a DG44 CHO cell line transfected with SMIP-016 cDNA with 400 μM castanospermine (CS). After purification, SMIP-016 and SMIP-016GV were analyzed by LC/MS via an ESI-TOF (Agilent Technologies) mass spectrometer detector.
Patient sample processing and cell culture
Blood was obtained from patients with informed consent in accordance with the Declaration of Helsinki and under a protocol approved by the Institutional Review Board (IRB) of The Ohio State University (Columbus, OH).Citation43 All patients had immunophenotypically defined CLL and had been without prior therapy for a minimum of 30 d at the time of collection. CLL cells were isolated from freshly donated blood with ficoll density gradient centrifugation (Ficoll-Paque Plus, Amersham Biosciences, catalog # 17–1440–03). Enriched CLL B cells were prepared with the use of the “Rosette-Sep” kit from StemCell Technologies (Catolog # 15024) according to the manufacturer's instructions. Isolated cells were incubated in RPMI 1640 media (Life Technologies, catalog # 12633–012) supplemented with 10% heat-inactivated fetal bovine serum (Sigma-Aldrich, catalog # F4135–500ml), 2 mM L-glutamine (Life Technologies, catalog # 25030–149) and 56 U/mL penicillin/56 µg/mL streptomycin (Life Technologies, catalog # 15140–122) at 37°C in an atmosphere of 5% CO2. Normal cells were obtained from either Red Cross partial leukocyte preparations or donors as part of a second approved exemption protocol. Natural killer (NK) cells were negatively selected with Rosette-Sep kits (StemCell Technologies, catalog # 15025) according to the manufacturer’s instructions. Monocytes were positively selected using MACS system (Miltenyi Biotec, catalog # 130–049–601). The purity of enriched populations of normal cells was routinely checked with the use of PE labeled CD19, CD14 and CD56 staining by flow cytometry. The Daudi and Raji cell lines were obtained from ATCC (ATCC# CCL-213 and CCL-86 respectively) and the 697 cell line was obtained from DSMZ, (catalog # ACC 42) and the cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum.
Assessment of apoptosis by flow cytometry
Cell viability was measured using Annexin V-FITC/PI staining followed by FACS analysis according to the manufacturer’s protocol (BD PharMingen, catalog # 556422 and 556463) as described previously.Citation8 Data were analyzed with CXP software package (Beckman-Coulter). At least 10,000 cells were collected for each sample and data were acquired in list mode. Results were expressed as the percentage of total Annexin V/PI negative cells over untreated control.
In vitro treatment of cells with antibodies
Primary CLL or ALL cells were suspended in complete media at a density of 1x107 cells/mL. SMIP-016 and SMIP-016GV were used a 5 µg/mL concentration unless otherwise noted. All antibodies (trastuzumab, rituximab and alemtuzumab) were used at a concentration of 10 µg/mL. The cross-linker, goat anti–human IgG (Fc specific) (Jackson Immunoresearch, catalog # 109–005–008) was added to the cell suspension 5 min after adding the primary antibodies at a concentration 5 times that of the therapeutic protein or antibody (i.e., 25 µg/mL for 5 µg/mL of SMIP-016). A group of samples with no treatment was collected as media control.
Immunoblot analysis
Whole cell extracts were prepared as previously described by our group.Citation44 Equivalent amounts of protein were separated on SDS-polyacrylamide gels and transferred onto nitrocellulose membranes. Following antibody incubations, proteins were detected with chemiluminescent substrate (Pierce SuperSignal, catalog # 34077). The following antibodies were used for detection: anti-phosphotyrosine (4G10) and anti-GAPDH (both from Millipore, catalog # 05–321 and #MAB374).
