
Abstract
Proprotein convertase subtilisin/kexin type 9 (PCSK9) is a serine protease that plays an important role in the regulation of serum low-density lipoprotein (LDL) cholesterol by downregulation of LDL receptor, and as such is considered a novel target in cholesterol lowering therapy. In support of the drug development program for Evolocumab, a fully human IgG2 antibody that targets PCSK9, a quantitative ELISA to measure free PCSK9 in human serum was developed. PCSK9 serves as a biomarker of pharmacological response during treatment, and measuring levels of the free ligand post-dosing was of interest as an aid to establishing the pharmacokinetic and pharmacodynamic properties of the therapeutic. Given the complexities associated with the measurement of free ligand in the presence of high concentrations of circulating drug, it was important to challenge the method with experiments designed to assess ex vivo conditions that have the potential to affect the binding equilibrium of drug and ligand within test samples during routine sampling handling and assay conditions. Herein, we report results of experiments that were conducted to characterize the assay in alignment with regulatory guidance and industry standards, and to establish evidence that the method is measuring the free ligand in circulation at the time serum was collected. A robust supporting data package was generated that demonstrates the method specifically and reproducibly measures the free ligand, and is suitable for its intended use.
Introduction
Monoclonal antibodies (mAbs) represent a rapidly growing class of biotherapeutic, and contribute substantially to the number of biologic products that have been approved or are currently in development.Citation1-Citation3 Therapeutic mAbs target a wide range of indications, including cancer, immune-mediated disorders, cardiovascular disease, organ transplantation, and infectious disease.Citation1,Citation2,Citation4 The characteristics of mAbs that make them highly desirable as a biotherapeutic include their high selectivity and specificity, limited off-target toxicity profiles,Citation5 natural bivalency, high affinity target binding through noncovalent reversible interactions,Citation3 and long serum half-lives.Citation5 The ligands targeted may be membrane-bound, shed cell surface receptors, or circulating soluble proteins. There are more than 20 Food and Drug Administration (FDA)-approved therapeutic mAbs currently on the market, all of which are of the immunoglobulin G (IgG) subclass,Citation4,Citation5 and ~25% of the approved antibodies target soluble ligands.Citation6 The cornerstone to successful development of therapeutic mAbs, as well as all therapeutics, is establishing safe and effective dosing regimens for patients. Establishing effective dosing regimens relies on nonclinical and clinical development programs to characterize the toxicological, pharmacokinetic (PK), and pharmacodynamic (PD) properties of the drug in vivo. Collectively these data provide an understanding of the exposure-response relationship of the therapeutic, and allow quantitative modeling of the PK-PD interactions that occur between drug and target for the primary purpose of informing dose selection. Success of the PK and PD characterization is highly dependent on the development of bioanalytical methods that are robust and reliable in the quantification of drug or target.Citation7 Appropriately designed and well-characterized bioanalytical methods serve to lay a solid foundation of data that will aid in the scientific and clinical decisions that need to be made within each individual drug development program.
Evolocumab, a human IgG2 antibody that targets proprotein convertase subtilisin/kexin type 9 (PCSK9) is currently in development for the treatment of hypercholesterolemia. PCSK9 is a serine protease that is produced predominately by the liver, secreted into plasma, and circulates at concentrations ranging from 100–1000 ng/mL.Citation8 PCSK9 plays a key role in the recycling pathway of hepatic low-density lipoprotein receptors (LDLRs), which are critical to maintaining cholesterol balance.Citation9-Citation12 The receptor recycling pathway involves the binding of low-density lipoprotein cholesterol (LDL-C) to the hepatic cell surface LDLR where the complex is internalized, and transported to the endosome. The LDL-C dissociates from the receptor and is catabolized and the receptor is recycled to the cell surface for continued clearance of serum cholesterol.Citation8,Citation11 PCSK9 affects the receptor recycling pathway by binding to the LDLR and causing degradation of the receptor within the endosome/lysosome compartment.Citation11,Citation13,Citation14 Degradation of the LDLR results in decreased clearance of serum cholesterol, and as a result a higher risk of hypercholesterolemia.Citation9 Human genetic studies have shown that “gain-of-function” (GOF) mutations in the PCSK9 gene can lead to a form of familial hypercholesterolemia with a higher risk of cardiovascular disease.Citation9,Citation11,Citation13,Citation15 In contrast, humans with “loss-of-function” (LOF) mutations in the PCSK9 gene have lower serum cholesterol levels and a lower incidence of cardiovascular disease.Citation9,Citation11,Citation13,Citation15 Clinical studies indicate that the current standard of care for hypercholesterolemia, treatment with statins, results in increased PCSK9 levels leading to decreased receptor expression and elevated LDL-C.Citation8,Citation15 Collectively the data suggests that the inhibition of PCSK9 may sustain lower LDL-C levels,Citation15 and this approach has promise as an additional pharmacologic intervention step in the treatment of hypercholesterolemia.Citation8,Citation13,Citation15
Nonclinical and clinical development of Evolocumab has been guided by PK-PD models that have characterized the in vivo interactions of the mAb and the endogenous soluble target. In general, PK-PD models describe the concentrations and rates of change across the possible molecular species that exist after dosing, based on the dose of drug administered, and the affinity and kinetic parameters of both drug and ligand.Citation6,Citation7 The models are used to link changes in target levels at a given dose of the therapeutic to downstream effects of the drug. These data can then be used to simulate and predict the potential outcome across clinical study designs,Citation7 thereby improving the likelihood of success in the drug development paradigm. For programs where the therapeutic targets a circulating ligand, drug and target will exist within the biological matrix in multiple forms post-dosing, i.e., free ligand, free drug, and drug:ligand complexes. Success of the PK-PD modeling effort relies on accurate measurements of the molecular species, and ideally this would include measurements of either total or free levels of both drug and ligand. Quantification of the free form of ligand is the most direct assessment of target modulation following treatment;Citation16 when incorporated into the PK-PD model, it provides valuable information toward selection of an efficacious dose.Citation7,Citation17
To support the development of Evolocumab, a quantitative ELISA to measure free human PCSK9 was developed (). PCSK9 is considered a biomarker, with the data being used as an exploratory endpoint to monitor target suppression during treatment. The method was characterized for performance parameters consistent with those established for quantitative PK methods as described within the bioanalytical FDA guidance document and industry literature.Citation18-Citation20 Considering the biomarker status of the method and the intended use of the data, the method characterization studies were conducted following fit-for-purpose principles that included evaluating performance characteristics central to biomarker methods.Citation21-Citation23 To confirm quantification of free PCSK9 within the method, empirical data was generated to assess the key ex vivo conditions that have potential to shift the binding equilibrium between drug and ligand within samples during routine sample handling and assay conditions. The experimental design used to evaluate each individual parameter, and the resultant data are presented.
