Abstract
While the concept of Quality-by-Design is addressed at the upstream and downstream process development stages, we questioned whether there are advantages to addressing the issues of biologics quality early in the design of the molecule based on fundamental biophysical characterization, and thereby reduce complexities in the product development stages. Although limited number of bispecific therapeutics are in clinic, these developments have been plagued with difficulty in producing materials of sufficient quality and quantity for both preclinical and clinical studies. The engineered heterodimeric Fc is an industry-wide favorite scaffold for the design of bispecific protein therapeutics because of its structural, and potentially pharmacokinetic, similarity to the natural antibody. Development of molecules based on this concept, however, is challenged by the presence of potential homodimer contamination and stability loss relative to the natural Fc. We engineered a heterodimeric Fc with high heterodimeric specificity that also retains natural Fc-like biophysical properties, and demonstrate here that use of engineered Fc domains that mirror the natural system translates into an efficient and robust upstream stable cell line selection process as a first step toward a more developable therapeutic.
Introduction
There is strong interest in the design of tailored bispecific therapeutics that can simultaneously bind two or more distinct targets or epitopes to achieve novel mechanisms of action and efficacy,Citation1,Citation2 yet the successful transition of such therapeutics into clinic settings has been limited. A major obstacle in the early development of bispecific antibodies has been the difficulty of producing materials of sufficient quality and quantity for both preclinical and clinical studies.Citation3,Citation4 To circumvent the problem, a plethora of alternative bispecific formats based on either antibody or other protein domains have been designed in recent years,Citation1 but issues related to a number of other fundamental drug-like properties of the designed therapeutic remain to be addressed. Aside from pharmacokinetics (PK) and pharmacodynamics (PD) aspects, other properties of the drug, including the manufacturability, which affects the cost of goods; ease of analytical characterization and comparability of the product for quality control purposes; homogeneity; and formulation and stability at the requisite drug concentrations, must be considered. Ideally, a quality by design initiative should address all these factors early in the development process to increase the chances of success in the preclinical and clinical testing stages.Citation5 We are moving toward an era of modular therapeutic design, and, given the multi-functional characteristics that antibody-based therapeutics can possess, a bispecific therapeutic scaffold that takes into consideration all of these aspects would substantially empower the drug developer in the design of best-in-class drug candidates and also potentially simplify the subsequent development process.
The current standard for an engineered heterodimeric antibody Fc domain is the knobs-into-holes (KiH) design,Citation6-Citation8 and such a design lends itself to the development of very novel therapeutic candidates.Citation9-Citation11 Klein and coworkers recently presented a detailed review of KiH designs and subsequent heterodimeric antibody solutions for the generation of bispecific molecule.Citation12 While the KiH design has been incorporated into clinical candidates,Citation9,Citation11,Citation13 large-scale expression and production of these engineered KiH Fc heterodimers and bispecific antibodies in mammalian cell lines have been reported to be challenging because of variable purity of the heterodimer product.Citation14-Citation16 To address this issue, an aglycosylated heterodimeric Fc expression system based on E. coli has been proposed and is used in the development of some of the therapeutic candidates based on the KiH design.Citation13,Citation15 Alternately, annealing-based approaches for producing bispecific antibodies by mixing two different antibody components are also being pursued.Citation10,Citation15,Citation17,Citation18 The annealing approach, however, is a multistep development and manufacturing processes that introduce extra elements of risk and potentially increase the cost of goods. Other protein engineering efforts to preferentially achieve Fc heterodimers using a number of strategies, including rational design and phage display, has also been accompanied by losses in stability of the resultant Fc relative to the native homodimeric system.Citation7,Citation19-Citation21 Among other attributes, a loss in thermal stability of a mutant relative to the native protein indicates potential effects on protein quality and expression levels. Notably, a protein mutant with decreased stability is often a source of problems in the context of downstream process optimization and poses developability challenges.Citation22-Citation24 While there are variants of the KiH design that introduce a disulfide bond at the CH3:CH3 interface in order to recover stability of the heterodimeric Fc,Citation7,Citation14 additional cysteines in a protein increase the potential risk of misfolding, aggregation, and chemical instability.Citation25 Along these lines, developability issues have been assessed by biophysical characterization of antibodies prior to initiation of process development activities.Citation26 There is also the realization that development challenges can be addressed by engineering antibodies for enhanced biophysical properties.Citation27 This recent trend looks beyond the traditional quality by design approaches that tend to address stability and heterogeneity challenges at the formulation stage. Based on this background, we have opined that an engineered heterodimeric Fc that retains wild type antibody-like biophysical properties and consistently achieves high levels of purity is particularly important while developing a protein scaffold to be used subsequently as a platform for the design of bispecific therapeutics.
Engineering of Heterodimeric Fc Domains for Stability
To design heterodimeric Fc variants with improved stability and purity, we employed an iterative process of computational design and experimental screening to select the most successful combinations of mutations addressing aspects of both positive and negative designs (). Negative design mutations maximize unfavorable interactions in potential homodimers while driving specificity between the heterodimeric species, while positive designs optimize the stability of the desired protein interactions, but rarely can achieve > 90% specificity as a stand-alone approach.Citation28 To date, primarily negative design strategies have been used to develop Fc heterodimers as specificity has been the goal, but mutations introduced at the core CH3 domain interface yield heterodimers possessing CH3 domain melting temperatures of 69°C or less, while the wild-type CH3 domain melts at 82°C.Citation29,Citation30
Figure 1.(A) The figure illustrates the total number of variants that were experimentally tested and the progress in achieving > 95% heterodimer purity and wild type Fc stability over the three design iterations. The heterodimer specificity was measured by LC-MS and the thermal stability is expressed by the melting temperature (Tm) of the CH3 domain, measured by differential scanning calorimetry. The detailed design process is described in the Methods and Supplemental Material. (B) Selected variants from the 3 iterations of the design process in (A), including the final lead variant ZW1. (C) Superposition of the ZW1 Fc crystal structure and a wild type Fc crystal structure (PDB ID: 2WAH) to illustrate the equivalence of the heterodimeric ZW1 and the homodimeric wild type CH3 domain structure. The close-up shows a stereo representation of the CH3 domain interface.