Antibody-dependent cellular cytotoxicity (ADCC) assay
ADCC activity was determined by standard 4-h 51Cr-release assay using methods previously reported by our group.Citation45
Antibody-dependent cellular phagocytosis (ADCP) assay
Monocyte-derived macrophages (MDMs) were derived from peripheral blood monocytes using monocyte-colony stimulating factor (R and D Systems, catalog #216-MC) for seven days. They were fluorescently labeled with Min-Claret dye (Sigma, catalog # PKH26GL). CLL cells were fluorescently labeled with PKH-67 (Sigma, catalog # MIDI67) and coated with antibody for 1 h at 4°C. MDM and CLL cells were co-incubated for 30 min at a 1:5 E: T ratio, then colocalization of CLL with MDM was scored using flow cytometry and verified using microscopy.
Complement dependent cytotoxicity (CDC) assay
This assay was performed on primary CLL cells as previously reported by our group.Citation8
Retroviral transductions
A CD37 construct was designed with the human CD37 gene inserted onto a pBABE (Promega) backbone. The huCD37-pBABE vector, an empty pBABE vector and a no vector 697 cell control were independently transfected into separate flasks of Phoenix AmphoTM cells (Orbigen (now Allele), catalog #ABP-RVC-10001) using a calcium phosphate-mediated transfection kit from Promega. The supernatant containing the retrovirus was collected from the Phoenix AmphoTM cells after two days and the 697 cells were resuspended in the viral supernatant along with polybrene (hexadimethrine bromide) for 8 h and then resuspended in fresh media. Puromycin was added as a selection agent after two days. The cells were grown under selection and then separated out in limited dilution cloning.
Assessment of antibody-binding and antigen surface density
Quantitative analysis of CD37 surface density was done using the Quantum Simply Cellular kit (Bangs Laboratories, catalog # 816), according to the manufacturer's instructions.
NK cell and monocyte in vitro stimulation and cytokine release assays
For in vitro NK-cell stimulation experiments, SMIP protein or antibody were immobilized on a plate. Normal donor NK cells were plated at 2 x 105 NK cells/well. CD107a-FITC or IgG1-FITC control was added to the suspension at the start of the 4 h incubation at 37°C. NK cells were harvested at the end of incubation period, stained with CD56-PE and analyzed by FACS for CD107a surface expression. For supernatant experiments, cell-free culture supernatants were harvested after 4 h and analyzed for levels of IFN- γ by a Quantikine Human IFN-γ ELISA performed according to the manufacturer’s instructions (R&D Systems, catalog # DIF50).
Primary monocytes were incubated on plated antibodies or SMIP proteins for 24 h and supernatant was harvested. TNF levels were measured using a Quantikine Human TNF-α ELISA, performed according to the manufacturer’s instructions (R&D Systems, catalog # DTA00C).
Live cell imaging
CLL cells were treated with 5 μg/mL of SMIP protein or a Type II antibody for 1–16 h. Images were captured every hour using a Zeiss CCD camera (AxioCam Mrm) in ApoTome Microscope (Axio Observer.Z1; Carl Zeiss MicroImaging GmbH) and were analyzed with Zeiss Axiovision (Vs40) image acquisition software.
Statistical methods
Since each patient’s cells were under all conditions of each experiment, linear mixed effect models were used to estimate unrestricted covariance structures and produce robust hypothesis tests.Citation46 Holm’s method was used to adjust for multiplicity.Citation47
| Abbreviations: | ||
| ADCC | = | antibody dependent cellular cytotoxicity |
| ADCP | = | antibody dependent cellular phagocytosis |
| ALL | = | acute lymphocytic leukemia |
| CDC | = | complement dependent cytototoxicity |
| CLL | = | chronic lymphocytic leukemia |
| FcγR | = | Fc gamma receptor |
| MDM | = | monocyte derived macrophages |
Acknowledgments
We thank the patients for providing research samples used in this study and members of the CLL Experimental Therapeutics laboratory for critical comments. We are grateful for research support from The Leukemia and Lymphoma Society, American Cancer Society, the National Cancer Institute, and National Institutes of Health (P50-CA140158, IRG-67-003-47, PO1-CA95426, and K12-CA133250). Mr. and Mrs. Michael Thomas, The Harry Mangurian Foundation and The D. Warren Brown Foundation also support this work.