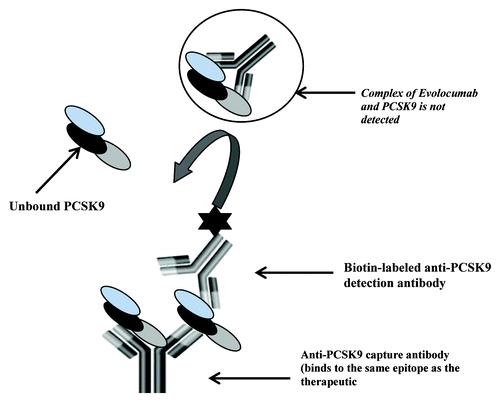
Figure 1. Schematic of the free human PCSK9 assay. The ELISA method utilizes an anti-human PCSK9 monoclonal that binds to the same epitope as the therapeutic antibody and a second biotin conjugated anti-human PCSK9 antibody for detection. The capture antibody by virtue of binding to the same epitope as the therapeutic should capture only the free form of PCSK9 present in samples.
Results
Performance of the rhPCSK9 standards and quality control (QC) samples in the precision and accuracy assessment was reproducible with inter-assay precision results showing coefficients of variation (CVs) of ≤4% for standards and ≤9% for QCs. The assay accurately measured the recombinant protein based on a %bias from nominal that ranged from –3 to 4% for standards and –2 to –10% for QCs. The total error for QCs was ≤19%, which was within the targeted acceptance criteria of ≤ 30%Citation18 (). Based on the data, the assay standard curve range was established at nominal concentrations that ranged from 15 (lower limit of quantitation) to 1200 ng/mL (upper limit of quantitation). The inter-assay precision of endogenous PCSK9 as measured in the serum controls (SCs) was a key performance characteristic to define within the method. As shown in , the CVs for both SC 1 and SC 2 were 11%, with the individual replicates having CVs that ranged from 0% to 9%. Cumulatively, these data demonstrate reproducibility of the method in quantification of both recombinant and endogenous PCSK9.
Table 1. Inter-assay precision and accuracy of rhPCSK9 standards and quality control samples
Table 2. Inter-assay precision of endogenous PCSK9 serum controls
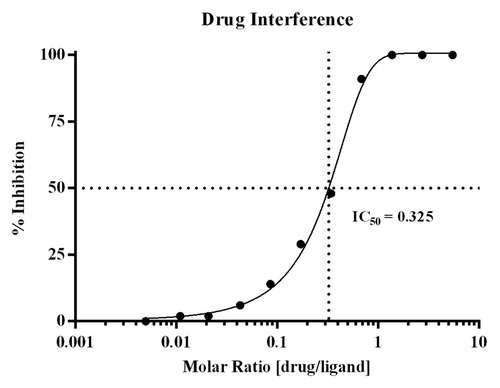
The drug interference assessment showed that when drug:ligand molar ratios reach ≥ 0.625, recovery of PCSK9 is inhibited by ≥91%. These data are not unexpected given the molecular weights of drug and ligand, which are 142 and 72 kiloDaltons, respectively, and the bivalent nature of the antibody. At this molar ratio, drug and ligand are at ~1.5 nM and 2.5 nM, respectively. Assuming bivalent binding of the drug, the drug would be nearly saturating in terms of capacity, thereby driving the equilibrium toward complex formation and a decrease in detection of free PCSK9. The test panel with the corresponding % inhibition results is presented in . displays the graphical titration curve with the calculated IC 50 that was determined to be a molar ratio of 0.325.
Table 3. Drug interference summary: ex vivo test panel
Figure 2. Graphical display of the drug interference titration curve. Drug interference within the method was evaluated using the ex vivo serum panel prepared in pooled human serum with endogenous PCSK9, and titrating levels of Evolocumab. The % inhibition was calculated across the drug:ligand molar ratios tested, and the IC 50 was calculated to be at a molar ratio of 0.325.
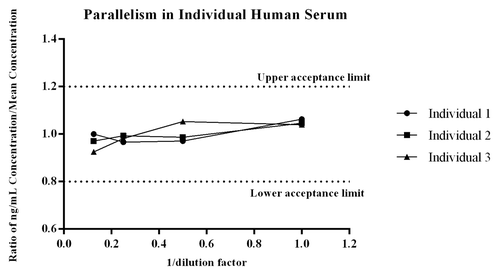
Parallelism determines the relative accuracy of recovery of the endogenous protein present in the biological matrix over multiple dilutions. In essence, it is a dilutional linearity evaluation of individual serum samples to determine if the method recognizes the endogenous ligand present in matrix similar to the recombinant ligand. Comparable results over the dilution series support use of the biomarker method as quantitative, while a lack of parallelism moves the assay to a quasi-quantitative state and directly affects how the data can be used.Citation22 Parallelism of the endogenous ligand within the method was observed across the three individual human donors because all values of PCSK9 across dilution points were within the targeted acceptance ratio of 0.8 – 1.2Citation22 (). The CV of measured PCSK9 within each dilution series for each donor was ≤8%, demonstrating the method measures the endogenous ligand reproducibility across individual serum donors.
