
Abstract
The use of antibodies to target their antigens in living cells is a powerful analytical tool for cell biology research. Not only can molecules be localized and visualized in living cells, but interference with cellular processes by antibodies may allow functional analysis down to the level of individual post-translational modifications and splice variants, which is not possible with genetic or RNA-based methods. To utilize the vast resource of available antibodies, an efficient system to deliver them into the cytosol from the outside is needed. Numerous strategies have been proposed, but the most robust and widely applicable procedure still remains to be identified, since a quantitative ranking of the efficiencies has not yet been done. To achieve this, we developed a novel efficiency evaluation method for antibody delivery based on a fusion protein consisting of a human IgG1 Fc and the recombination enzyme Cre (Fc-Cre). Applied to suitable GFP reporter cells, it allows the important distinction between proteins trapped in endosomes and those delivered to the cytosol. Further, it ensures viability of positive cells and is unsusceptible to fixation artifacts and misinterpretation of cellular localization in microscopy and flow cytometry. Very low cytoplasmic delivery efficiencies were found for various profection reagents and membrane penetrating peptides, leaving electroporation as the only practically useful delivery method for antibodies. This was further verified by the successful application of this method to bind antibodies to cytosolic components in living cells.
Introduction
When used as tools to interfere with cellular processes or to spatiotemporally track molecules in living cells, antibodies are extraordinarily valuable for research. First reports using this approach delivered the antibodies by microinjection into individual cells,Citation1-Citation5 thereby proving that an antibody, once completely folded, is stable and can still bind its antigen in the reducing environment of the cytoplasm. Further to microscopically track molecules in a living cell, antibodies may also be used to interfere with the function of cellular components, which would add a novel analysis level of functional genetics since even individual post-translational modifications like the role of one particular phosphorylation or splice variant could be assessed. Although there have been reports on the successful application of intracellular antibodies, called intrabodies if expressed in the cytosol, the use of this technology is very limited due to the failure of many antibodies to fold correctly in the cytosol.Citation6-Citation8 Various solutions for this problem have recently been provided by the use of various alternative scaffolds which fold correctly in the cytosol.Citation9-Citation12 Correct folding of antibodies is assured only in the endoplasmic reticulum (ER). Consequently, there are many examples where ER-retained antibodies have been used to generate antigen knockdowns,Citation13-Citation15 but this method is restricted to membrane proteins and secreted targets. However, both methods require access to the gene encoding the binder, thus they are not applicable for almost all commercially available research antibodies. To make use of this vast resource, efficient methods to deliver the antibody protein from the outside are needed.
There have been numerous reports on successful protein delivery into cells, including antibodies,Citation6,Citation16 but a systematic comparison is still missing. Reports on delivery of proteins by virus-like particles are promising, but not applicable to antibodies because this method requires cytosolic expression of the cargo protein.Citation17,Citation18
In this study, the different strategies as reviewed previouslyCitation6 have been compared. To do this, a standard method to assess the efficiency of cytosolic delivery that also discriminates between endosomal and cytosolic localization and is insensitive to known artifacts and misinterpretations had to be developed.
Results
Transbodies and Profection
This study’s final goal was to identify the best method to deliver functional antibody protein into living cells from the outside, and to prove that the delivered antibodies bind their antigen in the cytoplasm. In preliminary studies, we invested substantial efforts to employ the HIV-TAT47–57 peptide, as many studies describe the use of cell-penetrating peptides for cytoplasmic delivery.Citation19,Citation20 However, direct fusions to an scFv with HIV-TAT47–57 were non-producible in E. coli (Fig. S1A and B). In contrast, streptavidin::HIV-TAT47–57 peptide fusions were produced well and could successfully be complexed with biotinylated antibodies (data not shown) but were found to locate distinctively in a punctuate pattern, suggesting endosomal entrapment (Fig. S1D).
As the delivery by two profection reagents has been described to be 10–20 times more efficient than that of two protein transduction domains (PTDs),Citation21 we next analyzed protein transfection reagents that are described to release cargoes from endosomes by disturbing the endosome membrane or by a proton sponge effect.Citation22,Citation23 We first tried to compare the efficiencies of these methods using directly labeled antibodies.
Effect of Profection on cell viability
A critical parameter when using antibodies in living cells is viability. While the success of DNA delivery and cell viability is evident when a fluorescent protein is made, demonstrating a functional biosynthesis machinery, there is no information on cell viability if the readout is the fluorescence of a delivered protein. To test whether cells are still alive after profection, HeLa cells were subjected to live-death staining with propidium iodide (PI) after profection of a labeled antibody (IgG-FITC, OzBiosciences) with the lipid-based profection reagent “Pulsin” (Polyplus). Analysis of IgG-FITC-profected cells by flow cytometry revealed a high percentage of cells that were positive for IgG-FITC, as well as for PI (Fig. S2C). This indicates that many antibodies may have entered dead cells. This underlines the necessity to carefully monitor cell viability in profection studies.
Direct labeling of antibodies with fluorescent dyes
Fixation artifacts and misinterpreted localization of fluorescently-labeled proteins are well known from previous protein delivery studies.Citation24-Citation26 Nevertheless, positive controls provided with profection reagents often include fluorescently-labeled proteins. Therefore, scFv-Fc fusions of the anti-myosin antibody SF9 were chemically conjugated to the organic dye DyLight 488 and applied to HeLa cells. The degree of labeling was determined to be 2.1 DyLight 488 fluorophores per protein. When using fixed/permeabilized/myosin Dylight 488 stained cells for control, an evenly distributed fluorescence was detected covering the whole cell, while the specific filamentous pattern expected for myosin only appeared with a low signal-to-noise ratio (data not shown). The same was observed for an ATTO 488 conjugated anti-myosin antibody (clone SF9 produced and conjugated by Adipogen, data not shown).
Living cells incubated with the anti-myosin-Dylight 488 antibody without transfection reagent still showed fluorescence (Fig. S2A) evenly distributed over the whole cell, and additionally found in a spot-like pattern. These results suggested the labeled antibody might have attached to the cell surface unspecifically, and was then taken up by endocytosis.
To test whether it is the fluorescent labeling that leads to these results, HeLa cells were incubated on ice with labeled or unlabeled anti-myosin antibodies, which were then detected by a secondary antibody (without fixation). As incubation took place on ice, endocytosis was suppressed. Flow cytometric analysis revealed the absence of a fluorescent signal for cells that had been incubated with the unlabeled antibody and a positive fluorescent signal for cells that had been incubated with DyLight 488-labeled anti-myosin (Fig. S2B), suggesting that the fluorescent labeling led to the unspecific attachment. Therefore, conjugation to fluorescent dyes was not further considered for the generation of a probe for the systematic assessment of cytosolic delivery of antibodies because it may lead to misinterpretations and may hamper the use of directly labeled antibodies for live imaging.
Endosomal entrapment
Many previous profection studies have not assessed how efficiently proteins are released from endosomes after internalization. In order to monitor endosomal uptake of delivered proteins, the anti-myosin antibody was labeled with a pH-sensitive dye (CypHer5E), which exhibits increased fluorescence intensity with rising acidification in endocytic vesicles. HeLa cells were then incubated either with the anti-myosin-CypHer5E conjugate alone or in complex with a lipid-based profection-reagent (AbDeliverIN) and analyzed by flow cytometry. The fluorescence intensity of cells treated with anti-myosin-CypHer5E/AbDeliverIN complexes was higher compared with that of cells treated with anti-myosin-CypHer5E alone (Fig. S2D). This result suggests increased endosomal retainment in the presence of the profection reagent compared with its absence, while the extent of cytosolic delivery is still not assessable. These results are also in line with our observations for the TAT-peptide/antibody complexes, where endosomal entrapment was predominant while cytosolic concentrations were also not sufficient to provide detectable delivery to the cytosol.