Author's Contributions
S.R. designed and performed experiments, wrote the first draft of the manuscript, contributed to revisions of the paper and approved the final submitted version. C.C., J.P.B., G.L., N.K.J., R.L. provided input into experimental design, performed experiments and reviewed and approved the final version of the manuscript. D.J. and X.M. assisted in design of experiments, performed the statistical analysis reported, reviewed and approved the final version of the manuscript. S.T., J.J., J.M.F., L.A. and SD provided input into experimental design, patient samples and reviewed drafts of the manuscript and approved the final submitted version. J.M., M.M., R.L., B.S., A.S. and P.A. generated and provided reagents and reviewed and approved the final version of the manuscript. N.M. and J.C.B. obtained funding to perform the research, designed the experiments, participated in the analysis of the data, review of multiple drafts of the manuscript and approved the final version for submission.
Submitted
03/16/2013
Revised
06/01/2013
Accepted
06/05/2013
Disclosure of Potential Conflicts of Interest
J.M., M.M., R.L., B.S., A.S. and P.A. are employees of Emergent Biosolutions and have financial interests in TRU-016 and TRU-016GV development.
References
- Schwartz-Albiez R, Dörken B, Hofmann W, Moldenhauer G. The B cell-associated CD37 antigen (gp40-52). Structure and subcellular expression of an extensively glycosylated glycoprotein. J Immunol 1988; 140:905 - 14; PMID: 3257508
- Link MP, Bindl J, Meeker TC, Carswell C, Doss CA, Warnke RA, et al. A unique antigen on mature B cells defined by a monoclonal antibody. J Immunol 1986; 137:3013 - 8; PMID: 3489782
- Barrena S, Almeida J, Yunta M, López A, Fernández-Mosteirín N, Giralt M, et al. Aberrant expression of tetraspanin molecules in B-cell chronic lymphoproliferative disorders and its correlation with normal B-cell maturation. Leukemia 2005; 19:1376 - 83; http://dx.doi.org/10.1038/sj.leu.2403822; PMID: 15931266
- Press OW, Eary JF, Badger CC, Martin PJ, Appelbaum FR, Levy R, et al. Treatment of refractory non-Hodgkin’s lymphoma with radiolabeled MB-1 (anti-CD37) antibody. J Clin Oncol 1989; 7:1027 - 38; PMID: 2666588
- Kaminski MS, Fig LM, Zasadny KR, Koral KF, DelRosario RB, Francis IR, et al. Imaging, dosimetry, and radioimmunotherapy with iodine 131-labeled anti-CD37 antibody in B-cell lymphoma. J Clin Oncol 1992; 10:1696 - 711; PMID: 1403053
- Brown RS, Kaminski MS, Fisher SJ, Chang AE, Wahl RL. Intratumoral microdistribution of [131I]MB-1 in patients with B-cell lymphoma following radioimmunotherapy. Nucl Med Biol 1997; 24:657 - 63; http://dx.doi.org/10.1016/S0969-8051(97)00099-1; PMID: 9352537
- Heider KH, Kiefer K, Zenz T, Volden M, Stilgenbauer S, Ostermann E, et al. A novel Fc-engineered monoclonal antibody to CD37 with enhanced ADCC and high proapoptotic activity for treatment of B-cell malignancies. Blood 2011; 118:4159 - 68; http://dx.doi.org/10.1182/blood-2011-04-351932; PMID: 21795744
- Zhao X, Lapalombella R, Joshi T, Cheney C, Gowda A, Hayden-Ledbetter MS, et al. Targeting CD37-positive lymphoid malignancies with a novel engineered small modular immunopharmaceutical. Blood 2007; 110:2569 - 77; http://dx.doi.org/10.1182/blood-2006-12-062927; PMID: 17440052
- Furman R, Andritsos L, Flinn IW, Forero-Torres A, Foon KA, Pagel JM, et al. Phase 1 Dose Escalation Study of TRU-016, An Anti-CD37 SMIPTM Protein In Relapsed and Refractory CLL. [ASH Annual Meeting Abstracts]. Blood2010; 116:56; https://ash.confex.com/ash/2010/webprogram/Paper32343.html.