Figure 3. Parallelism of the endogenous ligand within the method was evaluated across three individual donors that were diluted at multiple dilutions and then quantitated against the recombinant PCSK9 standard curve. For each donor the concentration measured at each dilution, divided by the mean ng/mL concentration of the dilution series was assessed to determine if the ratio fell within the range of 0.8 -1.2.Citation22 As shown all values met the acceptance criteria, giving the assay validity as a quantitative biomarker method.
The data from the sample dilution dissociation experiment presented in shows that varying the dilution factor did not significantly alter the amount of free ligand that was quantified from test samples containing drug. The %bias from the mean ng/mL concentration of the sample dilution series was calculated for each test dilution, and all values were ≤20%, which is acceptable performance for a ligand binding assay (LBA).Citation18 A summary of the data are presented in . The difference in quantified levels of PCSK9 between the 1:10 and the 1:100 dilutions were all ≤18% for samples containing drug and ligand, as well as the control sample with no drug. As shown, there is a decrease in the level of PCSK9 measured at a 1:10 dilution across all drug:ligand molar ratios tested, including the control sample with no drug. The consistent decrease seen even within the control sample indicates the observation is most likely due to a matrix effect, and not due to liberation of PCSK9 from complex at the higher dilutions. Collectively, the data demonstrates sample dilution does not significantly liberate PCSK9 from the complex.
Figure 4. To assess the effect that sample dilution may have on the binding equilibrium, samples containing drug and ligand were diluted through the linear range of the method. The samples were diluted by factors of 10, 25, 50, 75, and 100 prior to addition to the plate. The samples tested were prepared with drug:ligand molar ratios of 0, 0.174, and 0.347. To normalize for matrix effects at the various dilutions each diluted test sample was quantitated against a dilution matched standard curve. As shown in there is a decrease in the level of PCSK9 measured at a 1:10 dilution, and this appears to be due to a matrix effect as the observation is consistent within the control sample with no drug. The difference in quantified levels of PCSK9 between the 1:10 and the 1:100 dilutions were all ≤ 18% for all samples. For each test dilution the %bias from the mean ng/mL concentration of the sample dilution series showed all values were within ± 20%. The data demonstrates that sample dilution does not significantly liberate PCSK9 from the complex.
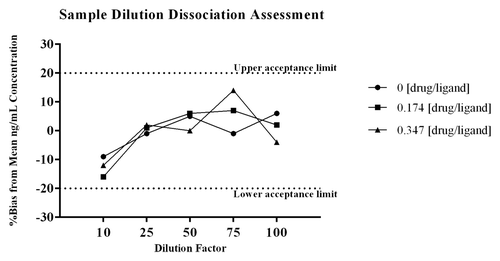
Table 4. Data summary from sample dilution dissociation assessment
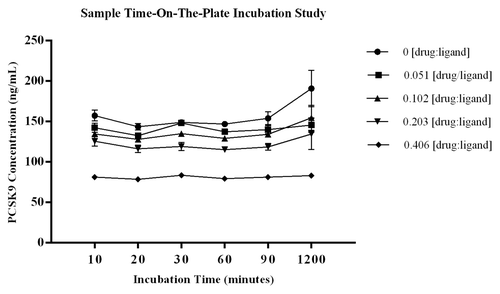
The study evaluating a time course of on-the-plate incubation of diluted samples showed there were no significant shifts in the level of PCSK9 quantified in test samples across the incubation times tested. Using the %bias data to assess increased levels of PCSK9 during sample incubation showed the greatest change to be 21.7%, and this occurred at the 1200 min time point within the control sample with no drug added. For the remainder of the test samples, the %biases from the mean PCSK9 concentration of each test series were all within ± 13.7% (CV for calculated means ≤ 11.1%). The CV of the three independent measurements analyzed for each test sample was ≤14.2% demonstrating robustness of the data sets. Data are presented graphically in with STD error bars around the three measurements. The method was established with standard sample incubation time of 60 ± 5 min, and the study results indicate the selected parameter is on a plateau where liberation of ligand from complex is not observed. The data also shows there is no change in measured PCSK9 from the 10 to 60 min incubation time, demonstrating that there is no early shift in the binding equilibrium during the assay capture step, which further supports the selected timeframe for capture of free ligand. These data indicate that a shift in the binding equilibrium does not occur with increased sample incubation time on-the-plate.
Figure 5. Graphical summary of the data from the study to evaluate the effect that sample incubation time on-the-plate may have on the equilibrium of free vs. bound ligand. Diluted samples were incubated on the plate for 10, 30, 60, 90, and 1200 min at room temperature. The %bias from the mean ng/mL PCSK9 for each test sample showed the largest %bias to be 21.7%, and this occurred at the 1200 min time point within the control sample with no drug added. The remainder of the test samples had %biases from the mean PCSK9 concentration of each test series that were within ± 9% (CV for calculated means ≤ 11.1%). The CV of the three independent aliquots analyzed for each test sample was ≤ 14.2% demonstrating robustness of the data sets. Data are presented graphically in with STD error bars from n = 3 independent measurements at each point.
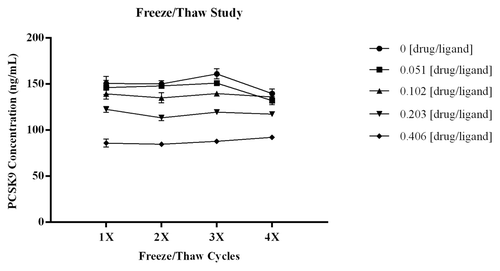
Multiple freeze/thaw cycles did not affect the amount of PCSK9 measured in the assay. As shown in , there was no significant change in the recovery of PCSK9 across the four freeze/thaw cycles and this was consistent across all drug:ligand molar ratios tested. The CV of the three independent measurements for each test point was ≤5.3%, demonstrating robustness of the data sets. The %bias from the mean ng/mL PCSK9 calculated for each test sample was within ± 8.5%, indicating there was no significant increase in free ligand. The CV for the calculated means across the test series were ≤5.8%.
