Abstract
About 60 percent of glioblastomas highly express the gangliosides 3′-isoLM1 and 3′,6′-isoLD1 on the cell surface, providing ideal targets for brain tumor immunotherapy. A novel recombinant immunotoxin, DmAb14m-(scFv)-PE38KDEL (DmAb14m-IT), specific for the gangliosides 3′-isoLM1 and 3′,6′-isoLD1, was constructed with improved affinity and increased cytotoxicity for immunotherapeutic targeting of glioblastoma. We isolated an scFv parental clone from a previously established murine hybridoma, DmAb14, that is specific to both 3′-isoLM1 and 3′,6′-isoLD1. We then performed in vitro affinity maturation by CDR hotspot random mutagenesis. The binding affinity and specificity of affinity-matured DmAb14m-IT were measured by surface-plasmon resonance, flow cytometry, and immunohistochemical analysis. In vitro cytotoxicity of DmAb14m-IT was measured by protein synthesis inhibition and cell death assays in human cell lines expressing gangliosides 3′-isoLM1 and 3′,6′-isoLD1 (D54MG and D336MG) and xenograft-derived cells (D2224MG). As a result, the KD of DmAb14m-IT for gangliosides 3′-isoLM1 and 3′,6′-isoLD1 was 2.6 × 10−9M. Also, DmAb14m-IT showed a significantly higher internalization rate in cells expressing 3′-isoLM1 and 3′,6′-isoLD1. The DmAb14m-IT IC50 was 80 ng/mL (1194 pM) on the D54MG cell line, 5 ng/ml (75 pM) on the D336MG cell line, and 0.5 ng/ml (7.5 pM) on the D2224MG xenograft-derived cells. There was no cytotoxicity on ganglioside-negative HEK293 cells. Immunohistochemical analysis confirmed the specific apparent affinity of DmAb14m-IT with 3′-isoLM1 and 3′,6′-isoLD1. In conclusion, DmAb14m-IT showed specific binding affinity, a significantly high internalization rate, and selective cytotoxicity on glioma cell lines and xenograft-derived cells expressing 3′-isoLM1 and 3′,6′-isoLD1, thereby displaying robust therapeutic potential for testing the antitumor efficacy of DmAb14m-IT at the preclinical level and eventually in the clinical setting.
Introduction
Over 17,000 cases of primary malignant brain tumors are diagnosed each year in the United States, and the incidence appears to be increasing. The most common type of primary brain tumor is glioblastoma multiforme, which has a median survival time of 40 to 50 weeks despite aggressive treatment.Citation1 Therefore, it is imperative that we develop novel approaches to treat glioblastoma and improve patients’ prognosis. Even though targeted monoclonal antibody (mAb) immunotherapy has substantial effects on medical care, many cancers are still resistant to mAb therapy alone.Citation2 To enhance therapeutic efficacy, many researchers have fused mAbs with cytotoxic drugs, radioisotopes, or toxins.Citation3
Gangliosides are a group of sialic acid-containing glycosphingolipids with the antigenic hydrophilic carbohydrate section extending extracellularly. The sequence of sugar residues that defines classes or “series” of gangliosides can also be found on glycoproteins. The appearance of novel or quantitative shifts in ganglioside phenotype following neoplastic transformation has been reported in tumors of diverse origins.Citation4,Citation5 Of greatest potential as glioma-specific oncofetal antigens and therapeutic targets are the surface gangliosides 3′-isoLM1 and 3′,6'-isoLD1. Within the central nervous system, 3′-isoLM1 and 3′,6'-isoLD1 marked areas of rapid fetal astroglial proliferation and migration; they also serve operationally as specific biomarkers of neoplasia in glioblastomas.Citation6,Citation7 The gangliosides 3′-isoLM1 and 3′,6′-isoLD1 have been identified as therapeutic targets in malignant gliomasCitation8,Citation9 due to their particular overexpression in glioblastomas.Citation10,Citation11 Since the expression of these molecules is normally restricted to human adult brain tissue and the central nervous system,Citation9,Citation12 they can potentially be used as diagnostic markers and targets for cancer immunotherapy.
We previously produced the monoclonal IgM antibodies DmAb14 (specific for both 3′-isoLM1 and 3′,6'-isoLD1), SL50 (specific for 3′-isoLM1) and DmAbs 21 and 22 (specific for 3′,6'-isoLD1).Citation8 These mAbs are exquisitely specific, but the IgM antibody class, which is common for anti-carbohydrate mAbs, is not optimal for preclinical studies or clinical trials. The distribution patterns of 3′-isoLM1 and 3′,6'-isoLD1 were studied with these specific mAbs by both immunohistochemistry and high performance liquid chromatography. The ganglioside 3′-isoLM1 was expressed in 48% of human gliomas,Citation13 and 3′,6'-isoLD1 (detected by DmAbs 21 and 22) was expressed in 68% of high-grade gliomas. Biochemical analysis of postmortem samples from glioma patients indicated that the gangliosides were expressed largely in the periphery of tumors instead of the central tumor massCitation12; a similar xenograft study supported these observations.Citation11 The potential role of these gangliosides in tumor cell migration/invasion makes it possible to utilize them as exquisitely specific cell-surface therapeutic targets to attack invasive and metastatic glioblastoma cells.
Among our ganglioside-targeting mAbs, DmAb14 exhibited high reactivity to both 3′-isoLM1 and 3′,6′-isoLD1,Citation14 but as an IgM,Citation13 the molecule is not ideal for clinical applications because of its large molecular size, poor tissue-penetrating ability, and complicated functioning mechanisms. Hence, an alternative strategy is use of genetically engineered single-chain variable fragments (scFv, VH and VL) fused with Pseudomonas exotoxin, which targets antigens specifically expressed by malignant gliomas. The constructed scFv unit can be engineered as a targeting guide to direct natural toxinsCitation15 or radioactive compoundsCitation16 to kill antigen-expressing tumor cells selectively. In addition, compared with the intact immunoglobulin molecules IgG or IgM, the smaller scFvs have a greater potential to clear rapidly from the blood and deeply penetrate tumors,Citation17 leading to enhanced therapeutic efficacy.Citation18
In this study, we first cloned the VH and VL fragment sequences from hybridoma DmAb14 to construct scFvs that retained the specificity against 3′-isoLM1 and 3′,6′-isoLD1. To yield high-affinity scFvs, in vitro affinity maturation of DmAb14-scFvs was performed by complementary-determining region (CDR) hotspot random mutagenesis. One of the best affinity-matured scFvs, Dmab14 min, was then converted into a recombinant immunotoxin (RIT) by fusing it with a genetically engineered Pseudomonas exotoxin, PE38KDEL, which carries a C-terminal with the last four amino acids of KDEL (lysine, aspartic acid, glutamic acid and leucine) to improve intracellular retention. This RIT, DmAb14m-PE38KDEL (DmAb14m-IT), developed to target the gangliosides 3′-isoLM1 and 3′,6′-isoLD1, showed matured affinity, robust cell internalization, and significant cytotoxicity, compared with the parental molecule and control RITs, indicating its potential utility for treatment of malignant brain tumors. An in vivo study will be organized in the future to test its therapeutic application.
Results
Construction of the VHCDR2 and VLCDR1 libraries
The affinity of DmAb14-scFv was improved through random mutagenesis based on hotspots within either the VHCDR2 region or the VLCDR1 region (). This approach is based on the fact that the DNA encoding the variable regions of antibodies contains mutational hotspots, which are nucleotide sequences where mutations are frequently concentrated during the in vivo affinity maturation process. Moreover, the mutation of hotspots that have significant contact with the antigens could alter the affinity of the antibodies. The amino acid sequences of VHCDR2 and VLCDR1 are shown in . The libraries introduced randomizations in the hotspot motif residues S75, N76, G77, K86, S87, and K88 for VHCDR2 and R25, A26, S27, S32, and Y33 for VLCDR1 in separate experiment sets as described in “Materials and Methods.” In the cloning step, three VH libraries were mixed and yielded 6 × 104 clones, and four VL libraries yielded 5 × 104 clones. Twenty clones were sequenced from the VH library and 14 from the VL library to verify that the construction of the library was appropriate. Sequencing data shows that each clone had different amino acid combinations in the region targeted for mutations. The size of the libraries ensured that most of the DNA sequences were represented.