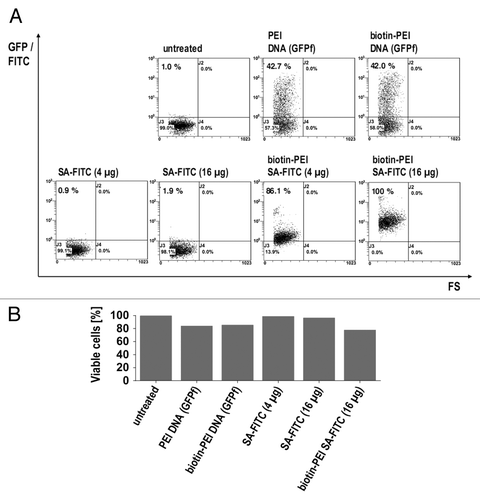
To obtain a rough estimate of actual cytosolic release, the commonly used DNA transfection reagent polyethylenimine (PEI) was biotinylated and subsequently employed for DNA and protein transfection. For this purpose, HeLa cells were treated with biotinylated PEI complexed with either farnesylated GFP (GFPf)-encoding DNA or the biotin-binding protein streptavidin (SA) with FITC as a fluorescent label. Analysis by flow cytometry revealed up to 100% of cells were fluorescent when treated with SA-FITC complexes, whereas only 42% of cells treated with DNA complexes were fluorescent (). The viability of cells treated with DNA or protein complexed with PEI was also assessed (). Although there were fewer fluorescent cells among those transfected with DNA, the fluorescence intensity of these cells reached clearly higher values compared with those reached by cells treated with SA-FITC complexes. This most likely reflects the signal amplification inherent in protein expression after successful delivery of a plasmid. Although DNA delivery efficiency may allow a rough estimate of actual cytosolic release, there was still no direct proof for cytosolic release of SA-FITC complexes, so we did not further pursue this approach.
Figure 1. Comparison of DNA-transfection (GFP encoding plasmid) and Protein-transfection (SA-FITC) by biotinylated PEI. (A) cells were analyzed by flow cytometry. (B) flow cytometric analysis of life death staining by propidium iodide.
Cre-Fc fusion proteins
In the previous experiments, numerous artifacts that impair the classical analysis methods based on microscopy and flow cytometry were observed, rendering any ranking of transfection efficiencies impossible. Cronican et al. used fusions of the recombinase Cre to supercharged proteins to follow their translocation into the cytoplasm.Citation27 To obtain a label-free method that does not compromise cell viability and that allows the discrimination between cytosolic and endosomal access, we generated N-terminal and C-terminal fusions of Cre to a human IgG1-Fc domain () as a probe. Once this antibody derivate is successfully delivered to the cytosol of reporter cell line SC1 REW22, the cells are expected to expresses GFP after the LoxP-dependent excision of a stop-cassette by the recombinase activity of the fusion protein (). In contrast to employing cellular responses as an indicator for protein delivery, which may be biased by cellular processes, a Cre-based reporter system ensures high specificity as Cre originates from a different organism (P1 Bacteriophage) and no LoxP recombination is observed in the absence of the Cre recombinase.
Figure 2. Cre-hIgG1-Fc fusion recombinase activity in vitro and in vivo. (A) schematic representation of Cre-Fc and Fc-Cre fusions and the reporter cell system based on the cell line SC1 REW22. (B) the plasmid LoxP2+ was incubated with Cre-Fc or Fc-Cre fusion proteins and analyzed for recombination by agarose gel electrophoresis. Cre recombinase served as a positive control (p.c.), the linearized plasmid LoxP2+ alone as a negative control (n.c.). (C) electroporation of Fc-Cre fusion protein and controls into reporter cell line SC1 REW22. 2x106 cells per sample were used for electroporation. Cells were analyzed by flow cytometry. The electroporation efficiency indicated by YFP+ cells reached up to more than 90%. The efficiency of electroporation and recombination together, indicated by GFP+ cells in samples electroporated with NLS-Cre DNA, reached up to more than 60%. Electroporation with Fc-Cre resulted in a maximum of almost 30% at the highest concentration of Fc-Cre protein. (D) effect of electroporation of DNA and proteins or empty electroporation on cell viability. Cells were analyzed by flow cytometry after life/death staining with propidium iodide. Electroporation of a plasmid encoding NLS-Cre was more toxic than electroporation of hIgG1-Fc-Cre fusion proteins. The average percentage of living cells from all timepoints is shown.
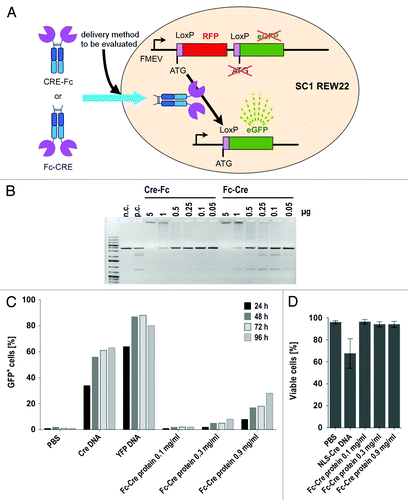
Both the Cre-fusions with N-terminal IgG1-Fc domain (Fc-Cre) and C-terminal IgG1-Fc domain (Cre-Fc) were efficiently expressed as secreted proteins in HEK293–6E cells and purified via protein A affinity chromatography. The recombinase activity of the fusion proteins was analyzed using a linearized plasmid containing LoxP sites (Loxp2+) in vitro. As expected, in the absence of recombinase activity, only a single fragment is seen on an agarose gel, while several additional fragments were seen in the presence of both Fc-Cre and Cre.-Fc fusion proteins (). The N-terminal fusion Fc-Cre was chosen for future experiments as the C-terminus of Cre was reported to be more critical for enzyme activity.Citation28
Fc-Cre reporter cell assay
To test whether Fc-Cre can exert its recombinase activity in the reporter cells by reaching the nucleus of these cells, Fc-Cre was delivered to SC1-REW22 cells via electroporation. For assessing delivery efficiency and maximum reporter activation at the given delivery efficiency, a plasmid encoding YFP and a plasmid encoding NLS-Cre were electroporated as controls. While NLS-Cre DNA served as positive control for reporter activation, the YFP DNA served as an indicator for electroporation efficiency. Reporter activation was analyzed by flow cytometry every 24 h for four days following electroporation. GFP+ cells were observed at the highest concentration of Fc-Cre protein (0.9 mg/mL) with a maximum of about 30% GFP-positive cells (). GFP+ cells increased with time after electroporation. As the various treatments were observed to affect cells in a different way, viability of cells was assessed by PI staining followed by flow cytometric analysis (). More than 90% of cells were viable in all samples except for the cells electroporated with the NLS-Cre encoding plasmid (50–80% viability), which might be caused by overexpression of NLS-Cre.
Comparison of protein delivery methods by the Fc-Cre reporter cell assay
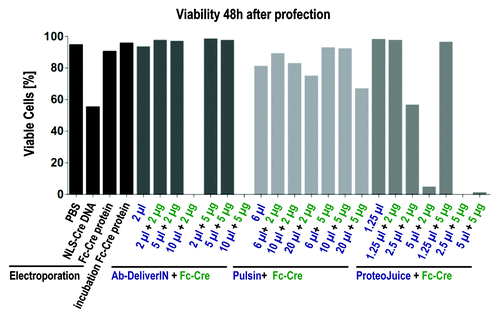
We tried to optimize the profection conditions individually using fluorescently labeled antibodies and Cre-Fc, but without ever observing specific binding patterns or more than 10% of Cre-Fc transfected. To compare all different methods “head-to-head,” Fc-Cre was delivered to SC1 REW22 cells by electroporation or by profection with four different profection reagents (). Profection of IgG-FITC and PE (as delivered for this purpose by the manufacturers) served as controls. Profection was performed with the standard ratios of protein and profection reagents as suggested by the manufacturers, as well as with 5 additional ratios each. Samples were analyzed by flow cytometry at four different time points from 24–96 h after electroporation or profection. As expected from the previous results, the conjugate IgG-FITC appeared to attach unspecifically to cells, as part of the cells (~5%) that had been incubated with IgG-FITC alone were found to be fluorescent. All samples profected with IgG-FITC or PE by different profection reagents were fluorescent, reaching up to more than 90% fluorescent cells. Although the percentage of fluorescent cells varied among samples treated with different profection reagents, each reagent in combination with IgG-FITC or PE resulted in a higher number of fluorescent cells than controls. If cells were profected with Fc-Cre, the percentage of GFP+ cells was above those of controls, but the fraction of positive cells was below 10% for all profection reagents tested. In contrast, electroporation of Fc-Cre, resulted in up to more than 70% of fluorescent cells. Viability of electroporated and profected cells was similar, although viability decreased at the highest concentrations of profection reagents (). For some profection reagents (Ab-DeliverIN, ProteoJuice), viability at the highest profection reagent concentration was so low that the remaining cells were not sufficient for measurement. Although the size of the Fc-Cre fusion protein and the lack of a NLS might impede translocation to the nucleus, the successful reporter activation in > 70% of all cells by electroporation suggests that this effect was minor. This experiment demonstrates large differences in the number of cells of a population that take up the antibody analog, depending of the transfection method. It does not, however, allow a quantification of the cytosolic delivery.