- Stavenhagen JB, Gorlatov S, Tuaillon N, Rankin CT, Li H, Burke S, et al. Fc optimization of therapeutic antibodies enhances their ability to kill tumor cells in vitro and controls tumor expansion in vivo via low-affinity activating Fcgamma receptors. Cancer Res 2007; 67:8882 - 90; http://dx.doi.org/10.1158/0008-5472.CAN-07-0696; PMID: 17875730
- Schuster M, Umana P, Ferrara C, Brünker P, Gerdes C, Waxenecker G, et al. Improved effector functions of a therapeutic monoclonal Lewis Y-specific antibody by glycoform engineering. Cancer Res 2005; 65:7934 - 41; PMID: 16140965
- Weitzman J, Betancur M, Boissel L, Rabinowitz AP, Klein A, Klingemann H. Variable contribution of monoclonal antibodies to ADCC in patients with chronic lymphocytic leukemia. Leuk Lymphoma 2009; 50:1361 - 8; http://dx.doi.org/10.1080/10428190903026500; PMID: 19562616
- Iida S, Misaka H, Inoue M, Shibata M, Nakano R, Yamane-Ohnuki N, et al. Nonfucosylated therapeutic IgG1 antibody can evade the inhibitory effect of serum immunoglobulin G on antibody-dependent cellular cytotoxicity through its high binding to FcgammaRIIIa. Clin Cancer Res 2006; 12:2879 - 87; http://dx.doi.org/10.1158/1078-0432.CCR-05-2619; PMID: 16675584
- Iida S, Kuni-Kamochi R, Mori K, Misaka H, Inoue M, Okazaki A, et al. Two mechanisms of the enhanced antibody-dependent cellular cytotoxicity (ADCC) efficacy of non-fucosylated therapeutic antibodies in human blood. BMC Cancer 2009; 9:58; http://dx.doi.org/10.1186/1471-2407-9-58; PMID: 19226457
- Saul R, Molyneux RJ, Elbein AD. Studies on the mechanism of castanospermine inhibition of alpha- and beta-glucosidases. Arch Biochem Biophys 1984; 230:668 - 75; http://dx.doi.org/10.1016/0003-9861(84)90448-X; PMID: 6424575
- Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 2002; 99:754 - 8; http://dx.doi.org/10.1182/blood.V99.3.754; PMID: 11806974
- Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol 2003; 21:3940 - 7; http://dx.doi.org/10.1200/JCO.2003.05.013; PMID: 12975461
- Anolik JH, Campbell D, Felgar RE, Young F, Sanz I, Rosenblatt J, et al. The relationship of FcgammaRIIIa genotype to degree of B cell depletion by rituximab in the treatment of systemic lupus erythematosus. Arthritis Rheum 2003; 48:455 - 9; http://dx.doi.org/10.1002/art.10764; PMID: 12571855
- Farag SS, Flinn IW, Modali R, Lehman TA, Young D, Byrd JC. Fc gamma RIIIa and Fc gamma RIIa polymorphisms do not predict response to rituximab in B-cell chronic lymphocytic leukemia. Blood 2004; 103:1472 - 4; http://dx.doi.org/10.1182/blood-2003-07-2548; PMID: 14563637
- Woyach JA, Lin TS, Lucas MS, Heerema N, Moran ME, Cheney C, et al. A phase I/II study of rituximab and etanercept in patients with chronic lymphocytic leukemia and small lymphocytic lymphoma. Leukemia 2009; 23:912 - 8; http://dx.doi.org/10.1038/leu.2008.385; PMID: 19225537
- Lapalombella R, Yeh YY, Wang L, Ramanunni A, Rafiq S, Jha S, et al. Tetraspanin CD37 directly mediates transduction of survival and apoptotic signals. Cancer Cell 2012; 21:694 - 708; http://dx.doi.org/10.1016/j.ccr.2012.03.040; PMID: 22624718