Figure 1. (A) PCR construction of mutant libraries. A primer fragment was first generated by using upstream DmAb14-scFv-F (NcoI) primer and downstream VHCDR2-aR, -bR, or -cR primers containing degeneracies (NNS) in the targeted VHCDR2 region. Another primer fragment was generated by using downstream DmAb14-scFv-R (NotI) and upstream VLCDR1-aF, -bF, -cF, or -dF primers containing degeneracies (NNS) in the targeted VLCDR1 region. DmAb14-scFv parental plasmid served as the template for amplification. The VHCDR2 fragments (280 bp) generated (panel A) and DmAb14-scFv-R(NotI) were paired and used as upstream and downstream primers (respectively) to generate the whole length of the scFv with VH mutation pool. Upstream DmAb14-scFv-F(NcoI) and downstream VLCDR1 (260 bp) primers, shown in panel A, were paired to generate a 750-bp scFv with a VL mutations library. (B) Different attacking mechanisms of IgM antibody and scFv-PE38KEDL RIT toward cancer cells. Left side of shows that IgM antibodies first form a pentamer to bind several identical epitopes on the cancer cells, and then need complements and/or phagocytes to fulfill the killing of the cancer cellsCitation19; (right side) displays the functioning mechanism of scFv-PE38KEDL RITs, which initializes by binding the surface epitope. Once the scFv-PE38KDEL RITs are internalized, the PE38KEDL toxin can eventually travel to the endoplasmic reticulum (ER) to cause the inactivation of eukaryotic translation elongation factor 2 (eEF2) by ADP-ribosylation, which results in translation inhibition and consequently cancer cell death.Citation33
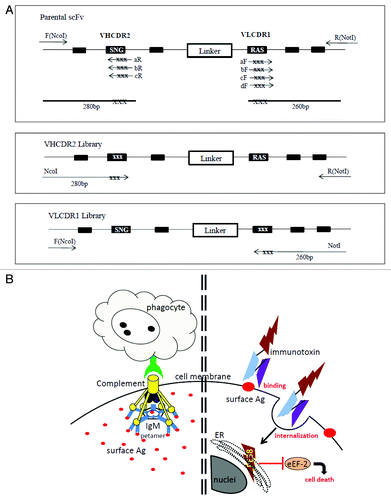
Table 1. Nucleotide and amino acid sequence of VHCDR2 and VLCDR1 and their mutants; IC50 and EC50 value tested on D54MG cells shown
Selection of VHCDR2 and VLCDR1 libraries and analysis of selected clones
Recombinant phages were rescued from 500 individual phage clones from each library, and these phages were randomly selected and tested for reactivity against purified gangliosides by phage ELISA. High signals of ELISA were observed in 10 clones from the VH library and 8 clones from the VL library. The selected clones were repeatedly tested with phage ELISA and sequenced. Two clones from the VH library, and three from the VL library, were found to constantly exhibit strong signals during reaction with gangliosides and contain full-length scFv without stop codons (). VHCDR2 mutant clones 86-MDS and 126-LPP retained the same residues as the parental VH at positions 86–88, but these two clones exhibited mutations at S75M, N76D, G77S and S75L, N76P, G77P, respectively. The VLCDR1 mutant clone 23-RA showed mutations at S27R and S29A, whereas the mutant clones 104-PWR and 184-LYG showed PWR and LYG respectively at positions 25–27 in place of RAS in the parental clone ().
Apparent affinity and cytotoxicity of VHCDR2 and VLCDR1 mutant immunotoxins
VHCDR2 and VLCDR1 mutant immunotoxin clones were prepared, and the cytotoxicity and binding affinity of each were measured by using purified RIT proteins on D54MG glioblastoma cells. The VHCDR2 mutants did not show improved apparent affinity. The affinities of mutant clone 86-MDS, with mutations at positions VH 75–77 and EC50 = 0.9 µM, and clone 126-LPP (EC50 = 28 µM) were not as good as that of the parental clone (EC50 = 0.45 µM) (). Of the three VLCDR1 mutants, clone 184-LYG showed better apparent affinity (EC50 = 0.2 µM) than both the parental and the VHCDR2 mutant clones. The other two VLCDR1 mutant clones, 23-RA (EC50 = 0.8 µM) and 104-PWR (EC50 = 4.7 µM), did not display improved affinity compared with the parental and VHCDR2 mutant clones (). Neither the VHCDR2 nor the VLCDR1 mutant immunotoxins showed significant cytotoxicity in the protein synthesis inhibition assay (data not shown).
Table 2. The combination mutant clones have mutations in both VHCDR2 and VLCDR1*
Construction of combined VHCDR2 and VLCDR1 mutant immunotoxins
Our goal was to obtain an scFv with increased affinity for gangliosides 3′-isoLM1 and 3′,6′-isoLD1 which, when converted to an immunotoxin, would improve cytotoxicity toward malignant brain tumor cells expressing these gangliosides. Neither VHCDR2 nor VLCDR1 affinity-matured mutants alone showed improved apparent affinity or significant cytotoxicity compared with the parent. The VH and VL mutants were therefore combined to form an scFv with mutations in both VHCDR2 and VLCDR1. Six recombinant clones were constructed as described in “Materials and Methods,” and the following immunotoxins incorporating scFvs were prepared: DmAb14–8623-PEKDEL, DmAb14–86104-PEKDEL, DmAb14–86184-PEKDEL, DmAb14–12623-PEKDEL, DmAb14–126104-PEKDEL, and DmAb14–126184-PEKDEL (). Each immunotoxin was purified to over 90% homogeneity and was eluted as a monomer by using TSK gel filtration chromatography. The purified immunotoxins were used in cytotoxicity assays and apparent affinity measured by flow cytometry.
Apparent affinity of combined mutant immunotoxins toward cell lines
The apparent affinity of the six affinity-matured DmAb14-scFv combined mutant immunotoxins toward D54MG cells is summarized in . These immunotoxins were found to be capable of binding to ganglioside-expressing cells using flow cytometric analysis. Two of the combined mutant immunotoxins, DmAb14–86184 and DmAb14–12623, showed an increased apparent affinity toward D54MG cells, about 7 to 35 times higher than that of the VHCDR2 or VLCDR1 mutant immunotoxins () and about 10 to 15 times higher than that of parental DmAb14-IT. The combined mutant DmAb14–86184-MDSLYG showed higher affinity, with an EC50 of 0.073 µM, than DmAb14–86-MDS, DmAb14–184-LYG, or parental DmAb14, with EC50 values of 0.9 µM, 0.2 µM, and 0.45 µM, respectively. The other combined mutant DmAb14–12623LPPREA showed the highest EC50 of 0.029 µM and had a similar pattern to that of DmAb14–86184-MDSLYG, with EC50 values higher than DmAb14–126-LPP (EC50 28 µM) and DmAb14–23-REA (EC50 0.8 µM). The other affinity-matured combined mutants did not show significantly improved apparent affinities.
Cytotoxicity of combined mutant immunotoxins on cell lines
The cytotoxicity of the combined mutant immunotoxins on ganglioside-expressing D54MG cells was then investigated. As shown in , only one of the six combined affinity-matured mutant immunotoxins, DmAb14–86184-PEKDEL, showed cytotoxicity on D54MG cells with an IC50 of 80 ng/mL (1.2 µM). The combined mutant DmAb14–12623-IT did not show any cytotoxicity, though it had the highest apparent affinity (). The positive control, D2C7-IT, is an anti-epidermal growth factor receptor (EGFR)wt/EGFRvIII scFv RIT and the negative control, anti-Tac-IT, is an anti-interleukin-2 (IL-2) receptor scFv RIT. The other combined mutant immunotoxins did not show significant cytotoxicity on D54MG cells, though DmAb14–12623-PEKDEL showed the highest apparent affinity to D54MG cells. This was the first time that an scFv RIT showed cytotoxicity on ganglioside-expressing cells. Therefore, the clone DmAb14–86184-PEKDEL was chosen and renamed DmAb14m-PE38KDEL (DmAb14m-IT) for future studies.
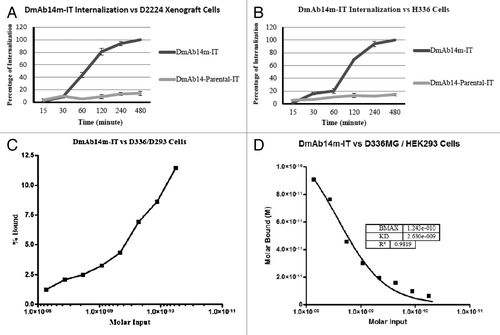
Figure 2. Cytotoxicity of immunotoxins on D54MG cells. Inhibition of protein synthesis was determined as percentage of [3H] leucine incorporation in D54MG cells, after 20 h of treatment with immunotoxins. The graph shows protein synthesis as measured by counts per minute of [3H] leucine incorporated into protein. The dashed line indicates 50% inhibition of protein synthesis, the point at which half of the [3H]-leucine has been incorporated in the absence of toxin. The DmAb14–86184-IT (DmAb14m-IT) IC50 on D54MG cells is 80 ng/mL (1.2µM) (A). The DmAb14–12623-IT IC50 on D54MG cells is none applicable (B). The value shown is the mean ± SD calculated from the means of three separate experiments. Cytotoxicity of DmAb14m-IT on cell lines and xenograft-derived cells. By using protein synthesis inhibition and cell death assays (WST-8), cytotoxic activity (IC50) of affinity-matured DmAb14m-PEKDEL was measured on different ganglioside-positive cell lines and xenografts (in ng/mL). DmAb14m-IT exhibited IC50 of 5 ng/mL (74.6 pM) on D336MG cells (C, D) and 0.5 ng/mL (7.5 pM) on xenograft D2224MG cells (E, F) in both measurements. DmAb14-scFv parental IT did not show any cytotoxic activity on those cells. Each assay was performed in triplicate, and at least three different assays were performed with each cell line. Mean values of three experiments ± SD are shown.