Figure 3. Protein delivery into SC1 REW22 reporter cells by electroporation or profection with ProteoJuice, Bioporter, Ab-DeliverIN or Pulsin. 4.4x105 cells per sample were used for electroporation and cells were analyzed by flow cytometry. (A) electrotransfer of NLS-Cre DNA, Fc-Cre protein and controls analyzed after 72h. Electrotransfer of NLS-Cre DNA resulted in two clearly separated populations while electrotransfer of Fc-Cre resulted in GFP+ cells with lower fluorescence intensity. Controls electropulsed in PBS (“empty”) or cells incubated with Fc-Cre without electropulse were not fluorescent. The electroporation efficiency indicated by YFP-DNA electroporated cells was above 90%. (B) electrotransfer: percentage of GFP+ cells over all timepoints. (C) negative controls for profection analysis. (D) profection of IgG-FITC and PE resulted in IgG-FITC or PE positive cells for all reagents, reaching maximal values of up to more than 90%. Incubation with only IgG-FITC resulted in a low percentage of IgG-FITC positive cells while cells incubated with PE alone were PE-negative. (E) profection of Fc-Cre. Fc-Cre was delivered as indicated by GFP+ cells, but for all reagents only in less than 10% of cells. If viability was so low it only allowed analysis of markedly less cells than for other samples this is indicated by an asterisk.
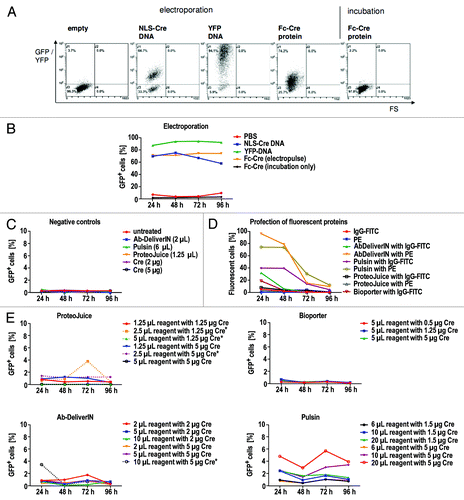
Figure 4. Viability of cells 48 h after treatment by profection or electroporation. Cells were analyzed by flow cytometry after life death staining with propidium iodide. At high concentrations of profection reagents viability decreased (Pulsin) or cells died so numerously leaving not enough cells for measurement (Ab-DeliverIN, ProteoJuice). For profection, cells had been incubated for 48 h with profection reagent and protein, the supernatant was substituted after 48 h by cell culture medium. Electroporation with proteins was performed using 0.9 mg/ml Fc-Cre.
scFv-Fc antibodies bind to their intracellular antigen after electrotransfer
To analyze whether a substantial amount of antibodies can be delivered into the cytosol, we analyzed electroporation for its suitability for in vivo imaging or functional interference in living cells. As the half-life of proteins within cells may depend on their tendency to aggregate, the scFv-Fc fusions to be used were first analyzed by size exclusion chromatography. Both the anti-myosin (SF9) and the anti-tubulin (F2C) scFv-Fc gave rise to a single peak at a volume corresponding to the size of a single scFv-Fc fusion protein (), indicating no aggregation tendency. In order to assess whether scFv-Fc fusions are also stable and functional in the reducing environment of the cytosol, a series of dilutions of the anti-Myosin (SF9) scFv-Fc antibody were electroporated into living HeLa cells. Myosin binding was analyzed later using a secondary antibody after fixation. To identify possible false-positive results due to surface attached antibodies that might be washed into cells after fixation and permeabilization, control cells were incubated with the same concentrations of the anti-myosin (SF9) scFv-Fc, but not electroporated. Clearly visible fiber-like structures were observed in cells electroporated with the two highest concentrations of the anti-myosin (SF9) scFv-Fc protein, while negative controls did not show the pattern expected for a myosin-staining at any of the protein concentrations used (). The staining pattern observed was specific and according to what has already been described for tubulin and myosin stainings.Citation29 Electroporation seems to have delivered enough antibody molecules to coat the cytoplasmic fiber networks in the living cell. None of the profection reagents was able to deliver this result (data not shown).
Figure 5. Size exclusion chromatography of anti-Myosin (SF9) and anti-Tubulin (F2C) scFv-Fc antibodies.100 µg protein in a volume of 200 µl were loaded to the column for each sample. The calculated molecular mass of the analyzed scFv-Fc fusions is 102 kDa and the mobility determined by SEC corresponds to a size of 122 kDa (anti-Myosin, 14.08 mL retention volume) and 118 kDa (anti-Tubulin, 14.16 mL retention volume).
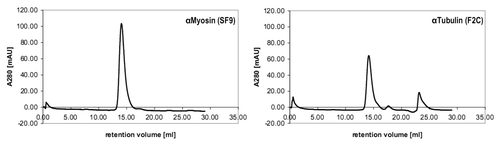
Figure 6. Electroporation of HeLa cells with different concentrations of anti-Myosin (SF9) scFv-Fc antibody. Cells were fixed, permeabilized and stained by a FITC-conjugated secondary antibody to detect the anti-Myosin scFv-Fc antibody. If not otherwise indicated, the exposure time for all images shown in this figure was 2s.

Stability of scFv-Fc antibodies in the cytoplasm
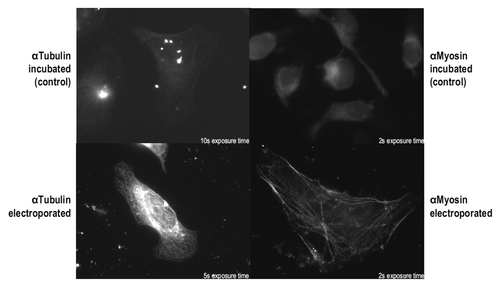
As proven by this experiment, the anti-myosin (SF9) scFv-Fc fusion is functional in the cytosolic environment after electroporation. However, this particular antibody is described to be functional if expressed in the cytosol,Citation30 so this result may be due to intrinsic stability in the absence of disulfide bonds.Citation31 We therefore used another antibody, anti-tubulin (F2C), which was described to be non-functional if expressed in the cytosol.Citation30 When the anti-tubulin (F2C) scFv-Fc was electroporated into HeLa cells followed by staining and microscopic analysis, a characteristic tubulin staining pattern was found. On the contrary, no tubulin-specific staining pattern was found in control cells that had been incubated with the antibody without electroporation ().
Figure 7. In vivo delivery of anti-Tubulin and anti-Myosin antibodies. HeLa cells were electroporated in the presence of the anti-Tubulin scFv-Fc antibody F2C (0.6 mg/mL) or the anti-Myosin scFv-Fc antibody SF9 (0.3 mg/mL). As a control, HeLa cells had been incubated with anti-Tubulin (1.1 mg/mL) or anti-Myosin (0.3 mg/mL). Cells were analyzed 24 h after electroporation or incubation, respectively.
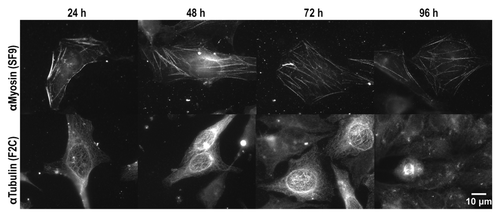
To determine the stability and functionality of the anti-myosin (SF9) scFv-Fc and the anti-tubulin (F2C) scFv-Fc antibody in the cytosol over prolonged time, measurements were extended to four days. HeLa cells were electroporated with anti-myosin (SF9) or anti-tubulin (F2C) at an optimal concentration based on the results from electroporation of the dilution series. Analysis by microscopy revealed fiber-like structures in cell samples from all time points ranging from 24 h to 96 h, although fluorescence signals seemed generally weaker in samples taken 96 h after electroporation (). The characteristic staining pattern expected for interphase-microtubule was observed for at least 72 h after electroporation of the anti-tubulin antibody. At time point 96 h, there were still few single cells with a staining pattern reminding of microtubule-bundles in metaphase or during cytokinesis, demonstrating a functionality of the scFv-Fc antibodies of at least four days in the living cell.
Figure 8. Intracellular stability of anti-Myosin (SF9) and anti-Tubulin (F2C) in HeLa cells. HeLa cells were electroporated in the presence of anti-Tubulin (0.6 mg/mL) or anti-Myosin (0.3 mg/mL) scFv-Fc antibodies and after electroporation, permeabilized and stained with a detection antibody. The exposure time was 10s for all images shown.
