Abstract
Bispecific IgG asymmetric (heterodimeric) antibodies offer enhanced therapeutic efficacy, but present unique challenges for drug development. These challenges are related to the proper assembly of heavy and light chains. Impurities such as symmetric (homodimeric) antibodies can arise with improper assembly. A new method to assess heterodimer purity of such bispecific antibody products is needed because traditional separation-based purity assays are unable to separate or quantify homodimer impurities. This paper presents a liquid chromatography-mass spectrometry (LC-MS)-based method for evaluating heterodimeric purity of a prototype asymmetric antibody containing two different heavy chains and two identical light chains. The heterodimer and independently expressed homodimeric standards were characterized by two complementary LC-MS techniques: Intact protein mass measurement of deglycosylated antibody and peptide map analyses. Intact protein mass analysis was used to check molecular integrity and composition. LC-MSE peptide mapping of Lys-C digests was used to verify protein sequences and characterize post-translational modifications, including C-terminal truncation species. Guided by the characterization results, a heterodimer purity assay was demonstrated by intact protein mass analysis of pure deglycosylated heterodimer spiked with each deglycosylated homodimeric standard. The assay was capable of detecting low levels (2%) of spiked homodimers in conjunction with co-eluting half antibodies and multiple mass species present in the homodimer standards and providing relative purity differences between samples. Detection of minor homodimer and half-antibody C-terminal truncation species at levels as low as 0.6% demonstrates the sensitivity of the method. This method is suitable for purity assessment of heterodimer samples during process and purification development of bispecific antibodies, e.g., clone selection.
Introduction
Bispecific antibodies, which unlike conventional monoclonal antibodies, can bind two different antigens and offer a novel therapeutic approach to the treatment of various malignant and autoimmune diseases.Citation1 The bispecific format promises greater efficacy, avoids the complicated and costly development of combination therapies, and is receiving increasing attention in the biopharmaceutical community.Citation1-Citation5 In 2009, catumaxomab became the first bispecific antibody to attain regulatory approval,Citation6 and other candidates are in clinical development.Citation7 Bispecific antibodies are poised to become the next generation of antibody-based drugs.
A variety of design strategies for bispecific antibodies have been investigated, including symmetric IgG-like fusion molecules and asymmetric antibodies.Citation1,Citation3 Asymmetric antibodies are based on heterodimerization between two different heavy chains which are selectively paired to two different light chains and is accompanied by unique challenges related to proper association of the individual heavy and light chains. The current work deals with the development of the intermediate heterodimeric antibodies comprising of two different heavy chains paired to two common light chains. Incorrect pairing of heavy chains and light chains leads to complicated heterogeneous antibody mixtures containing impurities such as homodimers (symmetric antibodies containing two common heavy chains and two common light chains). A number of elegant approaches including knobs-into-holes and electrostatic effects have been developed to address the challenges related to heterodimeric antibody assembly, and these approaches have been reviewed recently.Citation8-Citation10 The current work is based on novel alternate heterodimerization designs aimed at achieving asymmetric antibodies with improved purity and stability characteristics.
In spite of recent advances, a remaining challenge in developing heterodimeric antibodies is the lack of established purity assay methods for quantitative evaluation of heterodimer purity, when potentially a number of misrepaired or undesired species may exist in the expression product. A key challenge in analytical method development for bispecific antibodies is that the method must accurately and reproducibly detect impurities present at 2% or lower level relative to the main desired species. Detection and identification of these low percentages of impurities is important because of potential detrimental contamination in the final product. For some target receptors, even a small amount of the homodimeric impurity could exhibit a different mode of action and potential toxicity or immunogenicity compared to the heterodimeric bispecific antibody. In addition, the homodimeric impurities have a lower stability than the heterodimeric antibody and present a potentially higher risk for aggregation and immunogenicity. Purity assay methods need sufficient accuracy and resolution to detect and quantify fully assembled bispecific antibodies and their impurities. Evaluation of these antibody impurities is difficult due to similarities between structural and physicochemical properties of such impurities and the heterodimer. Traditional separation-based antibody purity assays such as electrophoresis- and high-performance LC (HPLC)-based methods lack the resolution needed to distinguish these antibody impurities from the desired product.