![Figure 2. Cytotoxicity of immunotoxins on D54MG cells. Inhibition of protein synthesis was determined as percentage of [3H] leucine incorporation in D54MG cells, after 20 h of treatment with immunotoxins. The graph shows protein synthesis as measured by counts per minute of [3H] leucine incorporated into protein. The dashed line indicates 50% inhibition of protein synthesis, the point at which half of the [3H]-leucine has been incorporated in the absence of toxin. The DmAb14–86184-IT (DmAb14m-IT) IC50 on D54MG cells is 80 ng/mL (1.2µM) (A). The DmAb14–12623-IT IC50 on D54MG cells is none applicable (B). The value shown is the mean ± SD calculated from the means of three separate experiments. Cytotoxicity of DmAb14m-IT on cell lines and xenograft-derived cells. By using protein synthesis inhibition and cell death assays (WST-8), cytotoxic activity (IC50) of affinity-matured DmAb14m-PEKDEL was measured on different ganglioside-positive cell lines and xenografts (in ng/mL). DmAb14m-IT exhibited IC50 of 5 ng/mL (74.6 pM) on D336MG cells (C, D) and 0.5 ng/mL (7.5 pM) on xenograft D2224MG cells (E, F) in both measurements. DmAb14-scFv parental IT did not show any cytotoxic activity on those cells. Each assay was performed in triplicate, and at least three different assays were performed with each cell line. Mean values of three experiments ± SD are shown.](/cms/asset/c164ec4f-9d98-4e36-9adc-80a3e0afa41b/kmab_a_10925860_f0002.gif)
Cytotoxicity of DmAb14m-IT on cell lines and xenograft-derived cells
The cytotoxicity of DmAb14m-IT was further tested on different glioblastoma cell lines and xenograft-derived cells known to express surface gangliosides. Cytotoxicity (IC50) was evaluated on the basis of both the protein synthesis inhibition assay ([3H] leucine incorporation) (, ) and the cell death assay (WST-8) (, ). The parental DmAb14-IT did not display any cytotoxicity on any ganglioside-expressing cells. In contrast, the affinity-matured DmAb14m-IT exhibited an IC50 of 5 ng/mL (74.6 pM) in the D336MG cell line (, ), and an IC50 of 0.5 ng/mL (7.5 pM) in the D2224MG xenograft cells (, ). The mutant immunotoxin was not cytotoxic to the ganglioside-negative cell line HEK293, a human embryonic kidney 293 cell line, indicating that the cytotoxic effect of this immunotoxin was selective to antigen-positive cells.
Internalization of DmAb14m-IT to human glioma D336MG cell line and D2224MG xenograft cells
Surface fluorescence was quenched by using anti-Alexa-488 antibody to determine the internalization fraction. Internalization was measured as a percentage of the total amount bound to ganglioside binders by comparing the Alexa-488 fluorescence of the quenched cells (intracellular compartment) to that of the unquenched cells (both intracellular and cell-surface compartments) using flow cytometric analysis. Unquenchable surface fluorescence (background) was estimated in parallel tubes on ice (no internalization at 0°C). The amount of unquenchable fluorescence of Alexa-488-labeled DmAb14m-IT and parental DmAb14-IT immunotoxins were about 24% and 8.5%, respectively, when incubated with D336MG cells, and 13% and 1.2%, respectively, when incubated with D2224MG xenograft cells. Each individual experiment was corrected for this “background fraction” before the internalization percentage was calculated.
As the duration of incubation at 37°C increased, the internalization percentage of DmAb14m-IT in D336MG cells also increased from 0.82% at 15 min, 14% at 30 min, 16% at 1 h, 30% at 2 h, and then dramatically to 98% at 4 h, eventually reaching 100% at 8 h.() In contrast, the internalization percentage of parental DmAb14-IT fluctuated between 6% and 12% throughout the incubation period (). Meanwhile, DmAb14m-IT internalization in D2224MG xenograft cells began increasing after 30 min and reached 43% at 1 h, 80% at 2 h, 94% at 4 h, and 100% at 8 h (). The parental DmAb14-IT did not show a significant internalization percentage in D2224MG xenograft cells ().
Figure 3. Time course of internalization of immunotoxins. Xenograft D2224MG (A) and cells D336MG (B) were incubated with 50 nM Alexa-488-labeled DmAb14m-IT or DmAb14-IT at 37°C for different times. After anti-Alexa-488 quenched the cell surface fluorescence, the internalized immunotoxins were measured. In both cells DmAb14m-IT showed significantly higher internalization rate than that of the DmAb14-IT. The data shown represent the mean ± SD of three similar experiments. Scatchard analysis. Defined affinity determination for 125I-labeled DmAb14m-IT vs. ganglioside-positive D336MG cells in contrast with gangliosides-negative HEK293 cells. The saturation curve was presented. (C) Scatchard plots for activity of the percentage bound. (D) Binding affinity graph showing KD = 2.6 × 10−9 M.
Binding affinity of DmAb14m-IT to D336MG cells by equilibrium binding titration
To accurately determine the equilibrium dissociation value of DmAb14m-IT, a cell-binding assay was performed by using a equilibrium binding titration curve, in which an isotope-direct-labeled immunotoxin reacted with a ganglioside-expressing D336MG cell line. DmAb14m-IT was labeled with 125I; the binding of DmAb14m-IT to live D336MG cells at 4°C was assayed as described in “Materials and Methods.” The data were plotted for Scatchard analysis (, ) in which the slope of the plot thus obtained was 0.38 nM−1, giving a KD of 2.6 nM, and a regression coefficient of 0.982. This KD value is 10 times lower than that measured by flow cytometry in the same D336MG cells.
Binding specificity of DmAb14m-IT to gangliosides by ELISA analysis
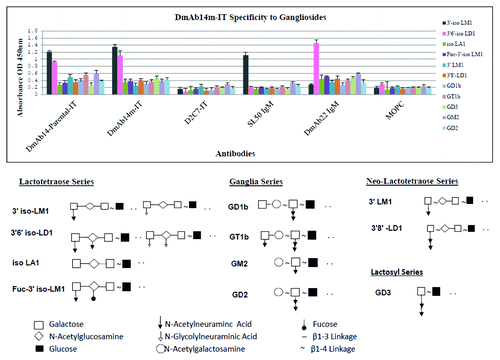
Binding specificity of DmAb14m-IT was further verified by ELISA analysis. The reaction of DmAb14m-IT against purified gangliosides including four series of gangliosides, shown in . Among the 11 gangliosides, two oligosialogangliosides 3′-isoLM1 and 3′,6′-isoLD1 were specifically recognized by parental DmAb14-IT and DmAb14m-IT compared with those shown by the structural control D2C7-IT, which is an RIT consisting of the anti-EGFRwt/EGFRvIII scFv and the PE38KDEL with the same construct as DmAb14m-IT. Also notable is the lack of recognition of the nine gangliosides isoLA1, Fuc-3′-isoLM1, 3′-LM1, 3′,8′-LD1, GD1b, GT1b, GD3, GD2 and GM2 by DmAb14m-IT. Different reaction systems were used for scFv IT and positive IgMs; nonetheless, significantly higher signals were obtained for SL50 IgM (specific mAb for 3′-isoLM1) and DmAb22 IgM (specific mAb for 3′,6′-isoLD1) than that of the negative control MOPC IgM on 3′-isoLM1 and 3′,6′-isoLD1, respectively, indicating that the gangliosides were properly coated.
Figure 4. DmAb14m-IT specificity to gangliosides by ELISA. Both parental DmAb14- IT and DmAb14m-IT showed significant reaction signal toward 3′-isoLM1 and 3′,6'-isoLD1 among the different series of gangliosides compared with negative structural control D2C7-IT. SL50 IgM and DmAb22 IgM served as positive controls for 3′-isoLM1 and 3′,6'-isoLD1, respectively, and MOPC served as the negative control IgM to confirm the ganglioside coating. DmAb14 immunotoxins specifically react with 3′-isoLM1 and 3′,6'-isoLD1 by the epitope of the N-acetylneuraminic acid and N-glycolylneuraminic acid side chain with N-acetylglucosamine linked by β1–3 of oligosialogangliosides. The mean values of three experiments (mean ± SD at 450 nm). The structure of all gangliosides tested in the ELISA were illustrated.
Immunohistochemical analysis
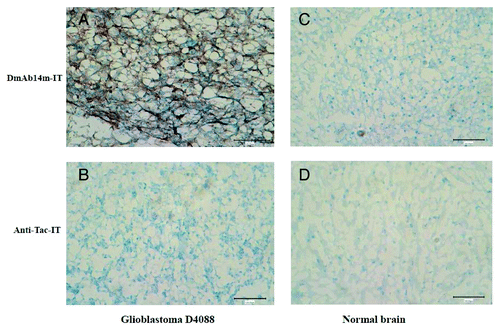
Since gangliosides such as 3′-isoLM1 and 3′,6′-isoLD1 are known to be expressed in glioblastoma tissues, immunohistochemistry was performed in glioblastoma and normal brain frozen tissue specimens (). Immunostaining by DmAb14m-IT demonstrated predominantly cell-surface patterns () in D4088 glioblastoma sections. Proliferating endothelial of normal brain cells were negative for DmAb14m-IT (). The structure-matched scFv-RIT negative control, anti-Tac-IT, did not react with D4088 glioblastoma cells (). Also, immunostaining by DmAb14m-IT and anti-Tac-IT in normal brain tissue did not show any significant reaction (, ). These results show that DmAb14m-IT specifically reacted with gangliosides in glioblastoma tissue.
Figure 5. Immunohistochemical analysis by DmAb14m-IT against glioblastoma and normal brain frozen tissues. Glioblastoma tissues glioblastoma-D4088 (A, B) and normal adult brain tissues (C, D) were stained by DmAb14m-IT (A, C) and structural negative control Anti-Tac-IT (B, D). Magnification: 200 × .
Discussion
As is known, IgM antibodies are not ideal for the immunotherapy of malignant gliomas due to several intrinsic drawbacks, such as high molecular weight, huge pentamer formation, poor tissue penetration, and indirect killing mechanisms (e.g., complement proteins and phagocytes are required).Citation19 Therefore, in this study, we developed an scFv RIT with low molecular weight, high binding affinity, high internalization rate, as well as direct and robust cytotoxicity to target malignant glioma cells that express gangliosides 3′-isoLM1 and 3′,6′-isoLD1. This scFv RIT is derived from our previously established DmAb14 IgM, which is a ganglioside 3′-isoLM1- and 3′,6′-isoLD1-specific antibody (). The approach used to increase the affinity of the parental DmAb14-scFv was to randomly mutagenize hotspot residues in VHCDR2 and VLCDR1 (). Hotspots are regions of DNA that are frequently mutated during the in vivo affinity maturation of antibodies.Citation20 Several different mutations that conferred increased affinity were identified,Citation21 and when the mutations in VHCDR2 and VLCDR1 were combined in a single construction, the antibody’s affinity was even higher.