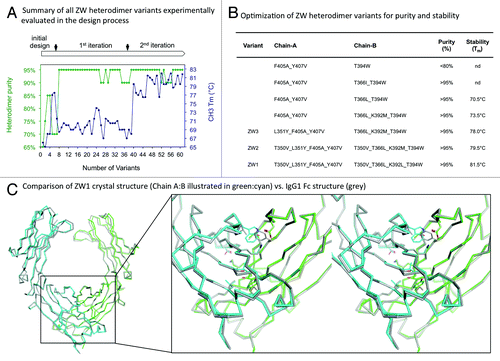
To design stable Fc domains with high heterodimer specificity, a rational structure-guided approach was employed and supported with computational algorithms relying on a combination of knowledge and physics based energy functions.Citation31-Citation35 The in silico approach can evaluate potential mutations by simulation and conformational sampling of amino acid side chains and backbones while tandem in vitro testing provides the necessary verification and feedback. Specifically, in the initial phase, different negative design Fc heterodimer variants engineered for specificity were proposed. This initial set of variants, while able to achieve high heterodimeric specificity, was confirmed to be less stable (< 71°C). From this initial phase, designs with greater than 90% purity and a melting temperature of ~68°C or greater, were selected for further optimization. In the two subsequent design iterations, the selected Fc heterodimer variants were modified to increase both stability and purity by using positive designs to maximize favorable interactions in the heterodimer without perturbing the effect of the negative design. The comprehensive structural and computational analysis provided a detailed understanding of the differences of each Fc variant compared to wild-type with respect to structural dynamics (including sites of local unfolding), interface hotspots, sites of asymmetry, cavities, and poorly packed regions. By using a rational and systematic approach to concurrently address purity and stability design requirements, we were required to perform in vitro evaluation for less than 70 total variants before achieving wild-type stability and Fc heterodimeric purity. The mutations, purity and stability characteristics of a select number of variants in the design progression are shown in . The lead heterodimeric Fc design variant (ZW1: (Chain A) T350V/L351Y/F405A/Y407V and (Chain B) T350V/T366L/K392L/T394W) has a thermal stability of 81.5°C and expresses to form pure heterodimers with no detectable homodimers.
In the early evaluation, the designed mutations were introduced in two different heavy chain CH3 domains of HER2/ErbB2-binding IgG1 trastuzumab (Herceptin®) while maintaining the native sequence for the VH, CH1 and CH2 domains on both chains. The two engineered heavy chains were co-expressed transiently, together with trastuzumab’s native light chain in HEK293 cells to screen for the heterodimeric Fc designs. The lead heterodimeric Fc design, ZW1, was identified and we have successfully solved the X-ray crystal structures of the Fc fragment at a resolution of 2.1 Å (PDB ID: 4BSW). The protein crystallized in a symmetric format analogous to native homodimeric Fc and the crystals are homogenous mixtures of the two possible orientations of the asymmetrical CH3/CH3 domain interface (). The mutated amino acid side chains are observed at a 50% occupancy level for each of the residues with respect to the two symmetrical orientations (). The RMSD of the variant CH3 domain relative to a reference wild type IgG1 Fc structure (PDB ID: 3AVE) is 0.35 Å for the backbone atoms and the RMSD relative to our computationally modeled structures is 0.70 Å. All mutated amino acids introduced in the Fc are fully buried in the CH3 interface and preserve the natural homodimeric symmetry of the wild- type CH3 domain and its surface characteristics. The designed Fc also retains the normal glycosylation pattern (Supplemental Material) and together with an unperturbed Fc surface, retained parental antibody-like binding affinity to the various Fcγ receptors and resultant effector activities such as antibody-dependent cell-mediated cytotoxicity, antibody-dependent cellular phagocytosis and complement-dependent cytotoxicity assay (data not shown). The heterodimeric Fc also retains typical IgG1 antibody-like FcRn binding features and this translates into parental antibody-like pharmacokinetic properties when evaluated in mice and cynomolgus monkey (data not shown). In silico analysis of the designed Fc sequences for potential T cell epitopes does not flag any new MHC ligands as a result of the mutations (data not shown).Citation36,Citation37
Stability of the designed Fc was determined using differential scanning calorimetry (DSC) and the thermal melting curves of ZW1 and other reference heterodimeric Fc-based antibodies is shown in . The thermograms of the full-length antibody molecules were fitted to four independent transitions to capture the differences in melting transitions of the Fab, CH2 and CH3 domains (see Material and Methods for details). The CH3 domain in the wild type IgG1 Fc has the midpoint of thermal unfolding (Tm) at 82.4°C and the CH2 domain has a Tm of 71.6°C.Citation38 Based on the first iteration in our design cycle, the F405A and Y407V mutations on chain A coupled with the T366L and T394W mutations on chain B yielded sufficient steric complementarity to favor a heterodimeric Fc with a CH3 Tm of ~72°C, about 10°C lower than the wild type CH3 domain and already comparable to the KiH variant ((ChainA) T366S/L368A/Y407V and (Chain B) T366W).Citation6 Subsequent optimization incorporating the L351Y mutation on chain A and K392M or K392L mutation on chain B improves the CH3 domain Tm close to 78°C. The ZW1 variant further includes the T350V mutation on both heavy chains and exhibits a Tm of 81.8°C for the CH3. The T350 position was previously reported as insignificant in the stabilization of the CH3:CH3 complex,Citation30 but we observe that the T350V mutation can be synergistically combined with other mutations via intra as well as inter-chain interactions to favor the stabilization of the CH3 domain. Similar to the KiH mutations,Citation6 all the lead designs developed in this study retain wild type-like stability of the CH2 domain.
Figure 2.(A) Differential scanning calorimetry (DSC) of ZW1 in comparison to wild type Fc and the Knob-into-hole (KiH) control. The melting temperature of the CH2 and CH3 domains are highlighted in green and blue respectively. (B) Forced degradation study of purified ZW1. Presence of high molecular weight (HMW) and low molecular weight (LMW) contaminants was determined by HPL-SEC. (C) Pre-formulation aggregation / turbidity screening of ZW1. The susceptibility of ZW1 to form visibly aggregates (precipitate) was evaluated under different buffer conditions by thermal melting and detection of the sample turbidity (see Supplementary Material and Methods for evaluation of pH conditions). (D) Analysis of purified ZW1 and trastuzumab at 100mg/ml by size exclusion chromatography.