Discussion
A large number of methods to bring antibodies into cells are described, most prominently cell-penetrating peptides and protein transfection (profection) reagents. In our study, all tested reagents yielded good viabilities (80–90%) for most samples and in different cell lines. The lower viability of HeLa cells after treatment with the reagent Pulsin compared with that of SC1 REW22 reporter cells might be due to a difference in sensitivity of different cell lines to the same reagent. While treatment with the highest concentration of Ab-DeliverIN or ProteoJuice induced cell death to such an extent that remaining cells were mostly insufficient for measurement, the highest concentration of Pulsin only led to a slightly decreased viability (around 70% living cells), but to the highest rate of reporter activation for all Fc-Cre profection samples compared. The increased permeability for PI may have implications for the mode of action of Pulsin, suggesting cell membrane perforation as a potential delivery mechanism.
In the reporter system, viability might also be impaired by the delivered protein itself. High levels of the recombinase Cre were reported to lead to cytotoxicity.Citation32,Citation33 However, the increased permeability of cells for PI may mainly originate from the effect of the profection reagent as the much higher amounts of Fc-Cre delivered by electroporation did not affect viability. Delivery of toxic amounts of Cre to the nucleus was only observed when transfecting the NLS-Cre encoding plasmid, which allows amplification of NLS-Cre by protein expression and active transport to the nucleus due to the NLS. The higher nuclear amount of Cre after transfection of the NLS-Cre encoding plasmid is also supported by the observed larger difference in intensity between negative and fluorescent cells.
Apart from toxic effects of the delivery methods, avoiding endosomal entrapment is of paramount importance for the use of antibodies in vivo. To distinguish delivery to the cytosol from internalization only to endosomes or the attachment to the cell surface, our test system uses an antibody analog constructed from an antibody Fc region and an enzyme with a calculated pI of 9.0 and a molecular mass of 132.8 kDa. Cre has a positive charge at neutral pH (theoretical pI 9.6) and there are controversial reports in literature on whether Cre itself can enter cells or not,Citation34-Citation36 While there have been reports claiming incubation with Cre alone leads to reporter activation in 1.5–17% of cells,Citation34,Citation36 other authors reported Cre did not enter cells in their experiments.Citation35 A possible explanation for this may be found in the different cell lines used in these studies or different amounts of Cre. In SC1 REW22 reporter cells with Fc-Cre without profection reagents, no reporter activation was detected, providing a good signal-to-noise ratio of the data.
A potential disturbance of profection agent/Fc-Cre complexes due to their similar charge may be another explanation for the observed low profection efficiency. However, the limited but successful profection with the cationic reagent Pulsin indicates that interactions between positively charged proteins and cationic reagents can take place or are not necessary for successful delivery. Significantly, in previous studies, the interaction between reagent and protein was described to be more dependent on protein surface area than charge.Citation37 Further, the delivered molecule contains not only the positively charged Cre, but also the four immunoglobulin domains of the human IgG Fc.
Cytosolic release seems to be the limiting factor for the protein delivery efficiency of PEI although PEI is described to promote endosomal release.Citation22 In our experiments, the treatment of HeLa cells with biotinylated PEI in complex with either SA-FITC or GFP-encoding DNA resulted in up to 100% of SA-FITC+ cells but only 42% of GFP+ cells, indicating that only a minor fraction of SA-FITC molecules may have reached the cytosol. A similar observation was made by Bieber et al.Citation38 when studying DNA-transfection by PEI. While all cells took up PEI-DNA complexes, indicating uptake is not the rate limiting step, only a small fraction of DNA reached its destination.Citation38 As transcription was found to be possible even if DNA was still attached to PEI, the bottleneck of delivery was attributed to the release process of PEI-DNA complexes from the lysosomal compartment.Citation38 The results obtained when using the Fc-Cre / SC1 REW22 reporter system support this conclusion. Profection of fluorescent proteins such as IgG-FITC or PE led to a high percentage of fluorescent cells for all tested profection reagents. However, the activation of the reporter that requires cytosolic release of at least some Fc-Cre was not observed for any of the reagents in higher than single digit percentages of cells. Given the assumption that not many CRE molecules are necessary to obtain a positive readout, this demonstrated a very limited efficiency of these delivery methods.
Further support for the hypothesis of endosomal entrapment as the bottleneck in protein delivery comes from recent studies on siRNA delivery, which also point to inefficient cytosolic release as the major limitation for delivery by lipid based transfection reagents.Citation39 Gilleron and colleagues found a delivery efficiency of only 1–2% when using lipid-based transfection reagents, which is similar to that observed here for most Fc-Cre profections.Citation39 Another study on siRNA delivery by lipid-based transfection reagents identified recycling and exocytosis as a further limitation to delivery efficiency.Citation40
A comparison of the results from the existing literature on profection is very difficult as the readouts for successful delivery vary broadly and systematic comparisons between profection methods are rare. Often, β-galactosidase was detected or fluorescent proteins were analyzed via microscopy or flow cytometry as the readout for delivery efficiency.Citation41-Citation48 As suggested by the numerous possible artifacts identified in our experiments or by others, these types of readout must be considered to be insufficient for indicating a true delivery to the cytoplasm. Another commonly used readout for protein delivery is cell death as an indicator for the delivery of toxic proteins.Citation41,Citation48-Citation50 However, the specificity of this readout has to be critically inspected considering the large variety of factors that may influence cell viability in addition to the toxicity that is associated with most transfection reagents.
A first attempt toward a systematic comparison of delivery methods included the evaluation of PTDs and Bioporter.Citation21 In that study, the readout for successful delivery of the DNA-binding domain of a transcription activator was luciferase activity. It should be noted that addition of the DNA-binding domain of the transcription activator alone already led to some activation of luciferase expression in a concentration-dependent manner. Nevertheless, Bioporter was found to activate luciferase expression above the expression levels of controls. While Bioporter-mediated delivery led to activation of the reporter in the report by Ye and colleagues,Citation21 no appreciable reporter activation was observed in the experiments presented here. A potential reason for this may be a difference of the reporter system’s sensitivities. While the signal of a single GFP molecule remains constant, the detection of luciferase benefits from signal amplification due to the enzyme activity. Also, the number of molecules that is required to either activate transcription or to induce recombination may differ. Additionally, the different origin of reporter cells may affect profection efficiency. Most importantly, only three different conditions were compared for Bioporter-mediated delivery. Although the evaluation system based on luciferase may be more sensitive, profection evaluation may not benefit very much from this sensitivity, as reagents that deliver only tiny traces of proteins are not suited for the potential applications with antibodies. In contrast to what has been found in this study, Todorova reported higher delivery efficiencies for profection by Bioporter (20%) than for electroporation (10%).Citation45 However, delivery efficiencies were only determined by flow cytometric analysis of cells for the presence of GFP, which is a method that cannot distinguish attached protein from cytosolic or endosomally-entrapped protein. The delivery efficiency of profection may therefore have been overestimated in this report, particularly as the profection reagent itself may lead to unspecific attachment of the protein to cells, while GFP alone does not bind to cells before electroporation.
As a conclusion, while profection exhibited generally low toxicity, its successful delivery requires a certain degree of membrane disruption, suggesting that there is no “gentle way” into the cell.
The evaluation method based on Fc-Cre avoids many pitfalls of previously described methods to assess antibody delivery efficiency. As living cells are assayed, there is no risk for fixation artifacts as seen in previous studies.Citation25 Cre comes from a different organism and is not involved in the cellular processes of the target cell, thus ensuring high specificity of the readout. Unlike cell death, proliferation or other cellular responses that may be biased by a plethora of factors, the proposed evaluation method’s results are not expected to be significantly influenced by other cellular processes. A crucial feature is the capability to distinguish cytosolic release from endocytosed or merely attached antibodies, while at the same time indicating viability of positive cells, since the readout acquires active translation.
We were not able to obtain significant cytosolic delivery using protein transduction domains. The efficiency of profection also was low and none of these methods ever provided detectable intracellular staining by the antibodies in the living cell. Furthermore, if profection relies on the association of the profection reagent with the protein, clumps of profection reagent with the protein may disturb imaging, as the results from Dalkara et al. suggest.Citation42 The formation of “clumps” may be irrelevant for some applications, but is critical for imaging.