Mass spectrometry is used during innovator or biosimilar antibody development to obtain accurate protein mass and primary structure information.Citation11-Citation13 Modern mass spectrometers such as electrospray ionization-quadrupole-time of flight (ESI-Q-TOF) instruments provide high resolution, high sensitivity, and accurate mass for protein and peptide analyses and are routinely used for evaluation of heterogeneity arising from modifications such as glycosylation and C-terminal lysine truncation. Although intact protein mass spectra have been used to establish the purity of bispecific antibodies,Citation14,Citation15 the abilities of the corresponding methods to detect homodimeric impurities and quantify relative differences between samples have not been systematically evaluated. Additionally, purity assessment of bispecific antibodies by mass spectrometry has been complicated by heterogeneity arising from N-linked glycans, and heterogeneity as a result of C-terminal lysine truncation.Citation14
This paper demonstrates an LC-MS-based assay using an advanced ESI-Q-TOF mass spectrometer that evaluates heterodimeric purity of MAb1, a prototype heterodimeric antibody, after deglycosylation by detecting and quantifying homodimeric and half-antibody (heavy chain + light chain) impurities. MAb1 is a proof-of-concept heterodimeric antibody based on the IgG1 trastuzumab and is an Fc heterodimer composed of two different half antibodies sharing a common light chain primary structure (data not shown). Heavy chains A and B have different amino acids at 6 sites in the Fc region (resulting in a 172 Da mass difference). MAb1 and the independently-expressed homodimeric standards (containing only the A or B heavy chain plus a common light chain) were first characterized by two complementary LC-MS techniques, intact protein mass analysis after deglycosylation and LC-MSE peptide mapping of Lys-C digests prepared from reduced and alkylated samples. Prior to the studies described herein, the homodimeric standards were purified by size-exclusion chromatography (SEC)-HPLC to separate out half-antibodies and enrich full-length homodimeric antibody in the sample. Deglycosylation was performed prior to intact protein mass analyses to reduce sample heterogeneity and simplify detection and quantification of heterodimer and homodimers. The available MAb1 heterodimer sample inherently showed no detectable homodimer impurities. To evaluate the capability of LC-MS to evaluate heterodimer purity, the independently expressed homodimeric standards were spiked into the MAb1 sample. Detection and relative quantification of homodimers and associated half-antibody impurities in heterodimer samples spiked with homodimeric standards in this manner were then demonstrated using the intact protein LC-MS method.
Results
Characterization of MAb1 and homodimeric controls by LC-MS
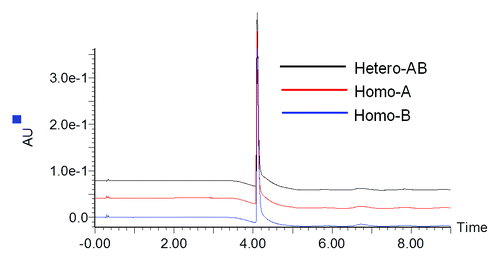
To evaluate molecular integrity and sample composition, heterodimer MAb1 (Hetero-AB) and independently expressed homodimer standards containing only heavy chain A or B (Homo-A and Homo-B) along with the common light chain were analyzed by reversed-phase LC-MS. Prior to analysis, deglycosylation of the antibodies was performed using PNGase-F to remove the heterogeneity due to N-linked glycans. As shown in , no differences in reversed-phase LC retention times were observed for components from samples Hetero-AB, Homo-A, and Homo-B even after optimization of the LC separation conditions.
Figure 1. UV280 chromatograms for Hetero-AB, Homo-A, and Homo-B.
The combined raw mass spectrum for the protein peak of Hetero-AB is shown in . Three components are apparent at the individual charge state levels of the spectrum ( inset). The MaxEnt1 deconvoluted spectrum for Hetero-AB () has three components with average masses suggesting the three possible C-terminal lysine truncation species of Hetero-AB. Hetero-AB with lysine truncation on both heavy chains is the predominant component observed. Details for all identified components, including relative abundances based on deconvoluted mass intensity, are listed in . Excellent mass accuracy (<15 ppm error) was obtained for the observed components.
Figure 2.(A) Raw and (B) MaxEnt1 deconvoluted mass spectra of Hetero-AB.
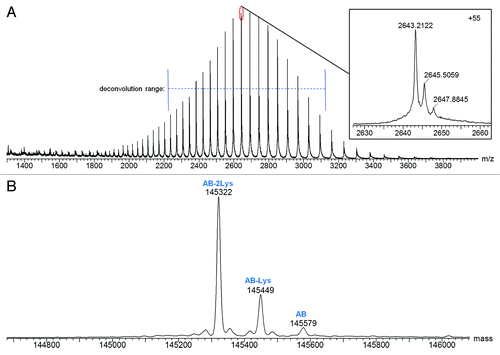
Table 1. Mass Spectral Characterization of Protein Components Detected in Hetero-AB
Combined raw mass spectra for the Homo-A and Homo-B standards are shown in and , respectively. To obtain the Homo-A and Homo-B standards for the heterodimer MAb1, heavy chains A and B were independently expressed with the common light chain and purified by Protein-A and SEC-HPLC. Expression of heavy chain A mainly resulted in half-antibody A, but SEC-HPLC allowed isolation and enrichment of the low level of the homodimeric antibody AA, used as standard Homo-A. Expression of heavy chain B mainly resulted in half-antibody B, and isolation of the low abundance homodimeric antibody BB from half-antibody B was not possible by SEC-HPLC; therefore, the Homo-B standard contains higher levels of half-antibody B. Due to the differences in half-antibody abundance of the homodimer standards, a second charge envelope corresponding to co-eluting half-antibody was observed for both Homo-A and Homo-B samples. Homo-B had much more half-antibody than Homo-A as observed in the mass spectra. The ratio of half-antibody to intact antibody in these samples appears to be stable under normal storage conditions, with no significant changes upon incubation at 4°C for up to two months (data not shown).
Figure 3. Mass spectra for Homo-A: (A) Raw mass spectrum; (B) MaxEnt1 deconvoluted spectrum for the intact antibody charge envelope; (C) MaxEnt1 deconvoluted spectrum for the half-antibody charge envelope.