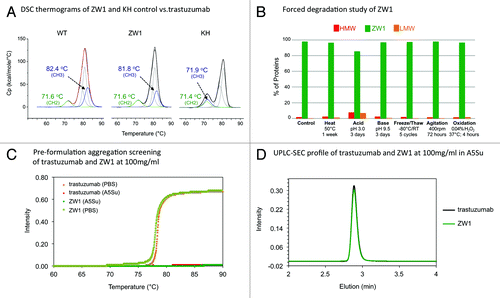
It is well-appreciated that the stability of the Fc is critical for obtaining a successful antibody therapeutic.Citation39,Citation40 Within the Fc structure, the CH2 domain melts at a lower temperature than the CH3 domain and this raises the questions of whether it is sufficient to retain thermal stability of the engineered heterodimeric antibody close to or just above the melting temperature of the CH2 domain and whether there is any particular advantage to recovering the wild type CH3 domain thermal stability in the engineered design. The CH2 domain is known to reversibly fold and the limiting factor in irreversible thermal unfolding of an antibody is likely the Fab, which in a number of clinically-developed antibodies has been engineered to have a Tm that is equivalent or higher than the CH3 melting temperature.Citation41,Citation42 The IgG1 structure has naturally evolved such that a stable CH3 domain is central to pairing of the heavy chains, and further, there is a strong coupling between the proper folded state of the CH3 domain with its stability supported by the correct formation of its internal disulfide bridge and dimer formation.Citation43-Citation45 We have opined that engineering of the heterodimer such that it retains its thermal stability should function as an indirect indicator that the engineered design is equivalent to the wild type Fc in the interchain pairing and stabilization. In addition, proper folding and stability of the CH3 domain could also contribute to the monomer-dimer equilibrium,Citation45 thus a stable CH3 heterodimer improves the homogeneity of the engineered antibody. From an application perspective, the next generation of antibody designs could very well involve bispecific antibodies that are themselves antibody drug conjugates,Citation46 and given that drug conjugation reactions tend to affect the stability and observed Tm of the parent antibody,Citation47 a more stable heterodimeric Fc can improve the manufacturability of these classes of therapeutics.
The favorable WT-like attributes of the ZW1 antibody was also confirmed in a number of other complementary pre-formulation stability studies. In forced degradation experiments, the formation of higher and lower molecular weight species of the heterodimeric antibody (at concentration of 10 mg/mL) was studied after the antibody was subjected to conditions of heat, acid, base, agitation, and oxidation ().Citation48 The short-term freeze-thaw and pH stability studies at concentrations of 10 mg/mL and 30 mg/mL showed no change in HPLC-SEC and CE-SDS profiles when comparing the antibodies before and after treatments (Table S2). Similar to wild-type IgG1 antibodies,Citation49 no detectable visible aggregates were observed in qualitative aggregation propensity studies using the Optimelt automated melting point instrument at concentrations of 30 mg/mL and 100 mg/mL (), as well as at pH of 4.5 and 4.8 and across the temperature range of 30°C to 85°C, while aggregation was detected when the pH is > 5.5 (Fig. S5). Moreover, aggregation was not observed in UPLC-SEC at the higher concentration ().
Purity Assessment of the Stable Heterodimeric Fc Domains
The LC-MS spectrum of the ZW1 variant relative to the control KiH design is shown in .Citation6 The DNA transfection ratios of 1.5:1:2.5, 1:1:2 and 1:1.5:2.5 for each of the two heavy chains A and B and the light chain, respectively, were evaluated during the transient transfection. The amount of light chain DNA ratio is equimolar to the sum of the two heavy chains employed in each of these transfections. The ZW1 variant does not present any detectable homodimeric species, unlike the KiH design, which leads to homodimeric pairing of one of the heavy chainsCitation6 at the increased DNA transfection level of chain A. The other lead variants ZW2 and ZW3 ( and Fig. S6) also present similar purity profiles. The transiently expressed samples of the lead designs, after protein A purification, were consistently observed to have greater than 95% heterodimer, the rest being monomeric (i.e., half antibody) species in multiple expression rounds. There is no detectable amount of either of the two homodimeric species in these samples. To further characterize the two chains and establish the limit of detection of the homodimeric and monomeric species, we independently expressed and characterized (by LC-MS) each of the heavy chains separately with the light chain.Citation50 In these independent expressions of both chain A and chain B, the product formed is greater than 70% monomers. This ability of the individual heavy chains to preferentially exist as stable monomers over homodimers is a critical design attribute and has important implication for the ability to select and develop a stable cell line producing the heterodimeric protein with minimal amount of contaminants.
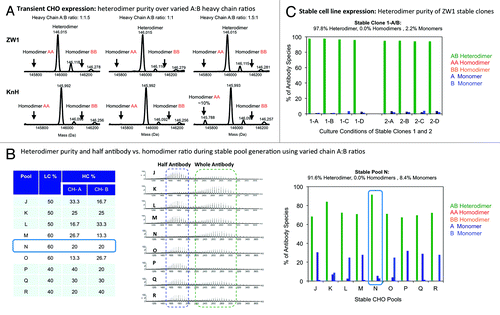
Figure 3. (A) Heterodimer purity of ZW1 in comparison to the knob-into-hole control. In order to estimate the robustness of heterodimer formation, heterodimers were produced by transient co-expression (three plasmids for light chain, heavy chain A and heavy chain B) using three different DNA ratios of the two heavy chains A and B (e.g., ratios A:B = 1:1.5; 1:1; 1.5:1). The heterodimer purity of the ProteinA purified product was determined by LC-MS following EndoS deglycosylation as described in detail in the Methods. As illustrated by the LC-MS spectra, ZW1 forms exclusively heterodimers over the tested expression range, while the KiH control forms close to 10% homodimers if chain A is slightly higher expressed than chain B. (B)-(C) Stable cell line generation of ZW1 and determination of heterodimer purity by LC-MS. The figure in (B) illustrates the generation of stable pools using different ratios of the light and heavy chains for co-transfection. The panel on the right shows the heterodimer purity of the different pools as determined by LC-MS. (C) LC-MS heterodimer purity of two stable clones. Two stable clones (1 and 2) were evaluated under different culture conditions in fed-batch cultures (conditions A-D) and the heterodimer purity of the ProteinA purified product was evaluated by LC-MS.