While the analysis of mutant clones selected by phage ELISA screening revealed the presence of a variety of different sequences, there was a pattern to the amino acid substitutions (). In the mutant clones, three amino acids of VHCDR2 (75–77) and three amino acids of VLCDR1 (25–27) were consistently different from the parental sequence, which indicates that these residues could be required for interacting with the antigens. After the binding affinity of each individual mutant had been tested, two of five individual mutants displayed a slight improvement (), but none of them showed any cytotoxicity. We combined the two mutations in VHCDR2 and the three mutations in VLCDR1 into one scFv construct, from which six combined mutants were derived (). Their binding affinities were tested against D54MG cells. Two (DmAb14–86184-IT and DmAb14–12623-IT) of the six combined mutants showed significant increased apparent affinity (72.6 nM and 28.8 nM, respectively), compared with the parental clone (450 nM) and other individual or combined mutants (). Furthermore, the cytotoxicity assays were tested on a D54MG, which is a classic ganglioside-expressing cell line, using both affinity-matured mutants (DmAb14–86184-IT, DmAb14–12623-IT), as well as the parental DmAb14-IT. The positive control, D2C7-IT,Citation22,Citation23 is an anti-EGFRwt/vIII scFv RIT and the negative control, anti-Tac-IT,Citation24 is an anti-IL-2 receptor scFv RIT. Most malignant gliomas overexpress EGFRwt or EGFRvIII proteins, while IL-2 receptor is rarely expressed, if at all, in malignant glioma cells. DmAb14–86184-IT was then found as the first RIT that targets gangliosides 3′-isoLM1 and 3′,6′-isoLD1 with significant cytotoxicity toward ganglioside-expressing cells with an IC50 of 80 ng/mL (1.2 µM), shown in . While DmAb14–12623-IT did not reveal any cytotoxicity on D54MG cells, it had the highest affinity (), which might indicate that binding affinity was needed for the cytotoxicity but not the most crucial factor. Therefore, we renamed DmAab14–86184-IT as DmAb14m-IT. Since there were many other glioma cell lines and xenograft-derived cells expressing gangliosides 3′-isoLM1 and 3′,6′-isoLD1, such as the D336MG cell line and the D2224MG xenograft cells, the cytotoxicity of DmAb14m-IT was then tested on the D336MG cell line and the D2224 xenograft cells. The IC50 of the DmAb14m-IT varied among the different cell lines and xenograft cells (), which was likely caused by the differing amount of gangliosides expressed on different cells. It is also possible that 3′-isoLM1 and 3′,6′-isoLD1 are present in different proportions of different cell types. Generally, 3′-isoLM1 is expressed in a much higher density than 3′,6′-isoLD1.Citation25 Xenograft D2224MG cells were screened by an anti-ganglioside mAb, specific for 3′-isoLM1Citation13 and 3′,6′-isoLD1,Citation25 which revealed that 88% of xenograft D2224MG cells were positive for 3′-isoLM1 and only 2% for 3′,6′-isoLD1. Hence, 3′-isoLM1 may be the main ganglioside expressed on xenograft D2224MG cells. In fact, the xenograft D2224MG cells were determined to have a higher ratio of 3′-isoLM1 to 3′,6′-isoLD1 than D336MG and D54MG cells. Since the DmAb14m-IT shows a more potent effect on xenograft D2224MG cells (IC50 of 0.5 ng/mL, 7.5 pM, ) than on D336MG cells (IC50 of 5 ng/mL, 74.6 pM, ), the DmAb14m-IT might have a greater binding affinity for 3′-isoLM1 and greater benefit for patients with 3′-isoLM1-dominant brain tumors. Plus, the toxic side effects might be avoided because 3′-isoLM1 is expressed in the human brain tissue only during fetal development.Citation26 In addition, the 3′-isoLM1 ganglioside is also expressed in other types of tumors, e.g., small cell lung carcinoma.Citation13 The immunohistochemical studies reported that the 3′,6′-isoLD1 was detected in CNS neoplasms of glial and non-glial origin. Of non-glial tumors, 62% of epithelial cancers such as breast, pancreas, and prostate cancers, were reactive with an anti-3′,6'isoLD1 mAb,Citation7 which suggests the current immunotoxin might be also useful in the treatment of those tumors.
Because of the promising cytotoxicity on ganglioside-expressing cells, the investigation of internalization became primary over other characterization studies. Many RITs are chimeric proteins used for cancer therapy, consisting of a targeting part linked to the catalytic domains of a natural toxin.Citation27 We selected Pseudomonas exotoxin A (PE38) as the toxin component. This potent bacterial toxin is composed of three domains,Citation28 which is able to enter tumor cells, inhibit protein synthesis, and eventually kill cells (). Thus, the internalization of the immunotoxin by the tumor cell becomes one of the crucial factors determining the cytotoxicity. With xenograft D2224MG cells and D336MG cells, DmAb14m-IT was found to be internalized significantly faster than the parental IT, with xenograft D2224MG cells () and D336MG () cells demonstrating internalization rates of 94% and 98%, respectively, within 4 h, while the parental IT had no significant internalization within the same time period (). The mutant IT internalized into D2224MG xenograft cells faster than D336MG cells, which may explain why DmAb14m-IT was more potent in xenograft D2224MG cells. This also shows that the significant internalization could be crucial for determination of significant cytotoxicity.
The parental DmAb14-IT and DmAb14m-IT differ only in the mutated amino acid residues in VHCDR2 and VLCDR1 of the single chain. Both showed specificity and binding affinity to the ganglioside-positive cells; however, only the mutant IT displayed cytotoxicity; again, the negative internalization event of parental DmAb14-IT could explain this issue.
For a better understanding of the equilibrium dissociation value, the binding affinity of DmAb14m-IT was tested on the D336MG cell line by using equilibrium binding titration curves. The data were then analyzed and presented in a Scatchard plot (, ), which revealed a more accurate equilibrium dissociation value of 2.6 × 10−9 M (). It is possible that the VH and VL loop may require all these residues to be present for proper conformational flexibility to achieve increased affinity, and to enable better structural conformation for internalization as compared with the parental IT and other mutant ITs.Citation14
Both the parental DmAb14-IT and DmAb14m-IT were confirmed to react with purified 3′-isoLM1 and 3′,6′-isoLD1, whereas D2C7-IT, an RIT specific to EGFRwt/vIII,Citation22,Citation23 did not display any binding ability among the series of gangliosides (). This indicates that DmAb14m-IT specifically recognizes, and binds to the epitope structure of the galactose with N-acetylneuraminic acid, the N-glycolylneuraminic acid side chain, and N-acetylglucosamine linked by β1–3 of oligosialogangliosides, but does not recognize or bind to other structures of the gangliosides.
In addition, the structure control D2C7-IT did not show any reactivity in ganglioside ELISA, but both the DmAb14m-IT and the D2C7-scFv-IT showed significant cytotoxicity against glioblastoma cells. This result clearly proved that these two immunotoxins targeted different tumor antigens on glioblastoma cells; therefore, D2C7-IT could be an excellent positive control for cytotoxicity in future studies.
Additional immunohistochemical analysis further supported the specificity of DmAb14m-IT to glioblastoma tissue () in comparison with normal brain tissue ().
In summary, an anti-ganglioside scFv immunotoxin with high cytotoxicity and glioblastoma specificity was developed. This goal was achieved by using intrinsic mutational hotspots as targets for in vitro antibody affinity maturation. The parental clone DmAb14-scFv-IT had the proper affinity but did not have any cytotoxicity, whereas the affinity-matured mutant, DmAb14m-IT, showed a much higher affinity and increased cytotoxicity to ganglioside-positive cell lines and glioma xenograft cells because of its higher internalization rate. DmAb14m-IT exhibited strong cytotoxic effects on the human glioblastoma D2224MG xenograft cells and no cytotoxic effects on the ganglioside-negative cell line HEK293, demonstrating the selectivity of the immunotoxin. Moreover, the increased cytotoxicity of the mutant immunotoxin may provide a clinical benefit with a low dose, which could decrease nonspecific toxicities. This study was, to the best of our knowledge, the first time that an scFv recombinant immunotoxin, derived from IgM secretion hybridoma, that targets 3′-isoLM1 and 3′,6′-isoLD1 gangliosides has been shown to have retained DmAb14 specificity, high binding affinity, and significant cytotoxicity on 3′-isoLM1 and 3′,6′-isoLD1 ganglioside-expressing tumor cells. This may not only be applied to glioblastoma cells, but also used to target other peripheral malignant tumors, such as lung, breast, pancreas, and prostate cancers.Citation20,Citation28 Compared with the monoclonal IgM (900 kDa), the lower molecular weight of the scFv immunotoxin (67 kDa) may confer advantages to the molecule, such as better penetration, faster circulation, and easier passage through the blood–brain barrier as this barrier may be compromised in the setting of high-grade brain tumors.Citation29 All of these advantages suggest great potential for applications in clinical therapy.
Unlike other common brain tumor immunotherapy targets such as EGFR, which is overexpressed in the brain tumor but also retains certain expression levels in normal brain tissue and other normal peripheral tissues, gangliosides are widely expressed in glioblastoma tissue exclusively. This restriction to the tumor cells makes gangliosides an ideal hbrain tumor target. The study of gangliosides, however, has been ongoing for over 25 years; it is truly a difficult antigen target to investigate, perhaps due to its complicated side-chain structure, and so far few successes have been reported. Our findings strongly suggest that the immunotoxin described here displays a robust potential for the clinical diagnosis and treatment for malignant glioma patients in the future.