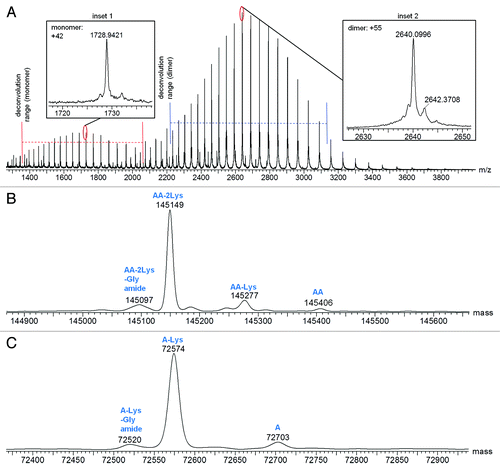
Figure 4. Mass spectra for Homo-B: (A) Raw mass spectrum; (B) MaxEnt1 deconvoluted spectrum for the intact antibody charge envelope; (C) MaxEnt1 deconvoluted spectrum for the half-antibody charge envelope.
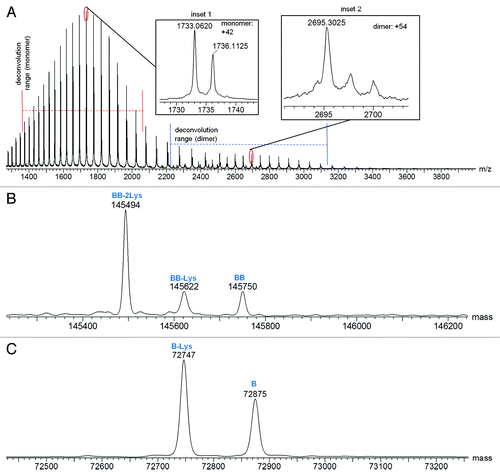
The intact and half-antibody charge envelopes were deconvoluted separately because each required different deconvolution parameters for optimal results. Deconvoluted spectra for the intact antibodies of Homo-A and Homo-B are shown in and , respectively, and deconvoluted spectra for the half antibodies are shown in and , respectively. Mass spectral details for identified components are listed in and . Relative intensities in and are calculated in relation to the combined intensities of all half-antibody and intact antibody components, but these relative intensities may not represent actual relative abundances of half antibodies and intact antibodies in the tested samples. Half antibodies likely have greater ionization efficiency than intact antibodies, possibly leading to under-representation of intact antibody abundance and over-representation of half-antibody abundance.
Table 2. Mass Spectral Characterization of Protein Components Detected in Homo-A
Table 3. Mass Spectral Characterization of Protein Components Detected in Homo-B
The deconvoluted spectra of homodimers AA and BB reveal differences in lysine truncation levels. AA has more lysine truncation than heterodimer AB, while BB has less lysine truncation than AB. A variation of homodimer AA with a mass of 145097 Da (72520 Da in the corresponding half-antibody) was confirmed by peptide mapping studies to be the fully lysine-truncated antibody with C-terminal amidated proline on one of the two heavy chains ().
Table 6. LC-MSE Characterization of Heavy Chain C-Terminal Truncations
The results for Homo-A shown in and demonstrate the capability of this method for detecting components of < 1% abundance. Half-antibody A with lysine truncation and C-terminal amidated proline was detected at 0.6%, while half-antibody A was detected at 0.9%.
To validate the amino acid sequences and C-terminal truncation species and to assess potential amino acid modifications, enzymatic digests of reduced and alkylated protein in Hetero-AB sample and Homo-A and Homo-B standards were examined by LC-MSE peptide mapping. The digests of reduced and alkylated Hetero-AB, Homo-A, and Homo-B were prepared by sequential Lys-C and trypsin digestion and analyzed using an alternating low and elevated collision energy LC-MS scan mode (LC-MSE).Citation16 Tryptic peptides were identified by precursor masses and confirmed by MSE fragment (b and y) ions. Sequence coverage was ≥ 98.2% overall for each of the three samples; chain-specific sequence coverage is presented in .
Table 4. Peptide Map Sequence Coverage
The six sites of sequence variation between heavy chains A and B were identified by the precursor masses of corresponding tryptic peptides and confirmed by MSE fragment ions. As an example, illustrates the determination of the amino acid difference at heavy chain position 369, which is part of tryptic peptide T36. Heavy chain A has a threonine at this position, while heavy chain B has a leucine at this position. LC elution and mass spectral identification for each of these two peptides are shown in . MSE fragment ion spectra confirming the sequence difference are show in . LC-MS details for all six sites of sequence difference between heavy chains A and B are shown in .
Figure 5. LC-MSE results identifying heavy chain A- and B-specific sequences for tryptic peptide T36: (A) Total ion chromatogram (TIC) with zoomed mass spectra shown in insets; (B) MSE fragment ions confirming the sequence difference.