Figure 6. To determine if multiple freeze/thaw cycles would affect the amount of PCSK9 measured in the assay, samples with drug:ligand at varying molar ratios were subjected to four freeze/thaw cycles. As shown in there was no significant change in the recovery of PCSK9 across the four freeze/thaw cycles for all samples tested. Three independent aliquots were analyzed for each sample and at each test point the CV s were all ≤ 5.3% demonstrating robustness of the data sets. Error bars within the graph indicate STD of the n = 3 independent measurements at each test point. The %bias from the mean ng/mL PCSK9 for each test sample was within ± 8.5% indicating there was no significant increase in free ligand. The CV for the calculated means across the test series were ≤ 5.8%.
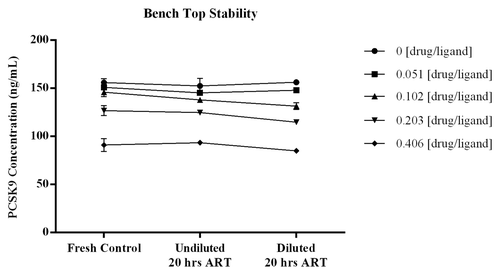
The stability of samples incubated on the bench at room temperature was demonstrated with the quantitated levels of free PCSK9 remaining consistent across the increased incubation times tested. displays the data graphically with errors bars around each point representing the STD of the three measurements. For all samples the %bias from the mean ng/mL concentrations calculated across the test series were within ± 6% (CV for calculated means ≤ 5.3%). The three independent assessments at each test point showed CVs of ≤7.4%, providing confidence in the measurements. The recovery of ligand from samples with increased time on the bench were comparable to values obtained from samples that were analyzed fresh, indicating PCSK9 is not significantly liberated from complex during room temperature sample handling procedures.
Figure 7. To determine if time at room temperature affects the binding equilibrium of PCSK9 and drug, samples prepared with drug:ligand molar ratios of 0, 0.040, 0.080, 0.160 and 0.320 were incubated, undiluted and diluted at 1:50, overnight (≥ 20 h) at room temperature. Test samples that were diluted and added immediately to the plate were included and served as fresh serum controls. The %bias from the mean ng/mL concentrations across the test series were all within ± 6% (CV for calculated means were ≤ 5.2%). Results from the n = 3 independent assessments at each test point showed CVs of ≤ 7.4% indicating the measurements were robust. Error bars within the graph represent the STD of the three independent measurements at each test point. The recovery of ligand from samples with increased time on the bench were comparable to values obtained from samples that were analyzed fresh indicating PCSK9 is not significantly liberated from complex during room temperature sample handling procedures.
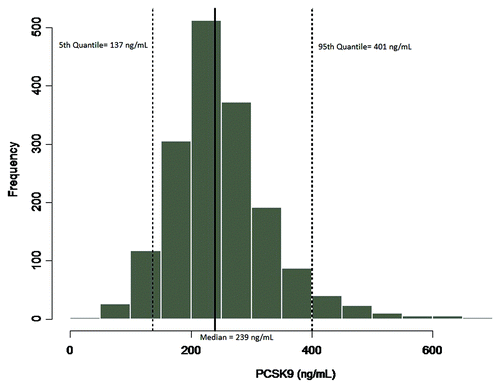
Implementation of the method in a study of the statin treatment, atorvastatin (Lipitor®), provided results that showed concentrations of PCSK9 were elevated in groups receiving atorvastatin treatment compared with normal subjects (). The median PCSK9 levels were 390, 668, and 705 ng/mL for normal subjects, 40mg/day, and 80 mg/day atorvastatin-treated subjects, respectively (). In this study, the subjects treated with atorvastatin showed an increase in median PCSK9 levels of 41–45% compared with the levels measured in normal subjects. In the second evaluation of the applied method, baseline levels of PCSK9 were determined across a large population of samples from a clinical study that was designed to evaluate the statin treatment pravastatin (Pravastatin Inflammation/CRP Evaluation [PRINCE]). The median PCSK9 concentration across the 1698 subjects was 239 ng/mL, with a minimum and maximum of 33.4 and 730 ng/mL, respectively, and a %CV of 33.7. The distribution profile of the PRINCE study samples is presented in . The values obtained for PCSK9 across both studies were consistent with the expected range cited within the literature of 100–1000 ng/mL,Citation8 providing a robust distribution profile in the target population and adding validity to the measurement of the endogenous ligand within the method.
Figure 8. To demonstrate utility of the assay, and generate internal data on the range and variability of PCSK9 a study was conducted in the target population; patients receiving the standard of care statin treatment for hypercholesterolemia. Three groups of subjects were included; Group 1 normal subjects receiving no statin treatment; Group 2 subjects receiving atorvastatin (Lipitor®) treatment at 40mg/Day; Group 3 subjects receiving atorvastatin at 80 mg/Day. As shown the concentrations of PCSK9 were elevated in groups receiving atorvastatin treatment as compared with normal subjects. The values obtained for PCSK9 in this study across the groups were consistent with the range cited within the literature of 100–1000 ng/mL.Citation8
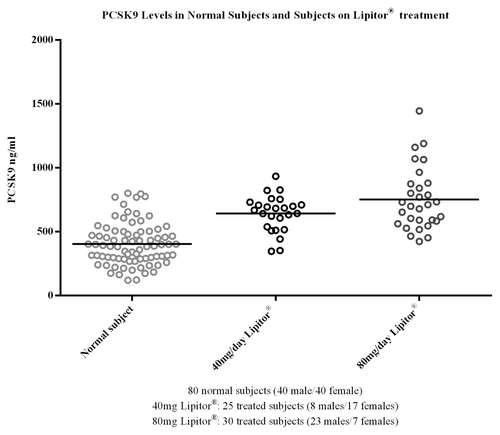
Table 5. Statistical summary of PCSK9 data in atorvastatin treated vs. normal subject
Figure 9. To characterize variability of the endogenous ligand, and establish an expected range of target in the patient population samples from the PRINCE clinical study, evaluating pravastatin (statin treatment), were evaluated for PCSK9 levels. A total of n = 1698 samples, 1229 male, 465 female and 4 with no assignment, were evaluated in the human free PCSK9 assay. The median value of the endogenous ligand was 239 ng/mL, with a minimum and maximum of 33.4 and 730 ng/mL respectively, and a CV of 33.7%. The PCSK9 levels measured in this study were also consistent with the range cited within the literature of 100–1000 ng/mL.Citation8 A distribution profile of the data are presented in , including the 90% confidence intervals.