The results presented here are an exciting first step toward live immunocytochemistry as imaging of HeLa cells electroporated with an anti-tubulin or an anti-myosin antibody exhibited characteristic staining patterns. In contrast to the early microinjection experiments,Citation5 the use of direct fluorescence labeling of the delivered antibodies is not recommended due to the observed artifacts. Here, alternative labeling strategies may be explored, such as direct fusions to GFP derivatives that allow a more directed and controlled way of labeling. Electrotransfer of scFv-Fc antibodies into living cells could soon well be a suitable delivery method for the purpose of live imaging, which is supported by a contemporary study using IgG antibodies by Freund and colleagues.Citation51
A parameter relevant to the general applicability of live cell immunocytochemistry is the stability of antibodies in the cytosol. Three main factors have to be considered to play a role here: proteolysis/protein turnover, VH-VL association stability in the scFv part and the maintenance of disulfide bonds. Proteolytic degradation of the antibodies obviously was slow, allowing some functional antibodies to survive for up to four days in the cytoplasm. The anti-myosin and anti-tubulin scFv-Fc antibodies used in this study were shown to have no detectable tendency for diabody or aggregate formation in vitro, so that no or only a small effect on their in vivo activity is expected from this form of denaturation. However, while opening up of the VH-VL association may not have been a factor in our experiments, individual analysis of other scFv or scFv-Fc antibody clones may be necessary to allow a general conclusion on whether a tendency to multimerize affects intracellular half-life of scFv-Fc antibodies. In respect of disulfide bonds, the initial findings with microinjected antibodiesCitation1-Citation5 suggested a negligible influence of the reducing milieu in the cytosol on the activity of mature antibodies, implying that the disulfide bonds remain intact. This has been confirmed in this study also for the scFv-Fc (yumab) format, as electroporation experiments demonstrated the presence of functional scFv-Fc antibodies for at least 3–4 d after delivery.
Conclusions and outlook
The identification of an efficient delivery method for antibodies into the cytosol raises hopes that the vast resource of available research antibodies may in the future be systematically assessed for their interference with cellular processes, for instance blocking of antigen function, apart from imaging. Because the anti-myosin clone SF9 binds to the coiled coil region,Citation30 its binding to myosin IIa is not expected to influence the function of the motor domain, so this effect could not be tested with the reagents used in this study. The epitope of the anti-tubulin clone used here remains to be analyzed,Citation30 but it obviously did not lead to depolymerization of microtubules. This effect, however, was observed after profection of another anti-tubulin antibody by Dalkara et al.,Citation37 where microtubule visualization was only possible after pre-treatment of cells with the microtubule stabilizing agent taxol before profection.
Besides the commercially available antibodies, rapidly increasing numbers of research antibodies targeting cytoplasmic proteins are currently delivered by international research consortia in the US and EU, e.g., AFFINOMICS (www.affinomics.org). Moreover, the use of in vitro antibody selection technologies like phage display allows for the construction of antibodiesCitation52,Citation53 with predefined fine specificity. For example, recombinant antibodies selected in vitro in the presence of a non-phosphorylated competitor allowed to follow the phosphorylation state at one single position on SMAD3 after stimulation of the cell with EGF.Citation54 It can be envisioned that the combination of the power of large scale in vitro research antibody generation with a generally applicable method for their immediate assessment for intracellular effects will help to move functional genetics to the next – the posttranslational - level.
Materials and Methods
Vector construction
For preparative digests, typically 3 µg of DNA were cut with 12–60 units of a restriction enzyme for 2 h at the temperatures and at buffer conditions recommended by the manufacturer. Vector backbones were dephosphorylated by either calf intestinal alkaline phosphatase (NEB) or Antarctic Phosphatase (NEB) according to the supplier’s instructions. For details of the vector constructions see Table S1. Ligation was performed over night at 16 °C with T4 Ligase (Promega) in a total reaction volume of 10 µL and the ligase was deactivated at 70 °C for 10 min before transformation of reaction products. Correct cloning was confirmed by sequencing.
Expression analysis of scFvs, scFv-TAT fusions and peptides in E. coli
Expression of scFvs or peptides from pOPE101 based vectors was performed in the E. coli strain XL1-Blue MRF’. Precultures were diluted to an OD600 of 0.05 in a final culture volume of 30 mL. Incubation of cultures took place at 37 °C and 250 rpm (Certomat BS-1, B. Braun Biotech International) and expression was induced at an OD600 of 0.4–0.6 by adding IPTG to a final concentration of 50 μmol/L. The temperature was lowered to 30 °C after induction and samples were taken every hour for four hours and overnight to monitor bacterial growth and protein expression.
Culture of mammalian cells
HeLa were maintained in Dulbecco’s modified eagle medium High Glucose (DMEM) from PAA supplemented with 1% Penicillin/Streptomycin and 8% fetal calf serum (all from PAA laboratories). SC1 REW22 reporter cells were maintained in DMEM High Glucose supplemented with 1% sodium pyruvate, 1% Penicillin/Streptomycin and 8% fetal calf serum (all from PAA laboratories). Cells were maintained in an incubator (HeraCell, Heraeus) at 37 °C and 5% CO2. Adherent cells were passaged every 2–3 d by adding 1 mL of Trypsin-EDTA (PAA) per 10 cm culture dish after removing traces of fetal calf serum (FCS) by washing with 5 mL PBS. Cells were incubated with Trypsin-EDTA for 2 min at 37 °C followed by adding 1 mL of FCS-containing fresh medium to stop the treatment and transferring cells to a new culture dish with fresh medium. HEK293–6E cells were maintained in FreeStyle F17 expression medium (Invitrogen) supplemented with 4 mM L-Glutamine, 0.1% Pluronic F-68 and 25 µg/mL G418 under constant shaking at 110 rpm, 37 °C and 5% CO2 in an Infors HT Minitron Incubator.
Production by secretion from mammalian cells
For secretion of recombinant proteins into the culture supernatant of mammalian cells, HEK293–6E cells were transfected with pCSE based vectors.Citation55 Typically 100 mL of cell suspension with a cell density of 1.5 x106 cells/mL were transfected with 100 µg of DNA complexed with 250 µg PEI. Cells were fed 48 h post transfection with additional medium corresponding to 50% of the total culture volume and tryptone N1 (Organotechnie) to a final concentration of 0.5%. Five days post transfection cells were sedimented at 1,000 x g for 10 min and salt and buffer conditions of the supernatant were adjusted to the binding buffer to be used for purification by adding an appropriate amount of a 10x concentrate of the binding buffer.
Purification of His-tagged proteins by IMAC under denaturing conditions
Proteins with a hexa-Histidine tag were purified by immobilized metal affinity chromatography (IMAC) either by employing Äkta prime (GE Healthcare) or Profinia (BioRad). For purification with Äkta prime the Ni2+ charged column (HisTrapCrude FF) was equilibrated with denaturing binding buffer (8 M urea, 0.5 M NaCl, 10 mM imidazol, 20 mM sodium phosphate buffer pH 7.4) before loading the filtered (0.45 µm filter) protein preparation. After loading the protein preparation, the column was washed with denaturing binding buffer until the absorption at 280 nm was below 10 mAU and the column was additionally washed with an imidazol gradient using denaturing washing buffer B (8 M urea, 0.5 M NaCl, 50 mM imidazol, 20 mM sodium phosphate buffer pH 7.4), which was added continuously until typically 60% of buffer B was reached in the buffer mixture. Proteins were subsequently eluted with denaturing elution buffer (8 M Urea, 0.1 M EDTA).
Purification of human IgG1 Fc-fused proteins by affinity chromatography
Purification by affinity chromatography was performed using an automated chromatography system (Profinia, BioRad) with a protein A column (Bio-Scale Mini UNOsphere SUPra Cartridge, BioRad) and a 10 mL Bioscale Mini BioGel P-6 Desalting Cartridge (BioRad). The column was equilibrated with Protein A binding/desalting buffer (8 g NaCl, 0.2 g KCl, 0.76 g NaH2PO4 and 1.1 g KH2PO4 per liter, pH 7.4), and also washed with this buffer after loading filtered (0.45 µm pore size) protein extracts. Elution was performed with 500 mM sodium citrate buffer (pH 3). Columns were cleaned with cleaning solution 1 (0.5 M NaCl, 50 mM Tris, pH 8) and cleaning solution 2 (0.5 M NaCl, 100 mM sodium acetate, pH 4.5) and stored in 4% benzylalcohol. Eluted and desalted proteins were in PBS and stored as frozen aliquots.
SDS-PAGE
Samples were boiled for 5–10 min after addition of Laemmli buffer (either with or without β-mercaptoethanol) at 95 °C on an Eppendorf Thermomixer. Gel electrophoresis took place at 200 V for 40–50 min. Protein mobility was compared with the Precision Plus Protein Unstained Standard (BioRad) or Precision Plus Protein Dual Color Standard (BioRad).
Quantification of proteins
The concentration of antibodies was determined by densitometric analysis of bands on Coomassie stained SDS-polyacrylamide gels compared with a reference sample loaded onto the same gel. The concentration standard was prepared from the scFv TM44-C7.3. Densitometric analysis was performed using the image analysis tool ImageJ (National Institutes of Health, USA). This method was particularly applied for concentration determination of protein preparations with low purity as this method allows determination of the concentration of a single protein in a protein mixture by selection of single bands for analysis. The concentration of proteins purified from productions in mammalian cell culture was determined by measuring the absorption at 280 nm (Nanodrop ND-1000 Libra S11, PeQLab).