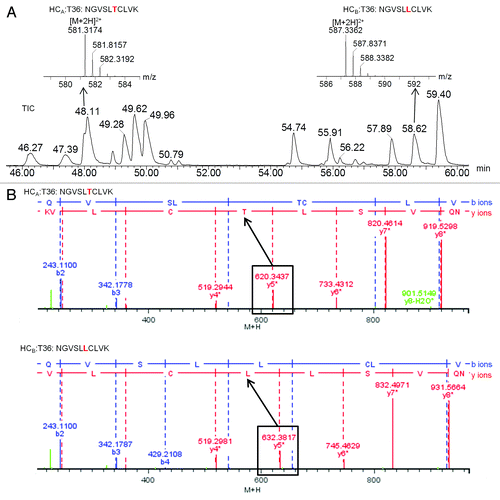
Table 5. LC-MSE Data for Amino Acid Residue Variations between Heavy Chains A and B
Analyses of the heavy chain C-terminal peptides confirmed the C-terminal truncation observed by intact protein LC-MS. contains the LC-MS details for the variations of the heavy chain C-terminal peptide. Components in Homo-A show the highest levels of C-terminal truncations, with 87.4% lysine truncation and with 5.6% C-terminal amidated proline after truncation of the last two residues lysine and glycine. C-terminal amidated proline is a result of enzymatic cleavage of glycine and has been previously reported.Citation17 Components in Homo-B have the lowest level of C-terminal truncations.
The peptide map results also permitted characterization of other amino acid residue modifications (data not shown). The detected modifications include low levels (< 5%) of N-deamidation, N-succinimide, M-oxidation, D-succinimide, and a DP cleavage. The observed modifications are typical for a monoclonal IgG1. Some of these modifications, such as N-deamidation and DP cleavage, may be artifacts of sample handling. The expected N-glycosylation in the conserved -NST- glycosylation motif was also observed, with several glycoforms, e.g., G0F, G1F, G2F, that are typical for an IgG1 being detected.
Detection and relative quantification of homodimer and half-antibody in heterodimer samples
After mass spectrometric structural characterization of the heterodimer and the homodimer standards, intact protein LC-MS analysis of deglycosylated antibody was selected to develop an assay for (1) assessment of heteodimer purity and (2) detection and relative quantification of homodimer and co-eluting half-antibody impurities in samples of heterodimer. To develop the assay, different amounts of deglycosylated Homo-A or Homo-B were spiked into deglycosylated Hetero-AB (MAb1) as simulated impurities. Mass spectra for the resulting samples were obtained.
Five samples of Hetero-AB containing 2–9% Homo-A (mole %) were prepared to examine detection of homodimer AA. Similarly, five samples of Hetero-AB containing 5‒30% Homo-B (mole %) were prepared to examine detection of homodimer BB. A higher range of Homo-B, which has a large fraction of half-antibody, was tested to obtain homodimer signals that were comparable to the samples containing Homo-A.
Combined raw mass spectra for heterodimer samples containing Homo-A or Homo-B show ions corresponding to the respective homodimer components (). Deconvoluted overlaid spectra showing the heterodimeric and homodimeric components at each concentration of Homo-A and Homo-B are shown in . These overlays, normalized to the most intense component (AB-2Lys), clearly show the homodimeric component in each spiked sample. Excellent MS resolution was obtained, with AB-Lys and BB-2Lys (theoretical mass difference of 45 Da) easily distinguished from each other (). Clear relationships between homodimer intensity response and concentration of homodimer standard Homo-A/Homo-B are apparent.
Figure 6. Raw mass spectra for Hetero-AB containing low levels of spiked homodimer standards, zoomed to show two charge states. (A) Hetero-AB containing 30% Homo-B standard. (B) Hetero-AB containing 9% Homo-A standard.
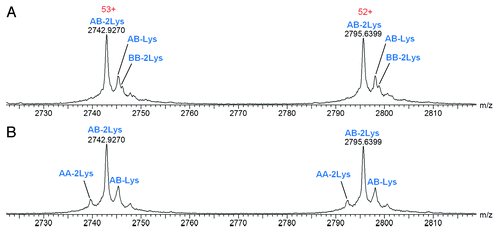
Figure 7. Deconvoluted mass spectra (intact antibody charge envelope) and homodimer intensity response curves for Hetero-AB mixed with varying amounts of Homo-A or Homo-B standards. (A) Overlaid spectra for Hetero-AB containing Homo-A, normalized to the AB-2Lys peak. (B) Overlaid spectra for Hetero-AB containing Homo-B, normalized to the AB-2Lys peak. (C) Plot of AA-2Lys intensity (relative to the sum of AA-2Lys and AB-2Lys intensities) vs. Homo-A relative concentration. (D) Plot of BB-2Lys intensity (relative to the sum of BB-2Lys and AB-2Lys intensities) vs. Homo-B relative concentration.
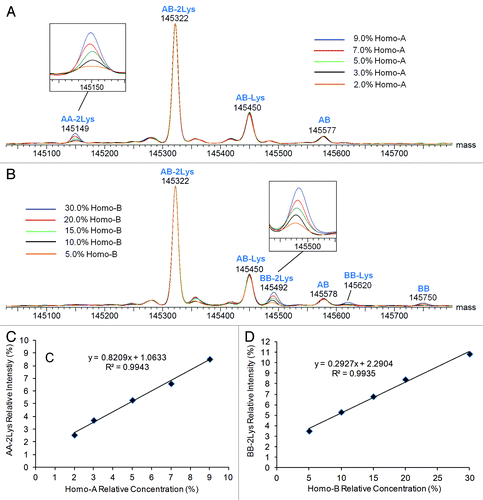
Plots relating the intensity of fully lysine-truncated homodimer (relative to all fully lysine-truncated intact antibodies) to the relative concentration of the respective standard Homo-A/Homo-B are shown in and . Linear responses for both homodimers were observed (R2 = 0.994). Standard deviations are estimated at ~0.15%, with most of the variability due to sample preparation. The observed relative intensities of homodimer AA are within 1% of the corresponding relative concentration of Homo-A, although the relative abundance of homodimer AA is estimated to be 2.5% for the sample spiked with 2.0% Homo-A (low end of relative concentration) and 8.5% for the sample spiked with 9.0% Homo-A (high end of relative concentration). The observed relative intensities of homodimer BB are significantly lower than the corresponding spiked concentrations of Homo-B because a large fraction of Homo-B is half-antibody B rather than homodimer BB.