Discussion
The objective of the work presented here was to characterize the human free PCSK9 ELISA method across performance parameters that align with current FDA guidance for PK assays, including parameters considered crucial to biomarker methods, and to provide an empirical data package that includes close examination of ex vivo conditions that could affect the measurement of the free ligand. The challenges associated with measuring free soluble target have been broadly discussed within the literature, and these are related to the thermodynamic and kinetic interactions of drug and ligand that affect the species that are measured in an immunoassay.Citation3 Due to the challenges associated with development of a method for quantification of free ligand in the presence of drug, it was important to rigorously test the stability of the drug:ligand complex across multiple conditions to characterize the species measured within the method. The experimental studies described here were conducted based on the intended use of the data, following fit-for-purpose principles that were linked to the scientific rigor applied to the characterization package.Citation21,Citation22
Precision and accuracy of the method to measure the recombinant protein in FBS across standards and QC samples was demonstrated with %CV and %bias values that were ≤15 and ≤20, respectively. These results show rhPCSK9 can be measured accurately and reproducibly within the method, thereby validating its use as an analytical standard. Of significant importance was the ability of the method to reproducibly measure endogenous PCSK9, and this is shown by the inter-assay CV of ≤11% for serum controls containing two levels of endogenous PCSK9. The other critical parameter evaluated was parallelism, which defines the biomarker method as quantitative or quasi-quantitative. The study included individual serum donor samples with endogenous PCSK9 that were subjected to multiple dilutions. The data across all donors demonstrated the assay recognizes the endogenous ligand similar to the recombinant ligand. The comparable results observed over the dilution series of these authentic samples supports use of the biomarker method as a quantitative assay. Results from the assessment of drug interference show that at a drug:ligand molar ratio of ≥0.6, detection of free PCSK9 is significantly inhibited (≥91%). Taking into account bivalent binding of the antibody, at this molar ratio the concentration of drug is nearly saturating and we would anticipate the equilibrium to shift toward complex formation. The IC50 was calculated to be at a molar ratio of 0.325, demonstrating the assay as designed detects the free ligand.
Cumulative results from the studies designed to evaluate the effect of varied ex vivo conditions showed there were no significant changes in the levels of endogenous PCSK9 that were measured across all parameters tested. There were no changes observed in measured PCSK9 when test samples were frozen and thawed over four cycles. The sample dilution study was central to characterizing the established minimum required dilution (MRD) in regards to its potential influence on the species measured in the method. The data from this study demonstrated that at 2-fold dilutions either side of the 1:50 MRD, there was no significant difference in the level of PCSK9 measured, and this was consistent across the concentrations of drug tested. Based on these data, the MRD is on a plateau where slight differences in sample dilution would not be expected to perturb the binding equilibrium. Equally important was the collective assessment of sample incubation time. The studies included extended incubation of diluted samples on the assay plate, and bench-top stability of diluted and undiluted samples. There were no significant changes in PCSK9 levels that were measured across all time points in both studies, showing that increased sample incubation time does not result in an equilibrium shift of free vs. bound ligand. There was no evidence that the capture antibody was able to liberate drug from the complex with increased incubation time on the plate, further supporting that, within the defined assay parameters, the method is measuring free PCSK9.
The comparative studies conducted using sera from patients receiving standard of care statin treatment for hypercholesterolemia demonstrated proof-of-concept, and generated internal data on the range and variability of PCSK9 in the target population. Results across both studies showed that the levels of PCSK9 measured in the assay were comparable to those reported in the literature, which added validity to the assay measurement of the biomarker.Citation8 Both studies provided robust data sets that served to characterize the variability of the endogenous ligand in the patient population, and helped to establish the expected range of target. Though not a pre-dose and post-dose comparative study, it was noted within the atorvastatin study that PCSK9 levels were increased in subjects receiving statin treatment. This observation was consistent with reports within the literature,Citation14,Citation15 further demonstrating proof-of-concept. Post-validation, the method has been used successfully in clinical studies and the data has allowed us to generate distribution profiles of PCSK9 levels in patients, establish the intra-subject variability of PCSK9 in the target population, and provide insight into biological impact, as shown by a strong correlation of decreased PCSK9 levels with the clinical endpoint of lowering LDL-C levels.Citation24 The clinical data has demonstrated the utility of the method in profiling PCSK9 concentrations over the course of therapeutic treatment.
Based on the binding principles of the method format, and the empirical data generated to assess the thermodynamic conditions that could perturb drug-ligand complex, the results provide evidence that the method measures the free form of the ligand within the variability of the ELISA method and the inherent variability associated with the circulating biomarker. The cumulative data set demonstrates that direct manipulation of the samples does not result in a significant change in the levels of PCSK9 detected, indicating that the equilibrium established in test samples is stable during routine sample handling or assay conditions. Quantification of free ligand in the presence of drug is difficult and anticipated to have challenges and limitations that are specific to the proteins involved and the associated biology, but it is not insurmountable. By establishing a scientifically sound assay design that utilizes highly specific reagents, implementing optimal assay conditions to minimize equilibrium shifts, and then applying scientific rigor to testing of the method, it is possible to quantify free ligand and provide confidence in the levels that are measured. The results of these studies demonstrate that the biomarker method is reproducible and robust in measuring free endogenous PCSK9 in human serum, and the body of work demonstrates the method is suitable for the intended use.