Conjugation of macromolecules with fluorescent dyes or biotin
Conjugation of antibodies to DyLight 488 was performed using the DyLight Antibody Labeling Kit from Thermo Scientific according to the manufacturer’s instructions. Briefly, purified scFv-Fc antibodies were concentrated using Vivaspin centrifugal concentrators and the final concentration was determined by measuring the absorption at 280 nm (NanoDrop). This solution was diluted in PBS to adjust to the recommended 2 mg/mL in a total volume of 0.5 mL. 40 µL of borate buffer (0.67 M) supplied with the kit were added to the protein solutions in PBS, and the entire solution was then transferred to a vial containing the DyLight 488 reagent (vials are supplied with an appropriate amount of fluorophore for labeling of 1mg of antibody). The reaction mix was incubated for 1 h at room temperature in the dark and free dye was removed using a purification resin supplied with the kit according to the manufacturer’s instructions. The degree of labeling was determined from the ratio of dye concentration to protein concentration calculated after measuring the absorptions at 280 nm and 493 nm using the molar extinction coefficient of 70,000 M−1cm−1 for the fluorophore. For calculation of the protein concentration, maximum absorption at 493 nm multiplied by a correction factor (correction factor 0.147 as indicated in the manual of the kit) was substracted from the absorption at 280 nm in order to correct for some minor absorption of the fluorophore at 280 nm. The degree of labeling for the scFv-Fc antibody clone SF9 conjugated to DyLight 488 was determined to be 2.1 dyes per protein.
Conjugation to the pH-sensitive dye CypHer5E was performed using CypHer5E mono NHS ester (GE Healthcare) according to the Cypher user manual (literature code 25–8010–12UM at http://www.gehealthcare.com/cypher5e). Briefly, proteins were concentrated by Vivaspin centrifugal concentrators and a 0.5 M sodium carbonate buffer pH 8.3 was added to the scFv-Fc antibody solution (1.19 mg/mL protein in PBS) in a 9:1 ratio of protein solution/0.5 M sodium carbonate buffer. CypHer5E was dissolved in DMSO and the concentration of the dissolved dye was calculated after measuring the absorbance of a 0.5 M sodium carbonate (pH 8.3) dilution of the dye at 500 nm. A 20-fold molar excess of dye was then added to the antibody solution and the mixture was left at room temperature for 1 h in the dark. Unconjugated dye was removed by dialysis at 4 °C overnight with PBS. The degree of labeling was determined as described above with the extinction coefficient of the CypHer5E dye being 40,000 M−1cm−1 and a correction factor of 0.16 for dye absorption at 280 nm. For determining the concentration of the dye, antibody-CypHer5E solutions were diluted in 0.5 M sodium carbonate buffer pH 8.3 and absorption of CypHer5e was measured at 500 nm. The degree of labeling for the scFv-Fc antibody clone SF9 conjugated to CypHer5E was determined to be 11.8 fluorophores per protein, which is within the range of dye to protein ratios indicated in the manual (dye to protein ratio 7–12). Labeled proteins were stored as frozen aliquots in brown tubes for protection from light.
Biotinylation of polyethylenimine (PEI) was performed with NHS-LC-Biotin from Pierce with 25 kDa linear PEI from Polysciences. The required amount of the NHS-LC-Biotin reagent was weighed, dissolved in DMSO and then added to a 1 mg/mL PEI solution in water as a 50-fold molar excess and the mixture was left to react for 3 h at room temperature. Unconjugated biotin was removed by dialysis against PBS at 4 °C overnight. Biotinylated PEI was stored as frozen aliquots.
Size exclusion chromatography (SEC)
Proteins were analyzed for aggregation and multimerization by size exclusion chromatography using Äkta purifier (GE healthcare) and a Superdex 200 10/300 GL gel filtration column. The relation of retention time to protein mass was determined by calibration of the column with proteins of defined mass. For sample analysis, the column was loaded with 100 µg protein in a volume of 200 µL each for the analysis of the scFv-Fc antibody clones SF9 and F2C.
Analysis of Cre-Fc and Fc-Cre for recombinase activity
The recombinant proteins Cre-Fc and Fc-Cre were analyzed for recombinase activity using the plasmid pLox2+ from New England Biolabs (NEB). Different amounts of Cre-Fc or Fc-Cre (5 µg, 1 µg, 0.5 µg, 0.25 µg, 0.1 µg, 0.05 µg) were allowed to react with 0.25 µg pLox2+ in 1x Cre reaction buffer (NEB) in a total volume of 50 µL at 37 °C for 30 min. The reaction mix was deactivated for 10 min at 70 °C before loading samples to a 1% agarose gel for analysis. As a positive control the reaction was also performed using 1 unit of Cre from NEB. The untreated plasmid pLox2+ served as a negative control.
Incubation of HeLa cells with antibodies
The antibody clone SF9 binds to the protein Myosin IIa, which is intracellular and therefore binding of SF9 to the cell surface is not expected. HeLa cells were incubated with labeled or unlabeled scFv-Fc antibody clone SF9 to determine whether it attaches to the cell surface unspecifically. HeLa cells, seeded on glass slides were incubated at 37 °C with 1.2 µg scFv-Fv antibody clone SF9 conjugated to DyLight 488 (SF9-DyLight 488) in a total volume of 100 µL cell culture supernatant. Cells were incubated for 4 h with the labeled protein and then prepared for fluorescence microscopy (PFA fixation).
In order to analyze whether SF9::DyLight 488 attaches unspecifically to the cell surface by flow cytometry, HeLa cells were incubated with unlabeled SF9 (scFv-Fc antibody) or SF9::DyLight 488 at 4 °C to prevent endocytosis. Incubation with 1, 2 or 3 µg of labeled or unlabeled protein in a total volume of 100 µL each took place for 1 h on ice. Cells were then washed twice with 3 mL buffer (0.5% BSA in PBS) and subsequently an anti-human IgG (Fc specific) APC conjugated antibody (Dianova) was added for detection and incubated for 30 min on ice.
Profection
Profection of cells was performed using the reagents Ab-DeliverIN (OZ Biosciences), Pulsin (Polyplus), ProteoJuice (Novagen) or BioPORTER (Genlantis) according to the manufacturer’s instructions. If different profection reagents were compared, small modifications were introduced to the protocols for better comparability. Because some profection reagents required serum-free conditions but others could be used with or without serum, all incubations were performed under serum-free conditions if the aim of the experiment was to compare profection reagents. In this case, FCS was added to the cells to a final concentration of 8% after 2–3 h of serum-free incubation with profection mixes. Incubation of proteins with the profection reagents was performed at room temperature for 20 min. For the comparison of profection reagents, samples were analyzed every 24 h and the cell culture supernatant containing the profection mix was replaced by fresh medium 48 h after profection had been started.
Profection with biotinylated PEI was performed by mixing FITC-conjugated streptavidin with biotinylated PEI in serum-free medium, incubating for 30 min at room temperature and subsequently adding the mixture to the serum-containing cell culture medium of HeLa cells. Protein and biotinylated PEI was mixed in polystyrene plates. As a control, 1 µg of DNA (plasmid CZ73) was transfected with biotinylated PEI. Cells were analyzed 24 h after profection with biotinylated PEI.
For flow cytometric analysis cells were detached with Trypsin/EDTA, washed once with PBS or FC-buffer and resuspended in 500 µL buffer for analysis. Viability of cells was determined by adding 10 µL of a 1 mg/mL PI solution to the sample directly before flow cytometric analysis.
Electrotransfer of DNA and Proteins into mammalian cells
Electroporation was performed using a Gene Pulser Xcell (BioRad). If not otherwise stated, 4.4 x 105 cells were mixed with 400 µL of a solution containing the protein or DNA to be delivered. Electroporation was performed in a 0.4 cm cuvette with the settings 650 µF and 300 V using an exponential pulse type. Different protein concentrations were used for electroporation as indicated for the respective experiments. For electroporation of DNA, 40 µg DNA was always used. Instantly after application of the electric pulse, 400 µL of prewarmed medium were added. For analysis by microscopy, cells were re-seeded on glass slides after they had been left to settle down for 3 h after electroporation in order to prevent any potential disturbance during imaging due to protein aggregates or by proteins that have attached to the bottom of the cell culture dish.