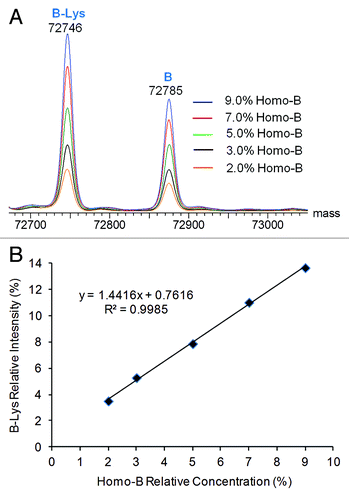
Detection and relative quantification of co-eluting half-antibody impurities were demonstrated in a similar fashion. Deglycosylated Homo-B, which contains high levels of half-antibody B, was mixed at levels of 9% or less (mole %) with deglycosylated Hetero-AB, and the mixtures were analyzed by LC-MS. The half-antibody and intact antibody charge envelopes were each independently deconvoluted. Overlaid half-antibody deconvoluted spectra are shown in , and a plot of relative half-antibody deconvoluted peak intensity (relative to the sum of half-antibody and heterodimer peak intensities) vs. the relative concentration of the Homo-B in the spiked samples is shown in . Only fully lysine-truncated species were used for calculation of relative half-antibody peak intensity. The LC-MS method reported about 2% excess intensity of the B half-antibody relative to the spiked amount of Homo-B at low concentrations of Homo-B. The deconvoluted mass peaks for both species of half-antibody B become more intense with corresponding increases in Homo-B concentration, resulting in a linear response curve.
Figure 8. Deconvoluted mass spectra from the half-antibody charge envelope and half-antibody intensity response curves for Hetero-AB mixed with varying amounts of Homo-B. (A) Overlaid half-antibody spectra for Hetero-AB containing Homo-B. (B) Plot of B-Lys intensity relative to the sum of B-Lys and AB-2Lys vs. Homo-B relative concentration.
Discussion
Bispecific antibodies are gaining popularity as biopharmaceutical drugs, and well-calibrated purity assays with sufficient resolution and sensitivity to evaluate heterodimeric purity are needed. We demonstrated detection and relative quantification of small levels of spiked homodimeric and half-antibody impurities of the heterodimeric antibody MAb1 using LC-MS analysis of deglycosylated proteins. Homodimeric impurities are particularly challenging to evaluate with traditional antibody purity assays because the similarities between homodimer and heterodimer lead to difficult, if not impossible, separations. An alternative assay that can assess purity without the need for separation of the heterodimer from impurities would be helpful for the development of bispecific antibodies. Modern MS technologies, such as the Q-TOF LC-MS demonstrated here, can serve as such an assay because they can distinguish intact antibodies and truncated or modified variants with small mass differences (e.g., 45 Da) and have large dynamic ranges (up to 104) and linear responses for quantification purposes.Citation18,Citation19
Before developing a heterodimer purity assay, it was important to characterize the structure and post-translational modifications of the antibodies. Our first step was to analyze the deglycosylated heterodimer and independently expressed homodimer standards by intact protein LC-MS analysis. These analyses permitted characterization of antibody heterogeneity and evaluation of MS capability to distinguish impurities that could be co-eluted with heterodimeric antibody. Deglycosylation to remove the heterogeneity related to N-linked glycoforms was critical for simplifying the data, especially in light of the C-terminal truncation heterogeneity. The second step, LC-MSE peptide map analysis, was then performed to confirm the protein sequences and the C-terminal truncation heterogeneity suggested by intact protein LC-MS. Following the prerequisite characterizations, samples containing mixtures of deglycosylated heterodimer and homodimer were prepared and relative levels of homodimeric (AA and BB) and half-antibody (B) impurities were evaluated by intact protein LC-MS.
Homodimeric impurities were easily detected by the intact protein LC-MS method, and relative quantification between samples was demonstrated with a linear deconvoluted MS intensity response in the range tested. Although the difference in mass between heterodimeric MAb1 and each of the homodimers is 172 Da, this method should be applicable to systems in which the heterodimer-homodimer mass difference is much smaller. Indeed, it was possible to distinguish fully lysine-truncated homodimer BB from partially lysine-truncated heterodimer ( and ). These two antibodies only differ by 45 Da. Actual limits of resolution depend on the specific instrumentation, but absolute limits are imposed by the approximately 25 Da isotopic envelope width of an antibody.Citation11 This method would not be applicable for systems in which the mass difference between the heterodimer and homodimer is too small (e.g., below 25 Da), but that scenario is not expected for most cases. The heterodimeric structure studied here with a common light chain on both arms is likely a more challenging example because the engineered mutations only in the CH3 domain result in small mass differences. Bispecific antibodies with two different light chains and likely differences in the CDR sequence composition in the two arms of the antibody would likely have more intrinsic differences in the masses of the two arms.