Materials
A purified anti-human PCSK9 monoclonal IgG2 antibody (capture reagent), that binds to the same epitope as the therapeutic, and a second purified anti-human PCSK9 monoclonal IgG2 antibody (detection reagent) conjugated to biotin, EZ-Link NHS-Chromogenic Biotin (Pierce), and recombinant human PCSK9 (rhPCSK9) were generated at Amgen Inc. Commercially-sourced pooled and individual human serum was acquired from Bioreclamation, Inc. Human serum samples collected as part of the “Pravastatin Inflammation/CRP Evaluation (PRINCE) study” were transferred from the Brigham and Women’s Hospital, Inc. to Amgen for assessment of PCSK9 levels only; all patient information was de-identified prior to transfer. Fetal bovine serum (FBS) and Dulbecco’s Phosphate-Buffered Saline (DPBS) were purchased from Gibco. Maxisorp Nunc-immuno 96-well microtiter plates were purchased from NalgeNunc International. Superblock® T20 and Streptavidin poly-horseradish peroxidase (HRP) were purchased from Thermo Scientific. Tetramethyl-benzedine (TMB) one component HRP microwell substrate was purchased from Surmodics, and 1 Normal (1N) sulfuric acid was purchased from Thermo-Fisher Scientific. Wash buffer was purchased from Kirkegaard and Perry Laboratories.
Methods
PCSK9 ELISA Format
The PCSK9 ELISA method was designed to measure the free form of ligand in test samples by utilizing a proprietary anti-human PCSK9 mAb that binds to the same epitope as Evolocumab as the capture antibody and a second biotin conjugated proprietary anti-human PCSK9 mAb for detection. The capture antibody, by virtue of binding to the same epitope as the therapeutic, should capture only the unbound form of PCSK9 present in samples (). The assay format is conducive to detection of free ligand; the capture antibody binds with high affinity to human PCSK9 (dissociation constant [Kd]) = 16 pM), making dissociation from circulating complex challenging.
ELISA Procedure
rhPCSK9 (Amgen Inc.) was used to prepare the standard curve with a range of 15–1200 ng/mL, and QC samples. 96-well Nunc Maxisorp plate wells were coated with 100 μL of anti-human PCSK9 coating antibody diluted to 1 μg/mL in coating buffer (1X DPBS) and incubated at 2–8 °C for 18 to 72 h. Plates were washed and blocked with 200 μL assay buffer (Superblock® T20) for 1 h. Standards, controls and samples were diluted 50-fold in assay buffer prior to assay. After washing, the diluted standards, controls and samples were added to the microplate wells and then incubated for 1 h at ambient room temperature (ART). After a wash step, the biotinylated secondary antibody was added to the wells and incubated for 1 h at ART. After another wash step, streptavidin poly-HRP solution was added to the microplate wells and incubated for 30 min at ART. After a final wash step, TMB substrate solution was added to the wells for color development, which was stopped after 10 min by the addition of 1 N sulfuric acid. The optical density was measured at 450 nm with a 650 nm reference wavelength subtraction, using a SpectraMax 384 Plus ELISA plate reader (Molecular Devices). Standard regression was established using a logistic (4-PL) curve fit with 1/yCitation2 weighting in the Watson laboratory information management system (Thermo Scientific, version 7.4).
Characterization Studies
Precision and accuracy
A total of six precision and accuracy assays were performed by three analysts over a period of two days. For the precision and accuracy evaluation, three independent sets of rhPCSK9 standard curve and QCs were prepared in 100% FBS. The nominal concentrations for the standards were 0, 15, 30, 75, 150, 300, 600, and 1200 ng/mL, and for QC samples 0, 15, 40, 200, 900, and 1200 ng/mL. Human serum controls containing endogenous PCSK9 at two levels, SC 1 and SC 2, were analyzed across six precision and accuracy assays in replicates for a total of 36 values each.
Drug interference
To evaluate drug interference within the method an ex vivo test panel was prepared in pooled human serum with 180 ng/mL endogenous PCSK9, spiked with titrating levels of Evolocumab at concentrations ranging from 1.8 to 1 000 000 ng/mL, incubated for ≥20 h at room temperature and then frozen. Samples were diluted 1:50 and analyzed within the ELISA method as described. The % inhibition was calculated based on the nominal value of PCSK9 measured in the pooled serum matrix.
Parallelism
Parallelism was evaluated by quantification of the endogenous PCSK9 levels in three individual human serum samples that were serially diluted 1:2 into FBS. Levels of endogenous PCSK9 measured in the three individual donors were 175, 227, and 306 ng/mL. Data was analyzed by calculating the individual PCSK9 concentrations at each dilution, dividing by the mean ng/mL concentration of the dilution series for each serum lot, and then plotting against 1/dilution factor. Acceptable performance was determined based on the ratios falling within an acceptance range of 0.8 to 1.2Citation22 (± 20%).
Sample dilution dissociation assessment
The PCSK9 method was established with a MRD of 1:50. To assess the effect that sample dilution may have on the binding equilibrium, samples containing drug and ligand were diluted through the linear range of the method. The samples were diluted in assay buffer by factors of 10, 25, 50, 75, and 100 (test series) prior to addition to the plate. To normalize for matrix effects at the various dilutions each diluted test sample was quantitated against a dilution matched standard curve (e.g., 1:10 dilutions were quantitated against a standard curve diluted at 1:10). The samples tested were prepared with drug:ligand molar ratios of 0, 0.174, and 0.347 corresponding to inhibitions of ~0, 30% and 50%, respectively (). For this study, samples were tested as single assessments with duplicate wells per test point. For each test sample, the mean ng/mL concentration of PCSK9 across test dilutions was calculated, and the %bias from the mean was determined at each dilution point to evaluate if there was an increase in measured ligand. The %bias was calculated as: [(observed -mean ng/mL PCSK9 of test series)/(mean ng/mL PCSK9 of test series)] × 100.