Materials and Methods
Cell culture
The human malignant glioma-derived cell lines D54MG and D336MG were used, which were established and maintained in our laboratory, as ganglioside-expressing cell lines. The human embryonic kidney cell line HEK293 was used as a negative control. All cell lines were cultured in Improved MEM Zinc Option (Richter’s zinc option; Invitrogen) supplemented with 10% FBS (Invitrogen) and passed at confluence with 0.25% trypsin-EDTA (Invitrogen).
Disaggregation of xenograft tumor samples
Xenograft tissues from malignant gliomas (D2224MG and BT56), obtained under sterile conditions from animals housed in the Duke Cancer Center Isolation Facility, were prepared for cell culture in a laminar flow hood using a sterile technique. Tumor material was finely cut with scissors and added to a trypsinizing flask containing approximately 10 mL of 100 µg Liberase (Roche). This mixture was stirred at 37°C for 10 min, and a cell-rich supernatant was obtained. The dissociated cells were filtered through a 100-µm cell strainer (BD Falcon, BD Biosciences), washed with complete medium, and pelleted at 1000 rpm for 5 min. The cell suspension was further treated with Ficoll-Hypaque to remove any red blood cells and was then washed once in a complete medium. The cells were cultured and passaged until sufficient numbers were obtained; they were then harvested with 0.25% trypsin-EDTA.
Cloning of variable heavy (VH) and variable light (VL) domains of DmAb14
Total cellular mRNA was isolated from 106 DmAb14-hybridoma cells by using the RNeasy Kit (Invitrogen). Primary VH and VL genes of the parental DmAb14 clone were amplified by RACE-PCR (Rapid Amplification of cDNA Ends-PCR) using a SMART 5′-RACE cDNA amplification kit (Clontech). The 3′ primers were murine heavy-chain (HC) and light-chain (LC) constant region sequences of the immunoglobulin (VH: 5′-GGC CAG TGG ATA GTC AGA TGG GGG TGT CGT TTT GGC-3′ and VL: 5′-GGA TAC AGT TGG TGC AGC ATC-3′). The primary VH and VL cDNA genes obtained from 5′-RACE were then used as templates to specifically amplify the VH and VL fragments respectively by using specific primers introduced at restriction enzyme sites and a (Gly4Ser)5 linker sequence for scFv assembly and subsequent subcloning.
The oligomers used for these reactions were as follows: DmAb14-H-F (NcoI), 5′-GCC GCC ACCA TG Gag GTC CAA CTG CAG-3′; DmAb14-H-R (linker), 5′–AGA TCC GCC ACC ACC GGA TCC CCC TCC GCC TGA GGA GAC GGT GAC-3′; DmAb14-L-F (linker), 5′–GGT GGT GGC GGA TCT GGA GGT GGC GGC AGC GGT AAC ATT GTG CTG-3′; and DmAb14-L-R (EcoRI), 5′–GCAGCC GAATT CAT TTT TAT TTC CAG CTT G-3′. The outcome-specific VH and VL sequences were aligned and verified according to the Kabat alignment scheme. DmAb14-scFv was constructed by using PCR splicing technology. In brief, the pre-mixed Advantage 2 DNA polymerase (Clontech) was mixed with 50 ng each of VH and VL PCR fragments at a 1:1 ratio in the presence of bovine serum albumin (BSA) in a 50 µL volume. The PCR mix was cycled by using the following profile: 1 cycle at 96°C for 5 min; followed by 5 cycles each at 94°C for 1 min, 55°C for 1 min, and 72°C for 1 min and 30 sec; and a final cycle at 72°C for 7 min. The program was paused at 4°C for 2 min to add premixed oligomers, namely, the forward and reverse primers that had been used to generate the VH and VL fragments. The reaction was then continued as follows: 1 cycle at 96°C for 5 min; followed by 25 cycles each at 94°C for 1 min, 55°C for 1 min, and 72°C for 1 min and 30 sec; and finally terminated at 72°C for 7 min. A 750-bp PCR product generated from this reaction was purified by using Qiagen Quick Spin columns (Qiagen, Inc.), and double digested with NcoI and EcoRI and subcloned into a T7 bacterial expression vector pET25b(+) (Novagen). The parental DmAb14-scFv sequence was verified by the dideoxy chain-termination method.
Preparation of recombinant immunotoxins
The DmAb14-scFv IT was generated by PCR using parental DmAb14-scFv plasmid as a template and primers introduced at the NdeI and HindIII restriction enzyme sites. After NdeI and HindIII digestion, the scFv fragment was inserted into pRB199 bacterial expression vector that had been engineered with the sequence for domains II and III of Pseudomonas exotoxin A (PE38KDEL) according to a previously described protocol.Citation30 The parental DmAb14-scFv IT was expressed under control of the T7 promoter in E. coli BL21 (λ DE3) (Stratagene). All recombinant proteins remained in the inclusion bodies. The IT proteins were then reduced, refolded, and further purified as monomers (67 kDa) by using ion-exchange and size-exclusion chromatography to greater than 90% purity.
Construction of DmAb14-scFv phagemid
The parental DmAb14-scFv phagemid was constructed by PCR amplification from parental DmAb14-scFv-pRB199 PE38KDEL. The following oligomers were used and were introduced at the NcoI and NotI restriction enzyme sites: DmAb14-scFv-F (NcoI), 5′-GCCGCCA CCATG GAG GTC CAA CTG CAG-3′, and DmAb14-scFv-R (NotI), 5′-ATG ATG TGC GGC CGC TTT TAT T TC CAG CTT G-3′. The PCR product was digested with NcoI and NotI and inserted into the phagemid vector pHEN2. The resulting parental phagemid DmAb14-scFv-pHEN2 was used as a template for further construction of mutant phage libraries.
Construction of VH mutant library
The VHCDR2 of DmAb14 consisted of 18 amino acids. DNA oligomers were designed to generate 3 libraries, each randomizing 9 nucleotides (3 consecutive amino acids). Degenerate oligomers were used with the sequence NNS for randomizing (N randomizing with all 4 nucleotides, and S introducing only C or G).Citation31 Parental DmAb14 phagemid was used as a template to introduce 3 amino acid randomizations in the CDR2 heavy chain in 3 separate 2-step PCRs ().
The following oligonucleotides were used: (a) DmAb14-VHCDR2a-R, 5′-GTT AGT ACG ACC GTT SNN SNN SNN AAT CTC TCC AAT CC-3′; (b) DmAb14-VHCDR2b-R, 5′-AAT ATA GTT AGT ACG SNN SNN SNN AGG ATT AAT CTC TCC-3′; (c) DmAb14-VHCDR2c-R, 5′-TAC AGT CAG TGT GGC SNN SNN SNN GAA CTT CTC-3′; (d) DmAb14-scFv-F (NcoI), 5′-GCCGCCA CCATG GAG GTC CAA CTG CAG-3′; and (e) DmAb14-scFv-R (NotI), 5′-ATG ATG TGC GGC CGC TTT TAT T TC CAG CTT G-3′. In the first PCR, 50 pg of the phagemid DmAb14-scFv-pHEN2 was used as a template in 3 separate reactions involving 20 pmol of DNA oligomer DmAb14-scFv-F (NcoI), along with 20 pmol of DNA oligomer DmAb14-VHCDR2a-R, DmAb14-VHCDR2b-R, or DmAb14-VHCDR2c-R (). The template and oligonucleotides were mixed with Advantage 2 PCR Kit (Clontech) in a 50 µL volume and then cycled using the following profile: 1 cycle at 95°C for 5 min, followed by 30 cycles each at 94°C for 1 min, 55°C for 1 min, and 72°C for 1 min. This reaction generated 280-bp products that contained mutations. The products were purified by using Qiagen Quick Spin columns (Qiagen, Inc.) and then quantified by visualization on a 1% agarose gel. The purified products generated in the first PCR were used as primers in a second PCR.
In the second reaction, approximately 2 pmol of each of the 3 products containing mutations (obtained from the first reaction and the NcoI restriction enzyme site), along with 20 pmol of the DNA oligomer DmAb14-scFv-R (NotI) and 50 pg of the parental phagemid DmAb14-scFv-pHEN2 as a template to generate the whole length of the scFv-VH mutation pool (). The PCRs were set and cycled by using the profile described above. Each reaction generated 750-bp insert libraries. The PCR product was digested with NcoI and NotI and purified by using Qiaquick columns (Qiagen, Inc.). The purified PCR product (80 ng) was ligated with 150 ng of the phage display vector pHEN2 (predigested with NcoI and NotI) and desalted. Then, 40 ng of ligated products was used to transform E. coli TG1 (Stratagene) by electroporation. Three transformations were performed to create a library containing 6 × 104 clones. The phage libraries were then rescued from the transformed bacteria. Cells from each transformation were grown in 10 mL of 2 × YT (Bacto Yeast Extract and Bacto Tryptone) containing 2% glucose at 37°C with shaking at 250 rpm. After 1 h, ampicillin (100 µg/mL, final concentration) was added, along with 1 × 1011 plaque-forming units (pfu)/mL of VCSM13 Interference-Resistant Helper Phage (Stratagene, Agilent Technologies). The cultures were grown for 1 h, pelleted, resuspended in 10 mL of 2 × YT supplemented with ampicillin (100 µg/mL) and kanamycin (50 µg/mL), and grown for 16 h at 30°C with shaking at 250 rpm. The bacteria were pelleted by centrifugation in a Sorvall SS34 rotor at 8000 rpm for 20 min. The phage-containing supernatants were filtered through a 0.45 µm syringe filter unit. The phages were precipitated by adding 2 mL of PEG/NaCl (20% PEG8000 in 2.5 M NaCl [w/v]) and incubated on ice for 30 min. The precipitated phages were pelleted by centrifugation in a Sorvall SS34 rotor at 10,000 rpm for 20 min and resuspended in 1 mL of NTE [100 mM NaCl, 10 mM Tris (pH 7.5), and 1 mM EDTA]. The rescued phage libraries were titered and stored at 4°C.