Analysis of subunits of antibodies resulting from limited digestion using an enzyme such as FabRICATOR® (Ides)Citation20 may be a useful complementary method to the intact protein LC-MS method described here, when the goal is to quantify the amount of individual subunits, to confirm certain chain pairing tendencies, or to estimate relative amounts of subunit chains. Such a method may also be suitable for the rare case with a small (e.g., < 25 Da) difference in mass between the heterodimeric product and homodimeric impurities. Such subunit analyses, however, would require additional sample processing and the efforts to separate two minor-different subunits of heterodimeric and homodimeric antibodies would not be trivial (if not impossible). Extensive development may be required compared with the simple intact protein LC-MS method described here.
In addition to the detection of homodimeric impurities, LC-MS can simultaneously detect co-eluting half-antibody (heavy chain + light chain) impurities and provide relative quantification between samples. Although half-antibodies can be detected and quantified by purely separation-based methods such as SDS-PAGE, evaluation of half antibodies in conjunction with homodimer evaluation will save time and resources. Half-antibody was detected in both homodimer controls, and a linear signal response of half-antibody was demonstrated by adding Homo-B to samples of Hetero-AB. Ionization efficiencies of half-antibodies and intact antibodies are expected to differ substantially, and calibration curves created with pure half-antibody would be required for absolute half-antibody quantification.
Heterogeneity due to C-terminal truncation of the heterodimer, homodimers, and half-antibodies was present (‒). C-terminal truncation levels are related to the conditions of protein expression and the expression system employed, and truncation levels can vary from batch to batch.Citation21 The method described herein involves monitoring only the major C-terminal truncation variants. Minor C-terminal truncation species of a low abundance impurity such as a homodimer would be problematic to accurately quantify, so the major truncation species provides a marker that represents all variations. It is reasonable to assume that the truncation variant ratios of a heterodimer and its homodimeric impurities would be similar within a single sample.
To more accurately quantify the homodimers and half antibodies, the relative abundances can be corrected by calibration curves such as those illustrated in and . For the greatest accuracy, calibration curves would be created with pure homodimer or half-antibody. Both Homo-A and Homo-B standards contained a mixture of half-antibody and homodimer variants, and the ratios observed by mass spectrometry likely over-represent the abundance of half-antibody. For purposes of MAb1 process development activities, Homo-A was assumed to be mainly composed of intact antibody and the Homo-A intact antibody calibration curve was used for correction of homodimer abundances. Likewise, Homo-B was assumed to be mainly composed of half-antibody, and the Homo-B half-antibody calibration curve was used for correction of half-antibody abundances. Although relative quantification of homodimeric impurities among samples is the primary need, further optimization of this method is expected to provide absolute quantification of homodimeric impurities.
Limits of detection for homodimeric or half-antibody impurities are estimated at 2% based on spiking of standards into the pure heterodimer. Considering the multiple components in both Homo-A and Homo-B, such as homodimer heavy chain C-terminal lysine-truncation variants BB-2Lys, BB-Lys, and BB () and half-antibody variants B-Lys and B () in Homo-B, the actual limit of detection and limit of quantification for an individual homodimer or half-antibody should be below the lowest concentration of spiked Homo-A/Homo-B. This is consistent with the observation in the previously described Homo-A characterization (, ) that components with abundance of 0.6–0.9% were detected and quantified. Therefore, this assay should be able to detect and quantify ≥ 0.6% individual homodimer or half-antibody impurities in heterodimer samples.
In conclusion, the LC-MS methods developed herein permit rapid and accurate detection and quantification of heterodimeric antibody MAb1 and allow relative quantification of low abundance (0.6%) homodimeric and half-antibody impurities as determined using spiking experiments with standards. MAb1 was > 95% heterodimer with no homodimer impurities. These methods, which were invaluable for clone selection, process development, and purification activities of MAb1, will be applied to future development of bispecific antibodies based on the MAb1 scaffold.
Materials and Methods
Samples and materials
Antibodies were expressed and purified at NRC-BRI. Dithiothreitol (DTT), formic acid (FA), and trifluoroacetic acid (TFA) were obtained from Thermo Scientific. Iodoacetamide (IAM) was obtained from Sigma-Aldrich. Optima grade water, optima grade acetonitrile, Tris buffer, guanidine HCl, and EDTA were obtained from Fisher Scientific. Endoproteinase Lys-C was obtained from Waco Chemicals. PNGase-F was obtained from New England Biolabs. Trypsin gold was obtained from Promega.
Instrumentation
Intact protein LC-MS analyses were performed on a Waters Acquity UPLC H-Class in line with a Waters Xevo G2-S Q-TOF mass spectrometer. LC-MSE analysis of peptide maps were performed on a Waters Acquity UPLC H-Class in line with a Waters Xevo G2 Q-TOF mass spectrometer. Daily calibration of the mass spectrometers with NaCsI was performed, and [Glu1]-fibrinopeptide B was used in the lockspray channel.