Sample time on-the-plate incubation study
To evaluate if the time that samples incubate on the plate has an effect on the equilibrium of free vs. bound ligand, test samples prepared with drug:ligand molar ratios of 0, 0.051, 0.102, 0.203, and 0.406 were analyzed. These samples correspond to the targeted molar ratios where drug is not saturating and the inhibition of PCSK9 is expected to be between ~5 and 50% (). Three independent aliquots of each test sample were analyzed to evaluate reproducibility of the result. Samples were diluted in assay buffer and incubated on the plate for 10, 20, 30, 60, 90, and 1200 min (test series) at room temperature. The mean ng/mL concentration of PCSK9, STD, and %CV was calculated across the three independent measurements for each test sample at each time point. To assess changes in the concentration of PCSK9 measured across test parameters the %bias was calculated as: [(observed -mean ng/mL PCSK9 of test series)/(mean ng/mL PCSK9 of test series)] × 100.
Freeze/thaw study
To determine if multiple freeze/thaw cycles affects the concentration of free PCSK9 measured, samples prepared with drug:ligand molar ratios of 0, 0.051, 0.102, 0.203, and 0.406 were subjected to four freeze/thaw cycles (test series). Three independent aliquots of each sample were analyzed to evaluate reproducibility of the result. The mean ng/mL concentration of PCSK9, STD, and %CV was calculated across the three independent measurements. To assess changes in the PCSK9 concentration measured across freeze/thaw cycles, for each test sample the %bias was calculated as: [(observed-mean ng/mL PCSK9 of test series)/(mean ng/mL PCSK9 of test series)] × 100.
Bench top stability
The stability of diluted samples was evaluated to determine if time at room temperature affects the binding equilibrium of PCSK9 and drug. Samples with drug:ligand molar ratios of 0, 0.051, 0.102, 0.203, and 0.406 were analyzed. Three independent aliquots of each test sample were analyzed to evaluate reproducibility of the result. Samples were incubated on the plate, undiluted and diluted at 1:50 in assay buffer (test series), overnight (≥20 h) at room temperature. A set of test samples that were diluted and added immediately to the plate served as fresh controls. The mean ng/mL concentration of PCSK9, STD, and %CV was calculated across the three independent measurements for each test point. To assess changes in the PCSK9 concentration measured across incubation times, the %bias for each test sample was calculated as: [(observed-mean ng/mL of test series)/(mean ng/mL of test series)] × 100.
Implementation of the method (proof of concept)
Two studies were conducted to both demonstrate utility of the assay, and generate data on the range and variability of PCSK9 in the target population. In the first study, commercially-sourced human serum was obtained from three groups: Group 1 (80 healthy donors [40 male/40 female]; 24–65 y old) receiving no statin or any other lipid lowering treatment, Group 2 (25 subjects [8 males/17 females]; 34–89 y old) receiving 40 mg/day atorvastatin calcium (Lipitor®) treatment, and Group 3 (30 subjects [23 males/7 females]; 34–89 y old) receiving 80 mg/day atorvastatin treatment. All subjects fasted overnight prior to the blood draw. Samples were analyzed in the free human PCSK9 ELISA method. In the second study, human serum samples collected as part of the Pravastin Inflammation/CRP Evaluation (PRINCE) clinical study, a statin therapy study, were evaluated to determine blood levels of PCSK9. A total of 1698 subjects; 1229 male, 465 female and 4 with no assignment, were analyzed within the free PCSK9 method.
Ethical statement
The transfer of PRINCE study materials from Brigham and Women’s Hospital to Amgen was conducted through a Material Transfer Agreement. The agreement acknowledged the materials were collected and maintained in accordance with applicable laws including informed consent, and all applicable regulations and institutional policies. All patient information was de-identified prior to transfer.
| Abbreviations: | ||
| ambient room temperature | = | ART |
| minimum required dilution | = | MRD |
| gain-of-function | = | GOF |
| low density lipoprotein cholesterol | = | LDL-C |
| LDL receptor | = | LDLR |
| loss-of-function | = | LOF |
| proprotein convertase subtilisin/kexin type 9 | = | PCSK9 |
| recombinant human PCSK9 | = | rhPCSK9 |
Disclosure of Potential Conflicts of Interest
There is no potential conflict of interest. The authors are employed with Amgen Inc. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.