Stable Cell Line Development
A Chinese hamster ovary (CHO) stable cell line expressing the ZW1 heterodimeric antibody was achieved using a “straight-forward” conventional monoclonal antibody (mAb) development process as an illustration of quality by design, wherein the fundamental biophysical characteristics is shown to affect the developability of the putative drug scaffold.Citation5 Using the Selexis SURE Variant Screening Platform™,Citation51 we developed nine stable CHO-based cell pools in parallel, with each expressing the heterodimeric trastuzumab-based antibody at different heavy and light chain ratios (). The engineered antibody, across all 9 CHO pools, achieved average conventional mAb-like yields (~250 mg/L) and the product quality and quantity were verified using LC-MS, ELISA, and SDS-PAGE. It was observed that the optimal CHO pool was transformed with an excess of light chains over the net amount of the two heavy chains (60:20:20, respectively). The LC-MS profile of the Protein A purified material from the selected CHO pool (Pool N) confirmed the absence of homodimeric contaminants with greater than 91% heterodimers and the remainder being monomeric half antibodies (). Subsequently, clones from Pool N were selected using the standard ClonePIX™ platform proceduresCitation52 with the clones serially expanded and screened based on expression titer to arrive at 20 final candidate clones. The productivity and performance in fed-batch cultivation process indicate that stable clones yield greater than 900 mg/L antibody prior to feed and upstream process optimization, comparable to what can be achieved for a conventional mAb at this stage of process development. As originally envisioned, the heterodimers and monomers are the principal product components across all the clones (). The heterodimer purity of two of the leading clones (1 and 2) are between 95–97% (with 3–5% being a combination of the Chain-A and Chain-B half antibodies) and are not contaminated with any detectable amount of either homodimers. Such stable clones with high productivity and a product profile with little product-derived impurities are typically considered critical success criteria for manufacturing because these attributes directly correlate with reduced costs of goods and a highly pure product consistent with CFR standards for biologics, where purity is defined as freedom from extraneous matter in the finished product (CFR 600.3, part 610). Further, failure to control product quality attributes is recognized as a potential safety risk to patients.
While the data presented here is based on a molecule comprising two different heavy chains and a common light chain, we have employed the heterodimeric Fc as a scaffold to create a number of bispecific molecules based on the fusion of the designed heterodimeric Fc with different antigen-binding modules such as single chain Fv (scFv) and domain antibody. We have consistently observed greater than 95% bispecific product on expression of these in CHO and HEK293 cell lines, confirming that the heterodimeric Fc design can find a wide range of therapeutic applications (data not shown). Further, alternate antigen-binding formats can also be combined within the same molecules to achieve hybrid bispecific designs, e.g., scFv on one arm and Fab on the other, domain antibody with Fab, etc. To summarize, we report a structure-guided protein engineering approach addressing fundamental biophysical properties of a heterodimeric Fc molecule early in its design, as a strategy to enhance its quality attributes like purity and stability that can positively impact developability of the next-generation of bispecifics, drug conjugates and other complex therapeutic proteins.
Methods
Rational protein engineering
A two-stage approach that combined negative and positive design strategies as summarized in Figure S1A was employed. Specifically, in the initial phase the core interface positions in the Fc were computationally screened using different negative design strategies (Fig. S1B) with a total of 16 variants based on 4 core designs characterized in vitro. From this initial set, the Fc variant heterodimers with greater than 90% purity and a melting temperature of about 68°C or greater were selected for further optimization in the second design phase for improved stability and purity using positive design strategies.
Complete all atom models of the wild type Fc structure of an IgG1 antibody was used for all modeling activities. Molecular dynamics (MD) was employed to evaluate the intrinsic dynamic nature of the Fc homodimer and the variant CH3 domains in an aqueous environment. Mean field and dead-end elimination side-chain packing methods along with flexible backbones were used to optimize and prepare model structures for the variants being screened. Following packing optimization, we scored a number of features including contact density, clash score, hydrophobicity and electrostatics. The Generalized Born method allowed us to accurately model the effect of the solvent environment and contrast the free energy differences following mutation of specific positions in the protein to alternate residue types. Contact density and clash score provided a measure of complementarity, a critical aspect of effective protein packing. The screening procedure employed knowledge-based potentials as well as coupling analysis schemes relying on pair-wise residue interaction energy and entropy computations.
Optimization of initial variants and structural rationale
To improve the initial negative design Fc variants for stability and purity, the structural and computational strategies described above were employed. The in depth structure-function analysis of the initial negative design variant provided a detailed understanding for each of the mutations introduced.
For example, the initial design (termed Steric-1) that led to the final variants presented here was based on a steric complementarity design approach close to the core of the CH3 interface. Detailed modeling and simulation analysis of the initial Steric-1 variant showed that the important interface hotspots were lost with respect to wild-type homodimer formation, in particular the interactions of wild-type (chainA)F405-(chainB)K409, (chainA)Y407-(chainB)T366 and the packing of (chainAB)Y407-Y407 and –(chainA)F405 (Fig. S2A). The analysis also revealed that one strong wild-type hotspot (chainA)T366 was affected, but not contributing in the heterodimer formation, while likely still being present in the unpreferred homodimer. As shown in (main text) and illustrated in Figure S2A, the single amino acid change of (chainB)T366L increased the heterodimer purity of the initial design variants from ~80% to > 95%.
The molecular dynamics simulation of the initial heterodimer variant with low stability showed a large conformational difference in the loop region D399-S400-D401 (Fig. S2B) and the associated β-sheets at K370. This resulted in the loss of the interchain interactions K409-D399. In the WT IgG1 CH3 domain, these regions tether the interface at the rim and protect the hydrophobic core interactions. This analysis indicated an important factor for the lower stability of the initial heterodimer variant compared with WT stability.