LC-MS Analysis of deglycosylated proteins
Prior to LC-MS analysis, antibody samples were incubated with PNGase-F to remove N-linked glycans. Antibody samples (0.4 µg each) were injected onto a Waters Acquity UPLC BEH 300 C4 (1.7 µm particle size) 2.1 × 50 mm column (80°C column temperature). Antibodies were eluted from the column with a 4 min gradient (35‒80% B, 0.200 mL/min flow rate). Mobile phase A was 0.1% FA in H2O. Mobile phase B was 0.1% FA in acetonitrile. The Xevo G2-S Q-TOF mass spectrometer was run in positive ion, sensitivity mode with detection in the range of 600‒4000 m/z. Source parameters were as follows: capillary voltage, 2.00 kV; sampling cone voltage, 40.0 V; source offset 100 V; source temperature, 130°C; desolvation temperature, 530°C; cone gas flow: 25 L/hr; desolvation gas flow, 900 L/hr. The protein peak was deconvoluted by the MassLynx MaxEnt1 function according to the following parameters: m/z input range, 2220‒3131 for intact antibody and 1361‒2046 for half-antibody; output resolution, 1.0 Da/channel; output mass range, 140321‒150321 Da for intact antibody and 67700‒77700 Da for half-antibody; uniform Gaussian width at half height, 0.6 Da for intact antibody and 0.5 Da for half-antibody; minimum intensity ratios, 30% for left and right; maximum number of iterations, 16 for intact antibody and 6 for half-antibody.
Digests preparation and LC-MSE analysis of peptide maps
Protein samples were dried in a centrifugal evaporator and reconstituted in pH 7.5 Tris buffer containing 6 M guanidine. DTT was added, and the samples were incubated at 55°C for 20 min to reduce disulfide bonds. IAM was then added, and the samples were incubated at room temperature for 30 min to alkylate free cysteines. The samples were diluted with water, Lys-C (1:42 w/w) was added, and the samples were incubated at 37°C for 2 h. Trypsin (1:21 w/w) was then added, and the samples were incubated at 37°C for another 2 h. TFA (20% solution) was added to the samples to stop the digestion. Samples (9.8 µg each) were injected onto a Waters Acquity UPLC BEH 300 C18 (1.7 µm particle size) 2.1‒150 mm column (65°C column temperature). Peptides were eluted from the column with a 90 min gradient (1‒41% B, 0.200 mL/min flow rate). Mobile phase A was 0.05% TFA in H2O. Mobile phase B was 0.04% TFA in acetonitrile. The Xevo G2 Q-TOF mass spectrometer was run in positive ion, sensitivity mode with detection in the range of 100–2000 m/z. Source parameters were as follows: capillary voltage, 3.00 kV; sampling cone voltage, 28.0 V; extraction cone voltage, 4.0 V; source temperature, 100°C; desolvation temperature, 250°C; cone gas flow: 0 L/hr; desolvation gas flow, 500 L/hr. The low collision energy was 4.0 eV, and the elevated collision energy ramp was 20.0–50.0 eV. Raw data were processed by BiopharmaLynx 1.3 with the following searchable modifications: cysteine carbamidomethylation, C-terminal lysine truncation, C-terminal lysine and glycine truncation with C-terminal amidated proline, N-G0F, N-G1F, N-G0, N-G2F, M-oxidation, N-deamidation, N-deamidation succinimide, and D-succinimide. Mass tolerances were set at 10 ppm for singly charged precursors and 30 ppm for MSE fragment ions (also converted to singly charged fragments).
| Abbreviations: | ||
| LC-MS | = | liquid chromatography-mass spectrometry |
| LC-MSE | = | data independent acquisition LC-MS using an alternative low and elevated collision energy scan mode |
| ESI-Q-TOF | = | electrospray-quadrupole-time-of-flight |
| SEC-HPLC | = | size exclusion high performance LC |
| UPLC | = | ultra performance LC |
| TIC | = | total ion chromatogram |
| IgG | = | immunoglobulin G |
| mAb | = | monoclonal antibody |
| CDR | = | complementarity determining region |
| ppm | = | parts per million |
| DP cleavage | = | clip between amino acid residues D and P |
| Lys (K) | = | lysine |
| SDS-PAGE | = | sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| DTT | = | dithiothreitol |
| FA | = | formic acid |
| TFA | = | trifluoroacetic acid |
| IAM | = | iodacetamide |
| HCl | = | hydrogen chloride |
| EDTA | = | ethylenediaminetetraacetic acid |
Acknowledgments
The authors thank Prathima Acharya, Tim Kelly, Abhinav Shukla, and David Poon for helpful discussion and support. The authors would also like to acknowledge Karine Brault and Yves Durocher for the preparation of the antibody samples. KBI authors thank Joe McMahon for strong support and guidance. Zymeworks authors would like to thank John Kelly for useful discussions.