- Richards JO, Karki S, Lazar GA, Chen H, Dang W, Desjarlais JR. Optimization of antibody binding to FcgammaRIIa enhances macrophage phagocytosis of tumor cells. Mol Cancer Ther 2008; 7:2517 - 27; http://dx.doi.org/10.1158/1535-7163.MCT-08-0201; PMID: 18723496
- Uchida J, Hamaguchi Y, Oliver JA, Ravetch JV, Poe JC, Haas KM, et al. The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J Exp Med 2004; 199:1659 - 69; http://dx.doi.org/10.1084/jem.20040119; PMID: 15210744
- Herbst R, Wang Y, Gallagher S, Mittereder N, Kuta E, Damschroder M, et al. B-cell depletion in vitro and in vivo with an afucosylated anti-CD19 antibody. J Pharmacol Exp Ther 2010; 335:213 - 22; http://dx.doi.org/10.1124/jpet.110.168062; PMID: 20605905
- Tedder TF, Baras A, Xiu Y. Fcgamma receptor-dependent effector mechanisms regulate CD19 and CD20 antibody immunotherapies for B lymphocyte malignancies and autoimmunity. Springer Semin Immunopathol 2006; 28:351 - 64; http://dx.doi.org/10.1007/s00281-006-0057-9; PMID: 17091246
- Wang ZQ, Bapat AS, Rayanade RJ, Dagtas AS, Hoffmann MK. Interleukin-10 induces macrophage apoptosis and expression of CD16 (FcgammaRIII) whose engagement blocks the cell death programme and facilitates differentiation. Immunology 2001; 102:331 - 7; http://dx.doi.org/10.1046/j.1365-2567.2001.01171.x; PMID: 11298832
- Ashraf SQ, Umana P, Mössner E, Ntouroupi T, Brünker P, Schmidt C, et al. Humanised IgG1 antibody variants targeting membrane-bound carcinoembryonic antigen by antibody-dependent cellular cytotoxicity and phagocytosis. Br J Cancer 2009; 101:1758 - 68; http://dx.doi.org/10.1038/sj.bjc.6605355; PMID: 19904275
- Ziegler HW, Kay NE, Zarling JM. Deficiency of natural killer cell activity in patients with chronic lymphocytic leukemia. Int J Cancer 1981; 27:321 - 7; http://dx.doi.org/10.1002/ijc.2910270310; PMID: 6169660
- Le Garff-Tavernier M, Decocq J, de Romeuf C, Parizot C, Dutertre CA, Chapiro E, et al. Analysis of CD16+CD56dim NK cells from CLL patients: evidence supporting a therapeutic strategy with optimized anti-CD20 monoclonal antibodies. Leukemia 2011; 25:101 - 9; http://dx.doi.org/10.1038/leu.2010.240; PMID: 20975664
- Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci U S A 2006; 103:4005 - 10; http://dx.doi.org/10.1073/pnas.0508123103; PMID: 16537476
- Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem 2001; 276:6591 - 604; http://dx.doi.org/10.1074/jbc.M009483200; PMID: 11096108
- Desjarlais JR, Lazar GA. Modulation of antibody effector function. Exp Cell Res 2011; 317:1278 - 85; http://dx.doi.org/10.1016/j.yexcr.2011.03.018; PMID: 21459085
- Lifely MR, Hale C, Boyce S, Keen MJ, Phillips J. Glycosylation and biological activity of CAMPATH-1H expressed in different cell lines and grown under different culture conditions. Glycobiology 1995; 5:813 - 22; http://dx.doi.org/10.1093/glycob/5.8.813; PMID: 8720080
- Yamane-Ohnuki N, Kinoshita S, Inoue-Urakubo M, Kusunoki M, Iida S, Nakano R, et al. Establishment of FUT8 knockout Chinese hamster ovary cells: an ideal host cell line for producing completely defucosylated antibodies with enhanced antibody-dependent cellular cytotoxicity. Biotechnol Bioeng 2004; 87:614 - 22; http://dx.doi.org/10.1002/bit.20151; PMID: 15352059