Immunocytochemistry for Fluorescence Microscopy
Cells were fixed with either 4% PFA if myosin was to be detected, or with methanol if microtubule were to be detected. For fixation with PFA, samples were incubated with 4% PFA for 15 min at room temperature, followed by washing once with PBS and permeabilization with 0.1% Triton X for 5 min. For methanol fixation, the cells were washed twice with PBS, incubated with methanol (pre-cooled at -20 °C) for 4 min at -20 °C. The fixed cells were washed again with PBS. Detection of scFv-Fc antibodies was performed by incubating with a goat anti-human (Fc gamma specific) FITC conjugated antibody (Dianova 109–095–098) for 1 h at room temperature protected from light. Samples were then washed twice with PBS and nuclei were stained for 2 min with DAPI (0.1 µg/mL DAPI, a 100 µg/mL DAPI stock solution dissolved in 70% ethanol was used as a 1:1000 working solution in PBS). Samples were washed again twice with PBS, followed by washing with water and mounting samples in FLUORO-GEL (Electron Microscopy Sciences). Samples were left to dry overnight and stored in the dark at 4 °C until analysis. Image acquisition was performed with a Zeiss Axiovert 200, at 100x magnification with a 1.3 NA objective (oil immersion).
Additional material
Download Zip (983.6 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We gratefully acknowledge Dr. Elke Grassmann (Cincinnati Children’s Hospital Medical Center, Cincinnati, USA) for generously providing the reporter cell line SC1 REW22. For advice on culture conditions and sharing information and her experience with the reporter cell line SC1 REW22, we thank Dr. Melanie Galla (MH Hannover, Germany). T.S. was supported by the EU FP7 program “Affinomics.” F.P. is supported by the CNRS and the Institut Curie.
References
- Blose SH, Meltzer DI, Feramisco JR. 10-nm filaments are induced to collapse in living cells microinjected with monoclonal and polyclonal antibodies against tubulin. J Cell Biol 1984; 98:847 - 58; http://dx.doi.org/10.1083/jcb.98.3.847; PMID: 6538204
- Gawlitta W, Osborn M, Weber K. Coiling of intermediate filaments induced by microinjection of a vimentin-specific antibody does not interfere with locomotion and mitosis. Eur J Cell Biol 1981; 26:83 - 90; PMID: 7198976
- Kallajoki M, Harborth J, Weber K, Osborn M. Microinjection of a monoclonal antibody against SPN antigen, now identified by peptide sequences as the NuMA protein, induces micronuclei in PtK2 cells. J Cell Sci 1993; 104:139 - 50; PMID: 8449992
- Lin JJ, Feramisco JR. Disruption of the in vivo distribution of the intermediate filaments in fibroblasts through the microinjection of a specific monoclonal antibody. Cell 1981; 24:185 - 93; http://dx.doi.org/10.1016/0092-8674(81)90514-6; PMID: 7016335
- Warn RM, Flegg L, Warn A. An investigation of microtubule organization and functions in living Drosophila embryos by injection of a fluorescently labeled antibody against tyrosinated alpha-tubulin. J Cell Biol 1987; 105:1721 - 30; http://dx.doi.org/10.1083/jcb.105.4.1721; PMID: 3117804
- Marschall AL, Frenzel A, Schirrmann T, Schüngel M, Dübel S. Targeting antibodies to the cytoplasm. MAbs 2011; 3:3 - 16; http://dx.doi.org/10.4161/mabs.3.1.14110; PMID: 21099369
- Stocks MR. Intrabodies: production and promise. Drug Discov Today 2004; 9:960 - 6; http://dx.doi.org/10.1016/S1359-6446(04)03269-6; PMID: 15539139
- Stocks M. Intrabodies as drug discovery tools and therapeutics. Curr Opin Chem Biol 2005; 9:359 - 65; http://dx.doi.org/10.1016/j.cbpa.2005.06.003; PMID: 15979379
- Caussinus E, Kanca O, Affolter M. Fluorescent fusion protein knockout mediated by anti-GFP nanobody. Nat Struct Mol Biol 2012; 19:117 - 21; http://dx.doi.org/10.1038/nsmb.2180; PMID: 22157958
- Ernst A, Avvakumov G, Tong J, Fan Y, Zhao Y, Alberts P, Persaud A, Walker JR, Neculai AM, Neculai D, et al. A strategy for modulation of enzymes in the ubiquitin system. Science 2013; 339:590 - 5; http://dx.doi.org/10.1126/science.1230161; PMID: 23287719
- Grebien F, Hantschel O, Wojcik J, Kaupe I, Kovacic B, Wyrzucki AM, Gish GD, Cerny-Reiterer S, Koide A, Beug H, et al. Targeting the SH2-kinase interface in Bcr-Abl inhibits leukemogenesis. Cell 2011; 147:306 - 19; http://dx.doi.org/10.1016/j.cell.2011.08.046; PMID: 22000011
- Kummer L, Parizek P, Rube P, Millgramm B, Prinz A, Mittl PR, Kaufholz M, Zimmermann B, Herberg FW, Plückthun A. Structural and functional analysis of phosphorylation-specific binders of the kinase ERK from designed ankyrin repeat protein libraries. Proc Natl Acad Sci U S A 2012; 109:E2248 - 57; http://dx.doi.org/10.1073/pnas.1205399109; PMID: 22843676
- Böldicke T. Blocking translocation of cell surface molecules from the ER to the cell surface by intracellular antibodies targeted to the ER. J Cell Mol Med 2007; 11:54 - 70; http://dx.doi.org/10.1111/j.1582-4934.2007.00002.x; PMID: 17367501
- Strebe N, Guse A, Schüngel M, Schirrmann T, Hafner M, Jostock T, Hust M, Müller W, Dübel S. Functional knockdown of VCAM-1 at the posttranslational level with ER retained antibodies. J Immunol Methods 2009; 341:30 - 40; http://dx.doi.org/10.1016/j.jim.2008.10.012; PMID: 19038261
- Zhang C, Helmsing S, Zagrebelsky M, Schirrmann T, Marschall AL, Schüngel M, Korte M, Hust M, Dübel S. Suppression of p75 neurotrophin receptor surface expression with intrabodies influences Bcl-xL mRNA expression and neurite outgrowth in PC12 cells. PLoS One 2012; 7:e30684; http://dx.doi.org/10.1371/journal.pone.0030684; PMID: 22292018
- Postupalenko V, Sibler AP, Desplancq D, Nominé Y, Spehner D, Schultz P, Weiss E, Zuber G. Intracellular delivery of functionally active proteins using self-assembling pyridylthiourea-polyethylenimine. J Control Release 2014; 178:86 - 94; http://dx.doi.org/10.1016/j.jconrel.2014.01.017; PMID: 24476809
- Kaczmarczyk SJ, Sitaraman K, Young HA, Hughes SH, Chatterjee DK. Protein delivery using engineered virus-like particles. Proc Natl Acad Sci U S A 2011; 108:16998 - 7003; http://dx.doi.org/10.1073/pnas.1101874108; PMID: 21949376
- Voelkel C, Galla M, Maetzig T, Warlich E, Kuehle J, Zychlinski D, Bode J, Cantz T, Schambach A, Baum C. Protein transduction from retroviral Gag precursors. Proc Natl Acad Sci U S A 2010; 107:7805 - 10; http://dx.doi.org/10.1073/pnas.0914517107; PMID: 20385817
- Heitz F, Morris MC, Divita G. Twenty years of cell-penetrating peptides: from molecular mechanisms to therapeutics. Br J Pharmacol 2009; 157:195 - 206; http://dx.doi.org/10.1111/j.1476-5381.2009.00057.x; PMID: 19309362
- Mueller J, Kretzschmar I, Volkmer R, Boisguerin P. Comparison of cellular uptake using 22 CPPs in 4 different cell lines. Bioconjug Chem 2008; 19:2363 - 74; http://dx.doi.org/10.1021/bc800194e; PMID: 19053306
- Ye D, Xu D, Singer AU, Juliano RL. Evaluation of strategies for the intracellular delivery of proteins. Pharm Res 2002; 19:1302 - 9; http://dx.doi.org/10.1023/A:1020346607764; PMID: 12403066
- Behr JP. The Proton Sponge: a Trick to Enter Cells the Viruses Did not Exploit. Chimia (Aarau) 1997; 51:34 - 6
- Khalil IA, Kogure K, Akita H, Harashima H. Uptake pathways and subsequent intracellular trafficking in nonviral gene delivery. Pharmacol Rev 2006; 58:32 - 45; http://dx.doi.org/10.1124/pr.58.1.8; PMID: 16507881