References
- Reichert JM. Monoclonal antibodies as innovative therapeutics. Curr Pharm Biotechnol 2008; 9:423 - 30; http://dx.doi.org/10.2174/138920108786786358; PMID: 19075682
- Fischer SK, Yang J, Anand B, Cowan K, Hendricks R, Li J, Nakamura G, Song A. The assay design used for measurement of therapeutic antibody concentrations can affect pharmacokinetic parameters: Case studies. MAbs 2012; 4:623 - 31; http://dx.doi.org/10.4161/mabs.20814; PMID: 22820463
- Kuang B, King L, Wang HF. Therapeutic monoclonal antibody concentration monitoring: free or total?. Bioanalysis 2010; 2:1125 - 40; http://dx.doi.org/10.4155/bio.10.64; PMID: 21083212
- Patel R, Johnson K, Andrien BA, Tamburini PP. IgG Subclass Variation of a Monoclonal Antibody Binding to Human Fc-Gamma Receptors. Amercian Journal of Biochemistry and Biotechnology 2013; 9:206 - 18; http://dx.doi.org/10.3844/ajbbsp.2013.206.218
- Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther 2008; 84:548 - 58; http://dx.doi.org/10.1038/clpt.2008.170; PMID: 18784655
- Davda JP, Hansen RJ. Properties of a general PK/PD model of antibody-ligand interactions for therapeutic antibodies that bind to soluble endogenous targets. MAbs 2010; 2:576 - 88; http://dx.doi.org/10.4161/mabs.2.5.12833; PMID: 20676036
- Roskos LK, Schneider A, Vainshtein I, Schwickart M, Lee R, Lu H, Faggioni R, Liang M. PK-PD modeling of protein drugs: implications in assay development. Bioanalysis 2011; 3:659 - 75; http://dx.doi.org/10.4155/bio.11.28; PMID: 21417734
- Konrad RJ, Troutt JS, Cao G. Effects of currently prescribed LDL-C-lowering drugs on PCSK9 and implications for the next generation of LDL-C-lowering agents. Lipids Health Dis 2011; 10:38; http://dx.doi.org/10.1186/1476-511X-10-38; PMID: 21352602
- Alborn WE, Cao G, Careskey HE, Qian YW, Subramaniam DR, Davies J, Conner EM, Konrad RJ. Serum proprotein convertase subtilisin kexin type 9 is correlated directly with serum LDL cholesterol. Clin Chem 2007; 53:1814 - 9; http://dx.doi.org/10.1373/clinchem.2007.091280; PMID: 17702855
- Banach M, Rizzo M, Obradovic M, Montalto G, Rysz J, Mikhailidis DP, Isenovic ER. PCSK9 inhibition - a novel mechanism to treat lipid disorders?. Curr Pharm Des 2013; 19:3869 - 77; http://dx.doi.org/10.2174/13816128113199990303; PMID: 23286435
- Chan JC, Piper DE, Cao Q, Liu D, King C, Wang W, Tang J, Liu Q, Higbee J, Xia Z, et al. A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc Natl Acad Sci U S A 2009; 106:9820 - 5; http://dx.doi.org/10.1073/pnas.0903849106; PMID: 19443683
- Do RQ, Vogel RA, Schwartz GG. PCSK9 Inhibitors: potential in cardiovascular therapeutics. Curr Cardiol Rep 2013; 15:345; http://dx.doi.org/10.1007/s11886-012-0345-z; PMID: 23338726
- Horton JD, Cohen JC, Hobbs HH. PCSK9: a convertase that coordinates LDL catabolism. J Lipid Res 2009; 50:Suppl S172 - 7; http://dx.doi.org/10.1194/jlr.R800091-JLR200; PMID: 19020338
- Welder G, Zineh I, Pacanowski MA, Troutt JS, Cao G, Konrad RJ. High-dose atorvastatin causes a rapid sustained increase in human serum PCSK9 and disrupts its correlation with LDL cholesterol. J Lipid Res 2010; 51:2714 - 21; http://dx.doi.org/10.1194/jlr.M008144; PMID: 20525997
- Careskey HE, Davis RA, Alborn WE, Troutt JS, Cao G, Konrad RJ. Atorvastatin increases human serum levels of proprotein convertase subtilisin/kexin type 9. J Lipid Res 2008; 49:394 - 8; http://dx.doi.org/10.1194/jlr.M700437-JLR200; PMID: 18033751
- Hansen RJ, Brown RM, Lu J, Wroblewski VJ. Qualification of a free ligand assay in the presence of anti-ligand antibody Fab fragments. MAbs 2013; 5:288 - 96; http://dx.doi.org/10.4161/mabs.23508; PMID: 23396084
- Lee JW, Kelley M, King LE, Yang J, Salimi-Moosavi H, Tang MT, Lu JF, Kamerud J, Ahene A, Myler H, et al. Bioanalytical approaches to quantify “total” and “free” therapeutic antibodies and their targets: technical challenges and PK/PD applications over the course of drug development. AAPS J 2011; 13:99 - 110; http://dx.doi.org/10.1208/s12248-011-9251-3; PMID: 21240643
- DeSilva B, Smith W, Weiner R, Kelley M, Smolec J, Lee B, Khan M, Tacey R, Hill H, Celniker A. Recommendations for the bioanalytical method validation of ligand-binding assays to support pharmacokinetic assessments of macromolecules. Pharm Res 2003; 20:1885 - 900; http://dx.doi.org/10.1023/B:PHAM.0000003390.51761.3d; PMID: 14661937
- Viswanathan CT, Bansal S, Booth B, DeStefano AJ, Rose MJ, Sailstad J, Shah VP, Skelly JP, Swann PG, Weiner R. Quantitative bioanalytical methods validation and implementation: best practices for chromatographic and ligand binding assays. Pharm Res 2007; 24:1962 - 73; http://dx.doi.org/10.1007/s11095-007-9291-7; PMID: 17458684
- US Department of Health and Human Services FaDA, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research. Guidance to Industry: Bioanalytical Method Validation 2001. 25 p.
- Lee JW. Method validation and application of protein biomarkers: basic similarities and differences from biotherapeutics. Bioanalysis 2009; 1:1461 - 74; http://dx.doi.org/10.4155/bio.09.130; PMID: 21083095
- Lee JW, Wu Y, Wang J. Fit-for-Purpose Method Validation and Assays for Biomarker Characterization to Support Drug Development. AAPS J 2010; 11:385 - 94
- Valentin MA, Ma S, Zhao A, Legay F, Avrameas A. Validation of immunoassay for protein biomarkers: bioanalytical study plan implementation to support pre-clinical and clinical studies. J Pharm Biomed Anal 2011; 55:869 - 77; http://dx.doi.org/10.1016/j.jpba.2011.03.033; PMID: 21530130
- Dias CS, Shaywitz AJ, Wasserman SM, Smith BP, Gao B, Stolman DS, Crispino CP, Smirnakis KV, Emery MG, Colbert A, et al. Effects of AMG 145 on low-density lipoprotein cholesterol levels: results from 2 randomized, double-blind, placebo-controlled, ascending-dose phase 1 studies in healthy volunteers and hypercholesterolemic subjects on statins. J Am Coll Cardiol 2012; 60:1888 - 98; http://dx.doi.org/10.1016/j.jacc.2012.08.986; PMID: 23083772