Submitted
05/17/2013
Revised
06/18/2013
Accepted
06/20/2013
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
References
- Kontermann RE. Dual targeting strategies with bispecific antibodies. MAbs 2012; 4:182 - 97; http://dx.doi.org/10.4161/mabs.4.2.19000; PMID: 22453100
- Holmes D. Buy buy bispecific antibodies. Nat Rev Drug Discov 2011; 10:798 - 800; http://dx.doi.org/10.1038/nrd3581; PMID: 22037028
- Müller D, Kontermann RE. Bispecific antibodies for cancer immunotherapy: Current perspectives. BioDrugs 2010; 24:89 - 98; http://dx.doi.org/10.2165/11530960-000000000-00000; PMID: 20199124
- Demarest SJ, Hariharan K, Dong J. Emerging antibody combinations in oncology. MAbs 2011; 3:338 - 51; http://dx.doi.org/10.4161/mabs.3.4.16615; PMID: 21697653
- Carter P. Improving the efficacy of antibody-based cancer therapies. Nat Rev Cancer 2001; 1:118 - 29; http://dx.doi.org/10.1038/35101072; PMID: 11905803
- Linke R, Klein A, Seimetz D. Catumaxomab: clinical development and future directions. MAbs 2010; 2:129 - 36; http://dx.doi.org/10.4161/mabs.2.2.11221; PMID: 20190561
- Chames P, Baty D. Bispecific antibodies for cancer therapy: the light at the end of the tunnel?. MAbs 2009; 1:539 - 47; http://dx.doi.org/10.4161/mabs.1.6.10015; PMID: 20073127
- Merchant AM, Zhu Z, Yuan JQ, Goddard A, Adams CW, Presta LG, et al. An efficient route to human bispecific IgG. Nat Biotechnol 1998; 16:677 - 81; http://dx.doi.org/10.1038/nbt0798-677; PMID: 9661204
- Gunasekaran K, Pentony M, Shen M, Garrett L, Forte C, Woodward A, et al. Enhancing antibody Fc heterodimer formation through electrostatic steering effects: applications to bispecific molecules and monovalent IgG. J Biol Chem 2010; 285:19637 - 46; http://dx.doi.org/10.1074/jbc.M110.117382; PMID: 20400508
- Klein C, Sustmann C, Thomas M, Stubenrauch K, Croasdale R, Schanzer J, et al. Progress in overcoming the chain association issue in bispecific heterodimeric IgG antibodies. MAbs 2012; 4:653 - 63; http://dx.doi.org/10.4161/mabs.21379; PMID: 22925968
- Zhang Z, Pan H, Chen X. Mass spectrometry for structural characterization of therapeutic antibodies. Mass Spectrom Rev 2009; 28:147 - 76; http://dx.doi.org/10.1002/mas.20190; PMID: 18720354
- Xie H, Chakraborty A, Ahn J, Yu YQ, Dakshinamoorthy DP, Gilar M, et al. Rapid comparison of a candidate biosimilar to an innovator monoclonal antibody with advanced liquid chromatography and mass spectrometry technologies. MAbs 2010; 2:379 - 94; PMID: 20458189
- Berkowitz SA, Engen JR, Mazzeo JR, Jones GB. Analytical tools for characterizing biopharmaceuticals and the implications for biosimilars. Nat Rev Drug Discov 2012; 11:527 - 40; http://dx.doi.org/10.1038/nrd3746; PMID: 22743980
- Chelius D, Ruf P, Gruber P, Plöscher M, Liedtke R, Gansberger E, et al. Structural and functional characterization of the trifunctional antibody catumaxomab. MAbs 2010; 2:309 - 19; http://dx.doi.org/10.4161/mabs.2.3.11791; PMID: 20418662
- Jackman J, Chen Y, Huang A, Moffat B, Scheer JM, Leong SR, et al. Development of a two-part strategy to identify a therapeutic human bispecific antibody that inhibits IgE receptor signaling. J Biol Chem 2010; 285:20850 - 9; http://dx.doi.org/10.1074/jbc.M110.113910; PMID: 20444694
- Xie H, Gilar M, Gebler JC. Characterization of protein impurities and site-specific modifications using peptide mapping with liquid chromatography and data independent acquisition mass spectrometry. Anal Chem 2009; 81:5699 - 708; http://dx.doi.org/10.1021/ac900468j; PMID: 19518054
- Johnson KA, Paisley-Flango K, Tangarone BS, Porter TJ, Rouse JC. Cation exchange-HPLC and mass spectrometry reveal C-terminal amidation of an IgG1 heavy chain. Anal Biochem 2007; 360:75 - 83; http://dx.doi.org/10.1016/j.ab.2006.10.012; PMID: 17113563
- Silva JC, Gorenstein MV, Li G-Z, Vissers JPC, Geromanos SJ. Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol Cell Proteomics 2006; 5:144 - 56; http://dx.doi.org/10.1074/mcp.M500230-MCP200; PMID: 16219938
- Doneanu CE, Xenopoulos A, Fadgen K, Murphy J, Skilton SJ, Prentice H, et al. Analysis of host-cell proteins in biotherapeutic proteins by comprehensive online two-dimensional liquid chromatography/mass spectrometry. MAbs 2012; 4:24 - 44; http://dx.doi.org/10.4161/mabs.4.1.18748; PMID: 22327428
- Deperalta G, Alvarez M, Bechtel C, Dong K, McDonald R, Ling V. Structural analysis of a therapeutic monoclonal antibody dimer by hydroxyl radical footprinting. MAbs 2013; 5:86 - 101; http://dx.doi.org/10.4161/mabs.22964; PMID: 23247543
- Dick LW Jr., Qiu D, Mahon D, Adamo M, Cheng K-C. C-terminal lysine variants in fully human monoclonal antibodies: investigation of test methods and possible causes. Biotechnol Bioeng 2008; 100:1132 - 43; http://dx.doi.org/10.1002/bit.21855; PMID: 18553400