Consequently, we identified the key residues and sequence motifs responsible for the low stability and the subsequent positive design engineering efforts were therefore specifically focused on stabilizing the loop conformation of positions 399–401 in a more “closed”-wild-type like conformation. To achieve this stabilization of the loop conformation of positions 399–401, the computational packing and modeling approach was used to evaluate different targeted design ideas. This strategy identified the single mutation difference K392M/L that leads to an increase in CH3 stability by about 4 °C (Table S1B, Fig. S2C).
Third, a cavity at the core packing positions T366, T394W and L368 was identified as a possible reason for the lower than wild-type stability (Fig. S2D). To improve the core packing, we computationally screened the positions at T366/L368 and we evaluated distal positions for stabilization of the core packing. This procedure identified the distal swap L351Y, which as a single mutation does not show any effect, but in combination with T366L and L368 gives an improved CH3 Tm of greater than 5°C, indicating a strong coupling effect of the distal change L351Y.
The employed engineering approach to improve the heterodimer stability is not limited to introducing mutations that increase complementarity across the two chains. We evaluated mutations of amino acids that are not directly contacting the complementary chain as a means to improve the stability of the Fc heterodimer. As an example, the second shell position T350 in the CH3 domain of IgG1 is buried with the threonine residue facing the interior of the CH3 domain (Fig. S2E). The distal second shell mutation T350V was identified by the described computational screening, and it improves the stability (Tm) of the Fc domain by about 2°C.
Transient expression and purification of heterodimer variants
The genes encoding the antibody heavy and light chains were constructed via gene synthesis using codons optimized for mammalian expression. The Fab sequences were generated from the HER2-binding trastuzumab and the Fc was an IgG1 isotype. The final gene products were separately sub-cloned into the mammalian expression vector pTT5. Mutations in the CH3 domain were introduced via site-directed mutagenesis of the template vectors. Transient co-expression in HEK293 or CHO was conducted using the 3 individual vectors encoding for the one light chain or the two heterodimer heavy chains A and B. The clarified culture medium was loaded onto a MabSelect SuRe (GE Healthcare) protein-A column and washed with 10 column volumes of PBS buffer at pH 7.2. The antibody was eluted with 10 column volumes of citrate buffer at pH 3.6 with the pooled fractions containing the antibody neutralized with TRIS at pH 11. The protein was then desalted using an Econo-Pac 10DG column (Bio-Rad) equilibrated in PBS.
Analysis of glycosylation profile
The trastuzumab-based ZW1 heterodimer was expressed and purified as described above. Glycans were analyzed with GlykoPrep™ Rapid N-Glycan Preparation with InstantAB™ (Prozyme) using the standard manufacturer protocol. The results are shown in Figure S3 and illustrate that the Azymetric heterodimer has a typical IgG1-like complex-type glycosylation pattern.
Differential scanning calorimetry
All DSC experiments were performed using a GE VP-Capillary instrument. The proteins were buffer-exchanged into PBS (pH 7.4) and diluted to 0.4 to 0.5 mg/mL, loaded into the sample cell and measured with a scan rate of 1°C/min from 20 to 100°C. Data was analyzed using the Origin software (GE Healthcare) with the PBS buffer background subtracted and normalized for the protein concentration.
To estimate the melting temperature (Tm) of the CH3 domain and to accurately compare variants, two different data analysis methods were employed. In one example the DSC thermograms were fitted to four independent transitions using the non 2-state model. For reliable Tm estimation, the transitions for the Fab and the CH2 domains were kept constant and equivalent for different variants (). To verify this Tm estimate, the variant thermograms were also subtracted from the wild type thermogram, which yielded a negative peak for the wild type CH3 domain and a positive peak for the variant peak (Fig. S4). The difference curve is flat with no peaks either in the positive or negative direction when the melting temperature of the variant is equal to that of the reference wild type protein domain.
Pre-formulation and stability assessment
The stability of the ZW1 heterodimer was assessed by incubation under forced degradation conditions. Purified heterodimer sample was concentrated to 150 mg/ml without signs of aggregation. The samples were diluted into the appropriate buffer and evaluated under forced degradation conditions as described in Table S1. The treated samples were analyzed by SDS-PAGE, CE-SDS and HPLC-SEC. The results are illustrated in and in Table S2.
In addition the stability of the sample was evaluated using a protein aggregation propensity assay to determine its susceptibility to visibly aggregate (precipitate) under various conditions, such as pH, temperature and salt ( and Fig. S5). Thirty μl of sample was placed in a glass capillary tube (1.5 × 90 mm) using a gel-loading tip. The capillary tubes were placed in the OptiMelt instrument (Stanford Research Systems) and the protein was forced to aggregate by linearly ramping the temperature (1°C/min from 30°C to 85°C), while the aggregation process itself was monitored by an increase in the sample turbidity.
Downstream purification assessment
Manufacturability assessment was performed to evaluate the behavior of the heterodimeric antibody using the industry-standard antibody purification processes combined with stability and product quality evaluations. This process involved a three-column step platform comprising Protein A affinity chromatography for capture, followed by cation exchange (CEX) chromatography for the removal of aggregates, leached protein-A and HCP. Finally, anion exchange (AEX) chromatography step in the flow-through mode is introduced to capture viruses, DNA and negatively charged contaminants. This assessment was used to identify potential manufacturing problems (e.g., process stability, product stability and quality) with a drug candidate(s) early in the research/development stage. Table S3 lists the major criteria used for the assessment of the sample, for example, step yields, HMW content, and elution volume and is summarized in Figure S6.