Screening of VH mutant library by phage ELISA
A diluted VHCDR2 mutant phage library was cultured and diluted. Recombinant phage-containing supernatants were rescued from individual clones and screened by antigen-based phage ELISA. Single colonies were selected from the diluted library plates and inoculated into 1 mL of 2 × YT medium supplemented with 2% glucose and 100 µg/mL of ampicillin. The colonies were cultured overnight at 37°C with shaking at 250 rpm. To one cultured colony preparation were added 450 µL of 2 × YT supplemented with 2% glucose and 100 µg/mL of ampicillin containing 2.5 × 108 pfu of VCSM13 Interference-Resistant Helper Phage (Stratagene) in 1.2-mL Cluster Tubes (Corning, Inc.). To another preparation, 50 µL of the overnight culture was added and then incubated the colony at 37°C for 2 h with shaking at 250 rpm. The cells were pelleted and resuspended in 500 µL 2 × YT containing 100 µg/mL ampicillin and 50 µg/mL kanamycin without glucose and then grown for 16 h at 30°C. The cells were again pelleted by centrifugation at 2800 rpm at 4°C, and the phage-containing supernatants were saved.
Purified gangliosides 3′-isoLM1 and 3′,6′-isoLD1 conjugated with BSA (purified from the D54MG xenograft, Sahlgrenska University Hospital, Gothenburg, Sweden) in PBS were immobilized overnight in 96-well plates (Nunc, Thermo Fisher Scientific) containing 50 ng/well at room temperature. The plates were blocked with 1% BSA/PBS at 37°C for 30 min and then washed 3 times with 0.05% Tween 20 in PBS by using a Bio-Rad Auto-Washer (Bio-Rad 1575 Immuno Wash Microplate Washer). Then, 100 µL/well of phage-containing supernatant was added for a 1 h incubation at 37°C and the plates were washed 3 times, as above. For phage ELISA, 100 µL/well of anti-M13 antibody-horseradish peroxidase (HRP) conjugate (Pharmacia Biotech) in blocking buffer was added, diluted according to the manufacturer’s instructions, to detect phage binders. For monoclonal IgM antibody, 100 µL/well of anti-mouse IgM HRP conjugate (µ-Chain Specific; Sigma) was added in blocking buffer (1:10,000 dilution). After 30 min of incubation at 37°C, the plates were washed; 100 µL/well of 3,3′,5,5′-tetramethyl benzidine (1-Step Ultra TMB-ELISA, Pierce) solution was used as a substrate for detection. After a blue color developed, 50 µL TMB Stop Solution (Pierce) was added to stop further color development. Absorption was measured at 450 nm. Clones reacting to gangliosides were referred to as positives.
Construction of VL mutant library
The VLCDR1 of DmAb14 consists of 15 amino acids. Parental DmAb14 phagemid DNA was used as a template in 4 separate 2-step PCRs that introduced randomizations in the hotspot located in the light-chain CDR1 ().
The following oligonucleotides were used: (a) DmAb14-VLCDR1a-F, 5′-ACC ATA TCC TGC NNS NNS NNS GAA AGT GTT GAG AGT TAT GGC-3′; (b) DmAb14-VLCDR1b-F, 5′-ACC ATA TCC TGC AGA NNS NNS NNS AGT GTT GAG AGT TAT GGC-3′; (c) DmAb14-VLCDR1c-F, 5′-ACC ATA TCC TGC AGA GCC NNS NNS NNS GTT GAG AGT TAT GGC-3′; (d) DmAb14-VLCDR1d-F, 5′-GCC AGT GAAAGT GTT NNS NNS NNS GGC AAT AAT TTT ATG CAC-3′; (e) DmAb14-scFv-F (NcoI), 5′-GCCGCCA CCATG GAG GTC CAA CTG CAG-3′; and (f) DmAb14-scFv-R (NotI), 5′-ATG ATG TGC GGC CGC TTT TAT T TC CAG CTT G-3′.
In the first reaction, 50 pg of the DmAb14-scFv phagemid DNA was mixed with 20 pmol of each pair of DNA oligomers in a 50 µL volume as follows: (a) + (f), (b) + (f), (c) + (f), and (d) + (f). The mixture was cycled by using the same profile used to generate the heavy-chain CDR2 mutant library. The reactions generated 260-bp products exhibiting randomization of the hotspot in the light-chain CDR1. After purification and quantification, 2 pmol of the 260-bp fragments generated in the first PCR, was subjected to a second PCR, along with 20 pmol of DNA oligomer (e) and 50 pg of parental DmAb14-scFv phagemid DNA as a template (). The PCR (50 µL) was performed by using an Advantage 2 PCR Kit (Clontech) and following the above-mentioned profile. The reactions generated a 750-bp library, which showed randomization of the hotspot in VLCDR1. The PCR products were then digested with the restriction enzymes NcoI and NotI, purified, and ligated with the pHEN2 phage vector as described for the heavy-chain CDR2 mutant libraries. The ligation was desalted, and one-tenth (40 ng) of the reaction was used to transform E. coli TG1. The VLCDR1 mutant phage library containing 5 × 104 clones was rescued as described in the heavy-chain CDR2 construction.
Selection of VLCDR1 library by phage ELISA
A diluted VLCDR1 phage library was cultured and plated. The technical procedures used were the same as those described in the VHCDR2 libraries selection.
Construction of VHCDR2 and VLCDR1 combination libraries
The heavy-chain CDR2 mutant was used as a template to generate the VH fragment. For this purpose, the primers DmAb14-H-F (NdeI), 5′-G GCG CAT ATG CAT GTC CAA CTG CAG-3′, DmAb14-H-R (linker), 5′-AGA TCC GCC ACC ACC GGA TCC CCC TCC GCC TGA GGA GAC GGT GAC-3′ in a 50 µL volume, and an Advantage 2 PCR Kit (Clontech) were used; a 420-bp product was obtained. The light-chain CDR1 mutant was used as a template to generate the VL fragment. The primers DmAb14-L-F (linker), 5′-GGT GGT GGC GGA TCT GGA GGT GGC GGC AGC GGT AAC ATT GTG CTG-3′, DmAb14-L-R (HindIII), 5′-C AAG CTG GAA ATA AAA A AA GCT TGG CAG C-3′ in a 50 µL volume, and an Advantage 2 PCR Kit (Clontech) were used to generate a 335-bp PCR product. Mutant VH and mutant VL DNA fragments were overlapped by a 15-amino acid linker sequence and connected by using the splicing PCR technique described above, except that the oligomers used in this reaction were DmAb14-H-F (NdeI) and DmAb14-L-R (HindIII), which are listed above, in this section. This reaction generated 750-bp products that contained mutations in both the VH and VL regions. The products were purified by using Qiagen Quick Spin columns (Qiagen, Inc.), digested with NdeI and HindIII, and cloned into a T7 expression vector pRB199 in which scFv was fused to a truncated version of Pseudomonas exotoxin A (PE38KDEL). The sequence was then verified.
Analysis of selected clone by DNA sequencing
Sequencing was performed by the Duke University DNA Sequencing Facility on an Applied Biosystems Dye Terminator Cycle Sequencing system (Life Technologies Corp.) by using AmpliTaq DNA Polymerase and ABI 3730 PRISM DNA sequencing instruments. Sequencing analysis was performed using the NCBI nucleotide BLAST program.
Cell-binding assay and constant EC50 by flow cytometry
A modification of the method reported previouslyCitation32 was used to detect scFv immunotoxin binding to ganglioside-expressing cells. To prevent internalization of the immunotoxin during the assays, all reagents and the buffer were kept on ice. In brief, cells to be tested for scFv immunotoxin binding were harvested by trypsinization, rinsed with 1% FBS in PBS, and portioned to assay tubes (approximately 106 cells per sample). The scFv immunotoxins were serial-diluted in 100 µL of 1% FBS-PBS, with an initial amount of 25.6 µg. The cells were incubated with 100 µL of various dilutions of the purified scFv immunotoxin or anti-Tac(Fv)-PE38KDEL, a negative control that does not bind to gangliosides, for 1 h on ice. The cells were then washed twice in 1% FBS-PBS and incubated with rabbit anti-Pseudomonas exotoxin A antibody (Sigma) for 30 min on ice. After the cells were washed twice, they were reacted with goat anti-rabbit IgG-FITC conjugate (Sigma) for 30 min on ice. The stained cells were again washed twice and then subjected to flow cytometry. An identical protocol was used for binding of the scFv immunotoxin in this study. All reagents were determined to be at saturation point, and binding reached equilibrium for the described conditions in all antibody-binding experiments. Equilibrium constants EC50 (apparent affinity) were determined by using the Graph Pad Prism Software (version 4.0) for 1-site binding and nonlinear regression analysis.
Determination of affinity constants (KD) by Scatchard analysis
Before the accurate KD value was determined by Scatchard analysis for the final combined mutant, the affinity determination used equilibrium binding titration curves to determine equilibrium dissociation constants KD.
The same number of cells was prepared for each individual tube, and the serial diluted Alexa-488 direct-labeled mutant-IT was added to each tube, with one tube for the negative control-IT. We incubated the samples on ice for 1 h. This allowed ample time for the binding reaction to reach 90% equilibrium for antibody clones. We washed the sample two times with 500 µL PBS buffer and then resuspended it in 500µL PBS and stored it on ice in the dark until analyzed by flow cytometry. We gathered 10,000 events to create a bivariate plot using the fluorescence channels that detect Alexa-488. We collected the statistical information from the given histogram of geometric mean, plotted the statistical data against the concentration of antibody to the GraphPad Prism4, and used nonlinear least-squares to fit the curve; the KD value was calculated.