- Fischer PM. Cellular uptake mechanisms and potential therapeutic utility of peptidic cell delivery vectors: progress 2001-2006. Med Res Rev 2007; 27:755 - 95; http://dx.doi.org/10.1002/med.20093; PMID: 17019680
- Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, Chernomordik LV, Lebleu B. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J Biol Chem 2003; 278:585 - 90; http://dx.doi.org/10.1074/jbc.M209548200; PMID: 12411431
- Schmidt N, Mishra A, Lai GH, Wong GC. Arginine-rich cell-penetrating peptides. FEBS Lett 2010; 584:1806 - 13; http://dx.doi.org/10.1016/j.febslet.2009.11.046; PMID: 19925791
- Cronican JJ, Beier KT, Davis TN, Tseng JC, Li W, Thompson DB, Shih AF, May EM, Cepko CL, Kung AL, et al. A class of human proteins that deliver functional proteins into mammalian cells in vitro and in vivo. Chem Biol 2011; 18:833 - 8; http://dx.doi.org/10.1016/j.chembiol.2011.07.003; PMID: 21802004
- Rongrong L, Lixia W, Zhongping L. Effect of deletion mutation on the recombination activity of Cre recombinase. Acta Biochim Pol 2005; 52:541 - 4; PMID: 15912212
- Moutel S, El Marjou A, Vielemeyer O, Nizak C, Benaroch P, Dübel S, Perez F. A multi-Fc-species system for recombinant antibody production. BMC Biotechnol 2009; 9:14; http://dx.doi.org/10.1186/1472-6750-9-14; PMID: 19245715
- Vielemeyer O, Nizak C, Jimenez AJ, Echard A, Goud B, Camonis J, Rain JC, Perez F. Characterization of single chain antibody targets through yeast two hybrid. BMC Biotechnol 2010; 10:59; http://dx.doi.org/10.1186/1472-6750-10-59; PMID: 20727208
- Tanaka T, Rabbitts TH. Functional intracellular antibody fragments do not require invariant intra-domain disulfide bonds. J Mol Biol 2008; 376:749 - 57; http://dx.doi.org/10.1016/j.jmb.2007.11.085; PMID: 18187153
- Loonstra A, Vooijs M, Beverloo HB, Allak BA, van Drunen E, Kanaar R, Berns A, Jonkers J. Growth inhibition and DNA damage induced by Cre recombinase in mammalian cells. Proc Natl Acad Sci U S A 2001; 98:9209 - 14; http://dx.doi.org/10.1073/pnas.161269798; PMID: 11481484
- Pfeifer A, Brandon EP, Kootstra N, Gage FH, Verma IM. Delivery of the Cre recombinase by a self-deleting lentiviral vector: efficient gene targeting in vivo. Proc Natl Acad Sci U S A 2001; 98:11450 - 5; http://dx.doi.org/10.1073/pnas.201415498; PMID: 11553794
- Lin Q, Jo D, Gebre-Amlak KD, Ruley HE. Enhanced cell-permeant Cre protein for site-specific recombination in cultured cells. BMC Biotechnol 2004; 4:25; http://dx.doi.org/10.1186/1472-6750-4-25; PMID: 15500682
- Wadia JS, Stan RV, Dowdy SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat Med 2004; 10:310 - 5; http://dx.doi.org/10.1038/nm996; PMID: 14770178
- Will E, Klump H, Heffner N, Schwieger M, Schiedlmeier B, Ostertag W, Baum C, Stocking C. Unmodified Cre recombinase crosses the membrane. Nucleic Acids Res 2002; 30:e59; http://dx.doi.org/10.1093/nar/gnf059; PMID: 12060697
- Dalkara D, Zuber G, Behr JP. Intracytoplasmic delivery of anionic proteins. Mol Ther 2004; 9:964 - 9; http://dx.doi.org/10.1016/j.ymthe.2004.03.007; PMID: 15194063
- Bieber T, Meissner W, Kostin S, Niemann A, Elsasser HP. Intracellular route and transcriptional competence of polyethylenimine-DNA complexes. J Control Release 2002; 82:441 - 54; http://dx.doi.org/10.1016/S0168-3659(02)00129-3; PMID: 12175756
- Gilleron J, Querbes W, Zeigerer A, Borodovsky A, Marsico G, Schubert U, Manygoats K, Seifert S, Andree C, Stöter M, et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat Biotechnol 2013; 31:638 - 46; http://dx.doi.org/10.1038/nbt.2612; PMID: 23792630
- Sahay G, Querbes W, Alabi C, Eltoukhy A, Sarkar S, Zurenko C, Karagiannis E, Love K, Chen D, Zoncu R, et al. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat Biotechnol 2013; 31:653 - 8; http://dx.doi.org/10.1038/nbt.2614; PMID: 23792629
- Chu CJ, Dijkstra J, Lai MZ, Hong K, Szoka FC. Efficiency of cytoplasmic delivery by pH-sensitive liposomes to cells in culture. Pharm Res 1990; 7:824 - 34; http://dx.doi.org/10.1023/A:1015908831507; PMID: 2172955
- Dalkara D, Chandrashekhar C, Zuber G. Intracellular protein delivery with a dimerizable amphiphile for improved complex stability and prolonged protein release in the cytoplasm of adherent cell lines. J Control Release 2006; 116:353 - 9; http://dx.doi.org/10.1016/j.jconrel.2006.10.005; PMID: 17097756
- Tinsley JH, Zawieja DC, Wu MH, Ustinova EE, Xu W, Yuan SY. Protein transfection of intact microvessels specifically modulates vasoreactivity and permeability. J Vasc Res 2001; 38:444 - 52; http://dx.doi.org/10.1159/000051077; PMID: 11561146
- Tinsley JH, Hawker J, Yuan Y. Efficient protein transfection of cultured coronary venular endothelial cells. Am J Physiol 1998; 275:H1873 - 8; PMID: 9815096
- Todorova R. Estimation of methods of protein delivery into mammalian cells--a comparative study by electroporation and bioporter assay. Prikl Biokhim Mikrobiol 2009; 45:493 - 6; PMID: 19764622
- van der Gun BT, Monami A, Laarmann S, Raskó T, Slaska-Kiss K, Weinhold E, Wasserkort R, de Leij LF, Ruiters MH, Kiss A, et al. Serum insensitive, intranuclear protein delivery by the multipurpose cationic lipid SAINT-2. J Control Release 2007; 123:228 - 38; http://dx.doi.org/10.1016/j.jconrel.2007.08.014; PMID: 17884225
- Weill CO, Biri S, Adib A, Erbacher P. A practical approach for intracellular protein delivery. Cytotechnology 2008; 56:41 - 8; http://dx.doi.org/10.1007/s10616-007-9102-3; PMID: 19002840
- Zelphati O, Wang Y, Kitada S, Reed JC, Felgner PL, Corbeil J. Intracellular delivery of proteins with a new lipid-mediated delivery system. J Biol Chem 2001; 276:35103 - 10; http://dx.doi.org/10.1074/jbc.M104920200; PMID: 11447231
- Samba-Louaka A, Nougayrède JP, Watrin C, Oswald E, Taieb F. The enteropathogenic Escherichia coli effector Cif induces delayed apoptosis in epithelial cells. Infect Immun 2009; 77:5471 - 7; http://dx.doi.org/10.1128/IAI.00860-09; PMID: 19786559
- Zassler B, Blasig IE, Humpel C. Protein delivery of caspase-3 induces cell death in malignant C6 glioma, primary astrocytes and immortalized and primary brain capillary endothelial cells. J Neurooncol 2005; 71:127 - 34; http://dx.doi.org/10.1007/s11060-004-1364-4; PMID: 15690127
- Freund G, Sibler AP, Desplancq D, Oulad-Abdelghani M, Vigneron M, Gannon J, Van Regenmortel MH, Weiss E. Targeting endogenous nuclear antigens by electrotransfer of monoclonal antibodies in living cells. MAbs 2013; 5:518 - 22; http://dx.doi.org/10.4161/mabs.25084; PMID: 23765067
- Bradbury AR, Sidhu S, Dübel S, McCafferty J. Beyond natural antibodies: the power of in vitro display technologies. Nat Biotechnol 2011; 29:245 - 54; http://dx.doi.org/10.1038/nbt.1791; PMID: 21390033
- Colwill K, Gräslund S, Renewable Protein Binder Working Group. A roadmap to generate renewable protein binders to the human proteome. Nat Methods 2011; 8:551 - 8; http://dx.doi.org/10.1038/nmeth.1607; PMID: 21572409
- Dübel S. Recombinant human antibodies for target discovery. Presentation and abstract. MIPTEC conference. Congress Centre Basel, 2012.
- Jäger V, Büssow K, Wagner A, Weber S, Hust M, Frenzel A, Schirrmann T. High level transient production of recombinant antibodies and antibody fusion proteins in HEK293 cells. BMC Biotechnol 2013; 13:52; http://dx.doi.org/10.1186/1472-6750-13-52; PMID: 23802841