LC-MS heterodimer quantification
The purified samples were analyzed as is or de-glycosylated with PNGase F (0.2U PNGase F/μg of antibody in 50 mM TRIS-HCl pH 8.0; overnight incubation at 37°C; final protein concentration was 0.45 mg/mL) or alternatively with EndoS (deglycosylation with EndoS using manufacturer protocol conditions, 50 mM TRIS-HCl pH 8.0 and incubation for 1 h at 37°C). The protein samples were analyzed by LC-MS using an Agilent 1100 HPLC system coupled to an LTQ-Orbitrap XL hybrid mass spectrometer (ThermoFisher Scientific) via a high-flow electrospray interface. The samples (2.5 μg) were injected onto a 2.1 × 10 mm Poros R2 column (Applied Biosystems) and eluted using a 2 mL/min gradient of 20–90% ACN, 0.1% FA over 3 min. The flow was split post-column to direct 100 μL/min into the electrospray interface. The column and solvent were heated to 80°C to improve protein peak shape. The LTQ-Orbitrap XL was calibrated using ThermoFisher Scientific’s LTQ Positive Ion ESI calibration solution (caffeine, MRFA and Ultramark 1621), and tuned using a 25 μg/uL solution of BSA. The cone voltage (source fragmentation setting) was 40 V, the FT resolution was 7,500 and the scan range was m/z 400–4,000. The protein spectra were deconvoluted using the Water MaxEnt software. The abundances of the hetero- and homodimer antibody species were determined directly from the resulting molecular weight profiles. The linearity of response was confirmed using defined mixtures of antibodies. The limit of detection was approximately 2%. An alternative purity assessment method using the Acquity UPLC – Xevo G2 QToF MS system was also developed.Citation50 Glycosylation of the heterodimeric antibodies was evaluated by trypsin digest followed by nano LC-MS/MS at the glycopeptide level. The LC-MS spectra of variants ZW2 and ZW3 are shown in the Figure S6.
Stable cell line development
Stable CHO cell lines expressing ZW1 were generated using Selexis’s proprietary SURE Variant Screening Platform™ employing one round of transfection and integration for pool generation followed by a single ClonePIX™ selection of stable clone candidates. One transfection series using different ratios of light chain and heavy chain A:heavy chain B was performed using monocistronic vectors (containing the puromycin resistance cassette), generating pools J, K, L, M, N, O, P, Q and R as illustrated in detail in . One single-cell derived colony picking round (ClonePIX™) was conducted to provide highly productive clonal cell lines. Approximately 100 clonal cell lines were screened using double-sandwich ELISA based selection process and the 20 best performing clones based on titer and growth characteristics were selected. The product profile for the pools and clone candidates was analyzed by SDS-PAGE and LC-MS (as described above).
| Abbreviations: | ||
| RMSD | = | root mean squared deviation |
| HPLC-SEC | = | high-performance liquid chromatography-size exclusion chromatography |
| CE-SDS | = | capillary electrophoresis-sodium dodecyl sulphate |
| SDS-PAGE | = | sodium dodecyl sulphate-polyacrylamide gel electrophoresis |
| DSC | = | differential scanning calorimetry |
| LC-MS | = | liquid chromatography-mass spectroscopy |
| ELISA | = | enzyme-linked immune sorbent assay |
| A5Su | = | acetate pH 5.0 sucrose buffer |
| Fc | = | fragment crystallizable |
| HCP | = | host cell protein |
| UPLC-SEC | = | ultra performance liquid chromatography-size exclusion chromatography |
Additional material
Download Zip (2.9 MB)Acknowledgments
We acknowledge valuable discussions with Mike Gresser, Tom Boone, Andreas Raibekas, Nessa Hawkins and Prathima Acharya. We thank Anders Ohrn and Greg Lakatos for useful discussions on application of molecular simulation techniques. We thank Luc Tessier and Anna Robotham for carrying out LC-MS experiments. X-ray diffraction data for both of the Azymetric structures were collected on beamline 08B1-1 at the Canadian light source, which is supported by the Natural Sciences and Research Engineering Council of Canada, the National Research Council Canada, the Canadian Institutes of Health Research, the Province of Saskatchewan, Western Economic Diversification Canada, and the University of Saskatchewan. We thank M. Fodje for assistance in data collection and beamline operation. MLDS is a Michael Smith Foundation for Health Research (MSFHR) Scholar. MJB is a Canada Research Chair and a MSFHR scholar. Author Contributions: SBD, GYKN, PIL, TSK, JK, YD, HX, PAG, MJB, DKYP designed the work. TSK, PIL, EEC, ID, KB, JK, JB, RJW, MHX, PAG, MS performed the work. SBD and TSK wrote the manuscript; all the authors reviewed the manuscript.
Disclosure of Potential Conflicts of Interest
TSK, EEC, PIL, ID, DKYP, GYKN, SBD are employees of Zymeworks Inc.; RJW and MHX are employees of KBI Biopharma Inc. and PAG is employee of Selexis SA.
Author Contributions
SBD, GYKN, PIL, TSK, JK, YD, HX, PAG, MJB, DKYP designed the work. TSK, PIL, EEC, ID, KB, JK, JB, RJW, MHX, PAG, MLDS performed the work. SBD and TSK wrote the manuscript; all the authors reviewed the manuscript.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/nucleus/article/25632.