For Scatchard analysis, affinity measurements were performed by using Iodogen-labeled scFv immunotoxin binding to cells positive and negative for ganglioside expression. The cells were seeded in a 24-well plate, cultured until confluence, fixed in 0.25% gluteraldehyde for 5 min at room temperature, washed 3 times with incubation buffer (115 mM PO4 [KH2PO4+K2HPO4] with 0.05% BSA and 0.05% gelatin), and frozen at -80°C until use. Negative control scFv immunotoxin and [125I]-labeled sample scFv were serially diluted starting from 8 µg/mL to 4 ng/mL, followed by the addition of various dilutions of [125I]-scFv immunotoxins to corresponding triplicate wells and incubation of the cell plates overnight at 4°C. The next day, the free radioactive antibody was removed and the cell plates were washed 4 times with the incubation buffer. Then, 500 µL/well of 2N sodium hydroxide was added, and the cell plates were incubated overnight at 37°C. The cells were completely suspended and transferred (including cell debris) into 2-mL screw-capped tubes, which were placed in a gamma counter, in order, and the cells were counted. The data obtained were analyzed by normalization based on standard curve and equilibrium constants, and a Scatchard plot was determined with Prism software (version 4.0) for nonlinear regression analysis.
Cytotoxicity assays
Cytotoxicity was measured by using both protein synthesis inhibition and cell death assays. In both assays, cells were plated in 96-well plates (Nunc) at a concentration of 1 × 104 cells/200 µL/well. Immunotoxins were serially diluted in PBS supplemented with 0.2% BSA starting from 10 ng/µL and 20 µL of each dilution was added to the corresponding wells, resulting in a maximum immunotoxin concentration of 1000 ng/mL. The plates were incubated for 20 h at 37°C.
Protein synthesis was measured on the basis of [3H] leucine incorporation. The cells were pulsed with 1 µCi/well of [3H] leucine in 20 µL of PBS supplemented with 0.2% BSA for 2.5 h at 37°C. Radio-labeled cells were captured on a FilterMate micro-harvester (PerkinElmer Life and Analytical Sciences) and counted with a Betaplate scintillation counter (Amersham Biosciences, GE Healthcare Bio-Sciences Corp.). Triplicate sample values were averaged, and the inhibition of protein synthesis was determined by calculating the percentage of incorporation compared with control wells without added toxin. The activity of the molecule was defined as the IC50.
Cell death was assessed by WST-8 conversion using Cell Counting Kit-8 according to recommendations from the supplier (Dojindo Molecular Technologies). The cells were seeded onto a 96-well plate (Costar, Corning, Inc.) at a concentration of 2 × 104 cells/100 µL/well. The immunotoxins were serially diluted in PBS supplemented with 0.2% BSA starting from 10 ng/µL, and 10 µL of each dilution was added to the corresponding wells. The cells were then incubated at 37°C for 20 h. To each well was added 10 µL of WST-8 (5 mM WST-8, 0.2 mM 1-methoxy-5-methylphenazinium methylsulfate, and 150 mM NaCl), and the incubation was continued at 37°C. The absorbance of the sample was measured at 450 nm every 20 min until the highest value for the negative control was obtained. Cytotoxicity was expressed as 50% inhibition of cell viability. All experiments were performed in triplicate. Statistical analyses were performed with Prism software (version 4.0) for Windows (GraphPad Software). Each immunotoxin was assayed at least twice, and critical immunotoxins were assayed more frequently.
DmAb14 min specificity to gangliosides by ELISA
DmAb14-scFv immunotoxins that specifically bind to gangliosides 3′-isoLM1 and 3′,6′-isoLD1 were assayed by ELISA. Eleven purified gangliosides (500 pM) were coated in PBS on a 96-well plate (Nunc), which was blocked with 1% BSA/PBS at 37°C for 1 h. Next, 500 ng of primary antibodies, parental DmAb14-IT, DmAb14m-IT and D2C7-IT, were applied in triplicate to observe ganglioside reaction, and the plates were incubated at 37°C for 30 min. D2C7-IT was used as an scFv structural negative control. SL50 IgM and DmAb22 IgM are positive controls for coating 3′-isoLM1 and 3′,6′-isoLD1, respectively, and MOPC was an IgM negative control. The plates were washed 3 times with 0.05% BSA and 0.05% Tween in PBS. After adding rabbit anti-PE (Sigma; dilution 1:30,000) to the scFv immunotoxins for 30 min and washing the plates, anti-rabbit IgG-HRP (Sigma, dilution 1:1,500) and anti-mouse IgM-HRP (Sigma, dilution 1:1,500) were applied to the scFv immunotoxins and IgM antibodies, respectively. The plates were incubated at 37°C for 30 min and washed 3 times with a washing buffer. Then, 100 µL of 1-Step Ultra-TMB ELISA (Thermal Scientific) substrate was added, and the reaction color turned blue within 3–5 min; stop solution was immediately applied to terminate the reaction and change the color to yellow. The ELISA plates were read at an absorbance of 450 nm.
Internalization assay
DmAb14-scFv immunotoxins were directly labeled with Alexa-488, and 50 nM Alexa-488-labeled DmAb14-scFv immunotoxin was incubated with 3 × 105 D336MG cells or xenograft D2224MG cells on ice for 1 h. After washing the cells twice with ice-cold PBS, the cells were resuspended in zinc option medium. Two portions of the reaction mix, one for measurement of background internalization and one portion for total binding measurement, were left on ice, the remaining reaction mix was incubated at 37°C for 15 min, 30 min, 1 h, 2 h, 4 h, and 8 h. The cells were washed twice with ice-cold PBS in a cold centrifuge. Except for the total binding portion, all cells were resuspended in 500 nM quenching anti-Alexa-488 antibody (Invitrogen) diluted in ice-cold PBS. All tubes were incubated for 1 h on ice. The cells were washed twice in ice-cold PBS and resuspended in 4% paraformaldehyde; the reaction samples were analyzed in a flow cytometer (FACSCalibur, BD Biosciences). Internalization was calculated by dividing the fluorescence of quenched cells (intracellular compartments only) by that of unquenched cells (both cell surface and intracellular compartments; total binding) after normalization against the background fluorescence. The cells incubated with Alexa-488-labeled immunotoxins on ice were used as a control while estimating the antibody’s quenching efficiency on each Alexa-labeled immunotoxin. Since internalization should not occur at 0°C, the fluorescence of these cells, measured after quenching with anti-Alexa-488, was considered to represent the unquenchable surface fluorescence (background). This amount was corrected when the immunotoxin internalization percentage was calculated.
Ethical statement
The xenograft tissues used in this study were obtained from animals housed in the Duke Cancer Center Isolation Facility. The animals involved were cared for in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol, Brain Tumors: Immunological and Biological Studies, was approved by the Institutional Animal Care and Use Committee (IACUC) at Duke University Medical Center (Protocol Number: A 107–11–04). Duke University and Duke University Medical Center maintain an animal program that is registered with the USDA, assured through the NIH/PHS, and accredited with AAALAC, International:
USDA: Customer Number: 83. Status: Current; No outstanding citations or non-compliances. Most recent site visit: Summer 2012.
NIH/PHS: Assurance Number: A3195–01. Status: Current through December 2013; No outstanding citations or non-compliances.
AAALAC: Accreditation Number: 363. Status: Continued Full Accreditation (Accredited continuously since 1976). Most recent site visit: Fall 2009.
| Abbreviations: | ||
| BSA | = | bovine serum albumin |
| CDR | = | complementarity-determining region |
| EC50 | = | Effective concentration 50% |
| EDTA | = | ethylenediaminetetraacetic acid |
| ELISA | = | enzyme-linked immunosorbent assay |
| FBS | = | fetal bovine serum |
| HC | = | heavy chain |
| HRP | = | horseradish peroxidase |
| IC50 | = | 50% inhibitory concentration |
| IgG | = | immunoglobulin G |
| IgM | = | immunoglobulin M |
| IL-2 | = | interleukin-2 |
| IT | = | immunotoxin |
| LC | = | light chain |
| LPP | = | leucine-proline-proline |
| mAb | = | monoclonal antibody |
| Mut (m) | = | mutant |
| PBS | = | phosphate buffered saline |
| PE38KDEL | = | Pseudomonas exotoxin with last four amino acids of lysine, aspartic acid, glutamic acid and leucine |
| PCR | = | polymerase chain reaction |
| RACE | = | rapid amplify c-DNA ends |
| RAS | = | arginine-alanine-serine |
| RIT | = | recombinant immunotoxin |
| scFv | = | single-chain variable fragment |
| VH | = | variable heavy-chain |
| VL | = | variable light-chain |
Additional material
Download Zip (243.3 KB)Acknowledgments
Thanks are due to Dr Carol J Wikstrand of the Department of Microbiology at the Saba University School of Medicine for the established DmAb14 hybridoma, as well as to Dr Michael Zalutsky of the Medical Physics Program and Biomedical Engineering at Duke University, for radio-labeling the DmAb14m-IT. We also thank Scott Szafranski and Ling Wang for technical support. This work was supported by the following NIH grants: NINDS grant P50 NS020023, NCI SPORE grant P50 CA108786, and NCI MERIT Award grant R37 CA011898.
Disclosure of Potential Conflicts of interest
No potential conflict of interest was disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/mabs/article/25860.