- Masuda K, Kubota T, Kaneko E, Iida S, Wakitani M, Kobayashi-Natsume Y, et al. Enhanced binding affinity for FcgammaRIIIa of fucose-negative antibody is sufficient to induce maximal antibody-dependent cellular cytotoxicity. Mol Immunol 2007; 44:3122 - 31; http://dx.doi.org/10.1016/j.molimm.2007.02.005; PMID: 17379311
- Niwa R, Sakurada M, Kobayashi Y, Uehara A, Matsushima K, Ueda R, et al. Enhanced natural killer cell binding and activation by low-fucose IgG1 antibody results in potent antibody-dependent cellular cytotoxicity induction at lower antigen density. Clin Cancer Res 2005; 11:2327 - 36; http://dx.doi.org/10.1158/1078-0432.CCR-04-2263; PMID: 15788684
- Thomas DA, O’Brien S, Kantarjian HM. Monoclonal antibody therapy with rituximab for acute lymphoblastic leukemia. [v.] Hematol Oncol Clin North Am 2009; 23:949 - 71, v; http://dx.doi.org/10.1016/j.hoc.2009.07.005; PMID: 19825447
- Laporte JP, Isnard F, Garderet L, Fouillard L, Gorin NC. Remission of adult acute lymphocytic leukaemia with alemtuzumab. Leukemia 2004; 18:1557 - 8; http://dx.doi.org/10.1038/sj.leu.2403422; PMID: 15229619
- Tibes R, Keating MJ, Ferrajoli A, Wierda W, Ravandi F, Garcia-Manero G, et al. Activity of alemtuzumab in patients with CD52-positive acute leukemia. Cancer 2006; 106:2645 - 51; http://dx.doi.org/10.1002/cncr.21901; PMID: 16688777
- Nijmeijer BA, van Schie ML, Halkes CJ, Griffioen M, Willemze R, Falkenburg JH. A mechanistic rationale for combining alemtuzumab and rituximab in the treatment of ALL. Blood 2010; 116:5930 - 40; http://dx.doi.org/10.1182/blood-2010-01-262006; PMID: 20844239
- Piccaluga PP, Arpinati M, Candoni A, Laterza C, Paolini S, Gazzola A, et al. Surface antigens analysis reveals significant expression of candidate targets for immunotherapy in adult acute lymphoid leukemia. Leuk Lymphoma 2011; 52:325 - 7; http://dx.doi.org/10.3109/10428194.2010.529206; PMID: 21077738
- Chevallier P, Robillard N, Houille G, Ayari S, Guillaume T, Delaunay J, et al. Simultaneous study of five candidate target antigens (CD20, CD22, CD33, CD52, HER2) for antibody-based immunotherapy in B-ALL: a monocentric study of 44 cases. Leukemia 2009; 23:806 - 7; http://dx.doi.org/10.1038/leu.2008.303; PMID: 18971949
- Keating M, Cheson B, Gribben JG, Robertson B, Nadler L. Innovative strategies for the treatment of CLL. Leuk Lymphoma 1996; 22:Suppl 2 53 - 64; http://dx.doi.org/10.3109/10428199609102703; PMID: 9021709
- Johnson AJ, Lucas DM, Muthusamy N, Smith LL, Edwards RB, De Lay MD, et al. Characterization of the TCL-1 transgenic mouse as a preclinical drug development tool for human chronic lymphocytic leukemia. Blood 2006; 108:1334 - 8; http://dx.doi.org/10.1182/blood-2005-12-011213; PMID: 16670263
- Rafiq S, Cheney C, Mo X, Jarjoura D, Muthusamy N, Byrd JC. XmAb-5574 antibody demonstrates superior antibody-dependent cellular cytotoxicity as compared with CD52- and CD20-targeted antibodies in adult acute lymphoblastic leukemia cells. Leukemia 2012; 26:1720 - 2; http://dx.doi.org/10.1038/leu.2012.40; PMID: 22333878
- Pan X, Jarjoura D. Robust Estimation of the Variance of a Contrast in Linear Mixed Models. Joint Statistical Meetings 2009.
- Holm S. A simple sequentially rejective multiple test procedure. Scand J Stat 1979; 6:65 - 70