References
- Kontermann R. Dual targeting strategies with bispecific antibodies. MAbs 2012; 4:182 - 97; http://dx.doi.org/10.4161/mabs.4.2.19000; PMID: 22453100
- Carter PJ. Introduction to current and future protein therapeutics: a protein engineering perspective. Exp Cell Res 2011; 317:1261 - 9; http://dx.doi.org/10.1016/j.yexcr.2011.02.013; PMID: 21371474
- Segal DM, Weiner GJ, Weiner LM. Introduction: bispecific antibodies. J Immunol Methods 2001; 248:1 - 6; http://dx.doi.org/10.1016/S0022-1759(00)00338-0; PMID: 11223064
- Carter P. Bispecific human IgG by design. J Immunol Methods 2001; 248:7 - 15; http://dx.doi.org/10.1016/S0022-1759(00)00339-2; PMID: 11223065
- Swann PG, Tolnay M, Muthukkumar S, Shapiro MA, Rellahan BL, Clouse KA. Considerations for the development of therapeutic monoclonal antibodies. Curr Opin Immunol 2008; 20:493 - 9; http://dx.doi.org/10.1016/j.coi.2008.05.013; PMID: 18586093
- Ridgway JB, Presta LG, Carter P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng 1996; 9:617 - 21; http://dx.doi.org/10.1093/protein/9.7.617; PMID: 8844834
- Atwell S, Ridgway JB, Wells JA, Carter P. Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library. J Mol Biol 1997; 270:26 - 35; http://dx.doi.org/10.1006/jmbi.1997.1116; PMID: 9231898
- Merchant AM, Zhu Z, Yuan JQ, Goddard A, Adams CW, Presta LG, et al. An efficient route to human bispecific IgG. Nat Biotechnol 1998; 16:677 - 81; http://dx.doi.org/10.1038/nbt0798-677; PMID: 9661204
- Kitazawa T, Igawa T, Sampei Z, Muto A, Kojima T, Soeda T, et al. A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat Med 2012; 18:1570 - 4; http://dx.doi.org/10.1038/nm.2942; PMID: 23023498
- Yu YJ, Zhang Y, Kenrick M, Hoyte K, Luk W, Lu Y, et al. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci Transl Med 2011; 3:84ra44; http://dx.doi.org/10.1126/scitranslmed.3002230; PMID: 21613623
- Schaefer W, Regula JT, Bähner M, Schanzer J, Croasdale R, Dürr H, et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Sci U S A 2011; 108:11187 - 92; http://dx.doi.org/10.1073/pnas.1019002108; PMID: 21690412
- Klein C, Sustmann C, Thomas M, Stubenrauch K, Croasdale R, Schanzer J, et al. Progress in overcoming the chain association issue in bispecific heterodimeric IgG antibodies. MAbs 2012; 4:653 - 63; http://dx.doi.org/10.4161/mabs.21379; PMID: 22925968
- Jin H, Yang R, Zheng Z, Romero M, Ross J, Bou-Reslan H, et al. MetMAb, the one-armed 5D5 anti-c-Met antibody, inhibits orthotopic pancreatic tumor growth and improves survival. Cancer Res 2008; 68:4360 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-07-5960; PMID: 18519697
- Klein C, Schaefer W. Bivalent, bispecific antibodies. USPTO. US2009/023811A1, 2009:para 268.
- Jackman J, Chen Y, Huang A, Moffat B, Scheer JM, Leong SR, et al. Development of a two-part strategy to identify a therapeutic human bispecific antibody that inhibits IgE receptor signaling. J Biol Chem 2010; 285:20850 - 9; http://dx.doi.org/10.1074/jbc.M110.113910; PMID: 20444694
- Wranik BJ, Christensen EL, Schaefer G, Jackman JK, Vendel AC, Eaton D. LUZ-Y, a novel platform for the mammalian cell production of full-length IgG-bispecific antibodies. J Biol Chem 2012; 287:43331 - 9; http://dx.doi.org/10.1074/jbc.M112.397869; PMID: 23118228
- Strop P, Ho WH, Boustany LM, Abdiche YN, Lindquist KC, Farias SE, et al. Generating bispecific human IgG1 and IgG2 antibodies from any antibody pair. J Mol Biol 2012; 420:204 - 19; http://dx.doi.org/10.1016/j.jmb.2012.04.020; PMID: 22543237
- Labrijn AF, Meesters JI, de Goeij BE, van den Bremer ET, Neijssen J, van Kampen MD, et al. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc Natl Acad Sci U S A 2013; 110:5145 - 50; http://dx.doi.org/10.1073/pnas.1220145110; PMID: 23479652
- Gunasekaran K, Pentony M, Shen M, Garrett L, Forte C, Woodward A, et al. Enhancing antibody Fc heterodimer formation through electrostatic steering effects: applications to bispecific molecules and monovalent IgG. J Biol Chem 2010; 285:19637 - 46; http://dx.doi.org/10.1074/jbc.M110.117382; PMID: 20400508
- Davis JH, Aperlo C, Li Y, Kurosawa E, Lan Y, Lo KM, et al. SEEDbodies: fusion proteins based on strand-exchange engineered domain (SEED) CH3 heterodimers in an Fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies. Protein Eng Des Sel 2010; 23:195 - 202; http://dx.doi.org/10.1093/protein/gzp094; PMID: 20299542
- Moore GL, Bautista C, Pong E, Nguyen DH, Jacinto J, Eivazi A, et al. A novel bispecific antibody format enables simultaneous bivalent and monovalent co-engagement of distinct target antigens. MAbs 2011; 3:546 - 57; http://dx.doi.org/10.4161/mabs.3.6.18123; PMID: 22123055
- Wang W. Protein aggregation and its inhibition in biopharmaceutics. Int J Pharm 2005; 289:1 - 30; http://dx.doi.org/10.1016/j.ijpharm.2004.11.014; PMID: 15652195
- Demarest SJ, Glaser SM. Antibody therapeutics, antibody engineering, and the merits of protein stability. Curr Opin Drug Discov Devel 2008; 11:675 - 87; PMID: 18729019
- Wen J, Jiang Y, Hymes K, Gong K, Nahri L. Using Differential Scanning Calorimetry in Understanding the Correlation Between Thermal Stability and Protein Stability: A Case Study. Microcal Application Note, 2011.