References
- Wong ET, Hess KR, Gleason MJ, Jaeckle KA, Kyritsis AP, Prados MD, et al. Outcomes and prognostic factors in recurrent glioma patients enrolled onto phase II clinical trials. J Clin Oncol 1999; 17:2572 - 8; PMID: 10561324
- Waldmann TA. Monoclonal antibodies in diagnosis and therapy. Science 1991; 252:1657 - 62; http://dx.doi.org/10.1126/science.2047874; PMID: 2047874
- Wu AM, Senter PD. Arming antibodies: prospects and challenges for immunoconjugates. Nat Biotechnol 2005; 23:1137 - 46; http://dx.doi.org/10.1038/nbt1141; PMID: 16151407
- Vukelić Z, Kalanj-Bognar S, Froesch M, Bîndila L, Radić B, Allen M, et al. Human gliosarcoma-associated ganglioside composition is complex and distinctive as evidenced by high-performance mass spectrometric determination and structural characterization. Glycobiology 2007; 17:504 - 15; http://dx.doi.org/10.1093/glycob/cwm012; PMID: 17293353
- Liu Y, Yan S, Wondimu A, Bob D, Weiss M, Sliwinski K, et al. Ganglioside synthase knockout in oncogene-transformed fibroblasts depletes gangliosides and impairs tumor growth. Oncogene 2010; 29:3297 - 306; http://dx.doi.org/10.1038/onc.2010.85; PMID: 20305696
- Svennerholm L, Fredman P, Jungbjer B, Månsson JE, Rynmark BM, Boström K, et al. Large alterations in ganglioside and neutral glycosphingolipid patterns in brains from cases with infantile neuronal ceroid lipofuscinosis/polyunsaturated fatty acid lipidosis. J Neurochem 1987; 49:1772 - 83; http://dx.doi.org/10.1111/j.1471-4159.1987.tb02435.x; PMID: 3681296
- Wikstrand CJ, Longee DC, McLendon RE, Fuller GN, Friedman HS, Fredman P, et al. Lactotetraose series ganglioside 3′,6′-isoLD1 in tumors of central nervous and other systems in vitro and in vivo. Cancer Res 1993; 53:120 - 6; PMID: 8416736
- Wikstrand CJ, Fredman P, Svennerholm L, Humphrey PA, Bigner SH, Bigner DD. Monoclonal antibodies to malignant human gliomas. Mol Chem Neuropathol 1992; 17:137 - 46; http://dx.doi.org/10.1007/BF03159988; PMID: 1384525
- Fredman P, Hedberg K, Brezicka T. Gangliosides as therapeutic targets for cancer. BioDrugs 2003; 17:155 - 67; http://dx.doi.org/10.2165/00063030-200317030-00002; PMID: 12749752
- Fredman P. Gangliosides associated with primary brain tumors and their expression in cell lines established from these tumors. Prog Brain Res 1994; 101:225 - 40; http://dx.doi.org/10.1016/S0079-6123(08)61952-4; PMID: 8029453
- Hedberg KM, Mahesparan R, Read TA, Tysnes BB, Thorsen F, Visted T, et al. The glioma-associated gangliosides 3′-isoLM1, GD3 and GM2 show selective area expression in human glioblastoma xenografts in nude rat brains. Neuropathol Appl Neurobiol 2001; 27:451 - 64; http://dx.doi.org/10.1046/j.1365-2990.2001.00353.x; PMID: 11903928
- Fredman P, von Holst H, Collins VP, Dellheden B, Svennerholm L. Expression of gangliosides GD3 and 3′-isoLM1 in autopsy brains from patients with malignant tumors. J Neurochem 1993; 60:99 - 105; http://dx.doi.org/10.1111/j.1471-4159.1993.tb05827.x; PMID: 8417168
- Wikstrand CJ, He XM, Fuller GN, Bigner SH, Fredman P, Svennerholm L, et al. Occurrence of lacto series gangliosides 3′-isoLM1 and 3′,6′-isoLD1 in human gliomas in vitro and in vivo. J Neuropathol Exp Neurol 1991; 50:756 - 69; http://dx.doi.org/10.1097/00005072-199111000-00007; PMID: 1748882
- Gottfries J, Mansson JE, Fredman P, Wikstrand CJ, Friedman HS, Bigner DD, et al. Ganglioside mapping of a human medulloblastoma xenograft. Acta Neuropathol 1989; 77:283 - 8; http://dx.doi.org/10.1007/BF00687580; PMID: 2922991
- Archer GE, Sampson JH, Lorimer IA, McLendon RE, Kuan CT, Friedman AH, et al. Regional treatment of epidermal growth factor receptor vIII-expressing neoplastic meningitis with a single-chain immunotoxin, MR-1. Clin Cancer Res 1999; 5:2646 - 52; PMID: 10499644
- Kuan CT, Reist CJ, Foulon CF, Lorimer IA, Archer G, Pegram CN, et al. 125I-labeled anti-epidermal growth factor receptor-vIII single-chain Fv exhibits specific and high-level targeting of glioma xenografts. Clin Cancer Res 1999; 5:1539 - 49; PMID: 10389943
- Yokota T, Milenic DE, Whitlow M, Schlom J. Rapid tumor penetration of a single-chain Fv and comparison with other immunoglobulin forms. Cancer Res 1992; 52:3402 - 8; PMID: 1596900
- Pastan IH, Archer GE, McLendon RE, Friedman HS, Fuchs HE, Wang QC, et al. Intrathecal administration of single-chain immunotoxin, LMB-7 [B3(Fv)-PE38], produces cures of carcinomatous meningitis in a rat model. Proc Natl Acad Sci U S A 1995; 92:2765 - 9; http://dx.doi.org/10.1073/pnas.92.7.2765; PMID: 7708720
- Murphy K, Travers P, Walport M, Janeway C. Janeway's immunobiology. New York: Garland Science, 2012.
- Chowdhury PS, Pastan I. Improving antibody affinity by mimicking somatic hypermutation in vitro. Nat Biotechnol 1999; 17:568 - 72; http://dx.doi.org/10.1038/9872; PMID: 10385321
- Hawley TS, Hawley RG. Flow cytometry protocols. Totowa, N.J.: Humana Press, 2004.
- Chandramohan V, Bao X, Keir ST, Pegram CN, Szafranski SE, Piao H, et al. Construction of an immunotoxin, D2C7-(scdsFv)-PE38KDEL, targeting EGFRwt and EGFRvIII for brain tumor therapy. Clin Cancer Res 2013; http://dx.doi.org/10.1158/1078-0432.CCR-12-3891; PMID: 23857604
- Hedegaard CJ, Pegram CN, Bigner DD, Poulsen HS. Internalization of the dual-specific immunotoxin D2C7-(scdsFv)-PE38KDEL in malignant glioma cell lines [abstract]. In: Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research; 2012 Mar 31-Apr 4. Chicago, IL, Philadelphia (PA): AACR; Cancer Res, 2012; 72(8 Suppl):Abstract nr 2732.
- Kreitman RJ, Chaudhary VK, Waldmann T, Willingham MC, FitzGerald DJ, Pastan I. The recombinant immunotoxin anti-Tac(Fv)-Pseudomonas exotoxin 40 is cytotoxic toward peripheral blood malignant cells from patients with adult T-cell leukemia. Proc Natl Acad Sci U S A 1990; 87:8291 - 5; http://dx.doi.org/10.1073/pnas.87.21.8291; PMID: 2236041
- Wikstrand CJ, Fredman P, Svennerholm L, Bigner DD. Detection of glioma-associated gangliosides GM2, GD2, GD3, 3′-isoLM1 3′,6′-isoLD1 in central nervous system tumors in vitro and in vivo using epitope-defined monoclonal antibodies. Prog Brain Res 1994; 101:213 - 23; http://dx.doi.org/10.1016/S0079-6123(08)61951-2; PMID: 7518092
- Molin K, Månsson JE, Fredman P, Svennerholm L. Sialosyllactotetraosylceramide, 3′-isoLM1, a ganglioside of the lactotetraose series isolated from normal human infant brain. J Neurochem 1987; 49:216 - 9; http://dx.doi.org/10.1111/j.1471-4159.1987.tb03417.x; PMID: 3585330
- Pastan I, FitzGerald D. Recombinant toxins for cancer treatment. Science 1991; 254:1173 - 7; http://dx.doi.org/10.1126/science.1683495; PMID: 1683495
- Allured VS, Collier RJ, Carroll SF, McKay DB. Structure of exotoxin A of Pseudomonas aeruginosa at 3.0-Angstrom resolution. Proc Natl Acad Sci U S A 1986; 83:1320 - 4; http://dx.doi.org/10.1073/pnas.83.5.1320; PMID: 3006045
- Provenzale JM, Mukundan S, Dewhirst M. The role of blood-brain barrier permeability in brain tumor imaging and therapeutics. AJR Am J Roentgenol 2005; 185:763 - 7; http://dx.doi.org/10.2214/ajr.185.3.01850763; PMID: 16120931
- Buchner J, Pastan I, Brinkmann U. A method for increasing the yield of properly folded recombinant fusion proteins: single-chain immunotoxins from renaturation of bacterial inclusion bodies. Anal Biochem 1992; 205:263 - 70; http://dx.doi.org/10.1016/0003-2697(92)90433-8; PMID: 1332541
- Ho M, Kreitman RJ, Onda M, Pastan I. In vitro antibody evolution targeting germline hot spots to increase activity of an anti-CD22 immunotoxin. J Biol Chem 2005; 280:607 - 17; PMID: 15491997
- Kadan MJ, Sturm S, Anderson WF, Eglitis MA. Detection of receptor-specific murine leukemia virus binding to cells by immunofluorescence analysis. J Virol 1992; 66:2281 - 7; PMID: 1312632
- Shapira A, Benhar I. Toxin-based therapeutic approaches. Toxins (Basel) 2010; 2:2519 - 83; http://dx.doi.org/10.3390/toxins2112519; PMID: 22069564