- Liu H, May K. Disulfide bond structures of IgG molecules: structural variations, chemical modifications and possible impacts to stability and biological function. MAbs 2012; 4:17 - 23; http://dx.doi.org/10.4161/mabs.4.1.18347; PMID: 22327427
- Yang X, Xu W, Dukleska S, Benchaar B, Mengisen S, Antochshuk V, et al. Developability studies before initiation of process development: Improving manufacturability of monoclonal antibodies. MAbs 2013; 5; http://dx.doi.org/10.4161/mabs.25269
- Buchanan A, Clementel V, Woods R, Harn N, Bowen MA, Mo W, et al. Engineering a therapeutic IgG molecule to address cysteinylation, aggregation and enhance thermal stability and expression. MAbs 2013; 5:255 - 62; http://dx.doi.org/10.4161/mabs.23392; PMID: 23412563
- Bolon DN, Grant RA, Baker TA, Sauer RT. Specificity versus stability in computational protein design. Proc Natl Acad Sci U S A 2005; 102:12724 - 9; http://dx.doi.org/10.1073/pnas.0506124102; PMID: 16129838
- Deisenhofer J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-A resolution. Biochemistry 1981; 20:2361 - 70; http://dx.doi.org/10.1021/bi00512a001; PMID: 7236608
- Dall’Acqua W, Simon AL, Mulkerrin MG, Carter P. Contribution of domain interface residues to the stability of antibody CH3 domain homodimers. Biochemistry 1998; 37:9266 - 73; http://dx.doi.org/10.1021/bi980270i; PMID: 9649307
- Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006; 65:712 - 25; http://dx.doi.org/10.1002/prot.21123; PMID: 16981200
- Jayaram B, Sprous D, Beveridge DL. Solvation Free Energy of Biomacromolecules: Parameters for a Modified Generalized Born Model Consistent with the AMBER Force Field. J Phys Chem B 1998; 102:9571 - 6; http://dx.doi.org/10.1021/jp982007x
- Vajda S, Sippl M, Novotny J. Empirical potentials and functions for protein folding and binding. Curr Opin Struct Biol 1997; 7:222 - 8; http://dx.doi.org/10.1016/S0959-440X(97)80029-2; PMID: 9094333
- Xiang Z, Honig B. Extending the accuracy limits of prediction for side-chain conformations. J Mol Biol 2001; 311:421 - 30; http://dx.doi.org/10.1006/jmbi.2001.4865; PMID: 11478870
- Davis IW, Arendall WB 3rd, Richardson DC, Richardson JS. The backrub motion: how protein backbone shrugs when a sidechain dances. Structure 2006; 14:265 - 74; http://dx.doi.org/10.1016/j.str.2005.10.007; PMID: 16472746
- De Groot AS, Martin W. Reducing risk, improving outcomes: bioengineering less immunogenic protein therapeutics. Clin Immunol 2009; 131:189 - 201; http://dx.doi.org/10.1016/j.clim.2009.01.009; PMID: 19269256
- Bryson CJ, Jones TD, Baker MP. Prediction of immunogenicity of therapeutic proteins: validity of computational tools. BioDrugs 2010; 24:1 - 8; http://dx.doi.org/10.2165/11318560-000000000-00000; PMID: 20055528
- Ionescu RM, Vlasak J, Price C, Kirchmeier M. Contribution of variable domains to the stability of humanized IgG1 monoclonal antibodies. J Pharm Sci 2008; 97:1414 - 26; http://dx.doi.org/10.1002/jps.21104; PMID: 17721938
- Li CH, Narhi LO, Wen J, Dimitrova M, Wen ZQ, Li J, et al. Effect of pH, temperature, and salt on the stability of Escherichia coli- and Chinese hamster ovary cell-derived IgG1 Fc. Biochemistry 2012; 51:10056 - 65; http://dx.doi.org/10.1021/bi300702e; PMID: 23078371
- Demarest SJ, Rogers J, Hansen G. Optimization of the antibody C(H)3 domain by residue frequency analysis of IgG sequences. J Mol Biol 2004; 335:41 - 8; http://dx.doi.org/10.1016/j.jmb.2003.10.040; PMID: 14659738
- Garber E, Demarest SJ. A broad range of Fab stabilities within a host of therapeutic IgGs. Biochem Biophys Res Commun 2007; 355:751 - 7; http://dx.doi.org/10.1016/j.bbrc.2007.02.042; PMID: 17321501
- Feige MJ, Walter S, Buchner J. Folding mechanism of the CH2 antibody domain. J Mol Biol 2004; 344:107 - 18; http://dx.doi.org/10.1016/j.jmb.2004.09.033; PMID: 15504405
- Thies MJ, Mayer J, Augustine JG, Frederick CA, Lilie H, Buchner J. Folding and association of the antibody domain CH3: prolyl isomerization preceeds dimerization. J Mol Biol 1999; 293:67 - 79; http://dx.doi.org/10.1006/jmbi.1999.3128; PMID: 10512716
- Thies MJ, Talamo F, Mayer M, Bell S, Ruoppolo M, Marino G, et al. Folding and oxidation of the antibody domain C(H)3. J Mol Biol 2002; 319:1267 - 77; http://dx.doi.org/10.1016/S0022-2836(02)00375-3; PMID: 12079363
- McAuley A, Jacob J, Kolvenbach CG, Westland K, Lee HJ, Brych SR, et al. Contributions of a disulfide bond to the structure, stability, and dimerization of human IgG1 antibody CH3 domain. Protein Sci 2008; 17:95 - 106; http://dx.doi.org/10.1110/ps.073134408; PMID: 18156469
- Reichert JM. Bispecific antibodies and ADCs: Once and future kings?. MAbs 2011; 3:329 - 30; http://dx.doi.org/10.4161/mabs.3.4.16589; PMID: 21654205
- Wakankar AA, Feeney MB, Rivera J, Chen Y, Kim M, Sharma VK, et al. Physicochemical stability of the antibody-drug conjugate Trastuzumab-DM1: changes due to modification and conjugation processes. Bioconjug Chem 2010; 21:1588 - 95; http://dx.doi.org/10.1021/bc900434c; PMID: 20698491
- Hawe A, Wiggenhorn M, van de Weert M, Garbe JH, Mahler HC, Jiskoot W. Forced degradation of therapeutic proteins. J Pharm Sci 2012; 101:895 - 913; http://dx.doi.org/10.1002/jps.22812; PMID: 22083792
- Raibekas AA. Estimation of protein aggregation propensity with a melting point apparatus. Anal Biochem 2008; 380:331 - 2; http://dx.doi.org/10.1016/j.ab.2008.05.023; PMID: 18544334
- Woods RJ, Xie M, von Kreudenstein T, Ng G, Dixit SB. LC-MS characterization and purity assessment of a prototype bispecific antibody. MAbs 2013; 5; http://dx.doi.org/10.4161/mabs.25488
- Girod PA, Le Fourn V. Rapid production of functional proteins of a combinatorial IgG library in CHO cells. Bioprocess Int 2012; 10:58 - 61
- Serpieri F, Inocencio A, de Oliveira JM, Pimenta AA Jr., Garbuio A, Kalil J, et al. Comparison of humanized IgG and FvFc anti-CD3 monoclonal antibodies expressed in CHO cells. Mol Biotechnol 2010; 45:218 - 25; http://dx.doi.org/10.1007/s12033-010-9269-2; PMID: 20336495