Abstract
Over the last decade, cytomegalovirus (CMV) has been suggested to promote the development of glioblastoma multiforme (GBM). Recent evidence demonstrates that CMV contributes to the progression of GBM in the context of oncosuppressor gene mutations. This finding provides further insights into the mechanisms whereby CMV exacerbates the malignancy of GBM.
The presence of cytomegalovirus (CMV) gene products in glioblastoma multiforme (GBM) was first documented in 2002.Citation1 This finding was significant for at least 2 reasons. First, the influence of viruses in the biology of cancer cells has been well described in other malignancies, such as cervical cancer and Kaposi’s sarcoma. Thus, the identification of CMV products in GBM lesions opened the possibility that CMV would promote malignant transformation and/or tumor progression. Second, in spite of standard of care therapy, GBM, which represents the most common and aggressive form of glioma, is associated with poor prognosis. Promising progresses toward an improved clinical management of glioma patients have been made with the advent of targeted therapies, including gene delivery approaches, angiogenesis inhibitors, oncolytic viruses, and various immunotherapeutic regimens. The observation that CMV selectively infects glioma cells identifies another means by which GBM may be targeted with biological anticancer agents.Citation2
By introducing the concept of oncomodulation, Michaelis, Cinatl, and colleagues have provided a framework to the elucidation of the active role played by CMV in tumor progression.Citation3 Oncomodulation is a process whereby CMV may infect cancer cells, hence affecting their functions, without being directly involved in malignant transformation. In this setting, neoplastic cells provide the molecular and genetic milieu whereby disturbances in tumor suppressors, transcription factors, and signal transduction pathways allow CMV to exert an oncomodulatory effect.
Price et al. have discovered some of the oncomodulatory effects of CMV in the setting of oncosuppressor gene mutations.Citation4,Citation5 An initial study demonstrated that Trp53+/− mice infected perinatally with murine CMV (MCMV) developed rhabdomyosarcoma (RMS) at an earlier age and exhibited a shorter survival than control animals. Similar to human RMS, these murine tumors were found to express CMV proteins including pp65 and immediate-early 1 (IE-1). More recently, 2 distinct mouse models of oncosuppressor mutations commonly associated with glioma were employed to determine the effect of perinatal MCMV infection on tumor progression and overall survival. In particular, GFAP-cre:Nf1loxP/+:Trp53+/− Mut3 mice and a syngeneic orthotopic model in which Nf1:Trp53:Pten mutated GBM cells were injected into the striatum of wild-type mice were used. Mut3 mice spontaneously develop high-grade astrocytomas with near-to-complete penetrance by adulthood. The localization of MCMV to malignant cells was confirmed and glioma-associated deaths were scored in survival analyses. In both these models, MCMV-infected mice showed an approximate decrease of 20% in cancer-related survival as compared with mock-infected animals.
The activation of signal transducer and activator of transcription 3 (STAT3) was identified as a potential molecular mechanism underpinning the decrease in survival of MCMV-infected mice, although it is possible that other, hitherto unidentified, mechanisms might be operational. An increase in STAT3 phosphorylation was detected in vivo, both in MCMV-infected Mut3 neural stem cells before tumor onset and in orthotopic GBM cells 4 wk after inoculation in wild-type mice. This was associated with an increase in proliferating cell nuclear antigen (PCNA) positivity, probably representing the proliferative effect that STAT3 activation has on GBM cells.Citation6 Human GBM tumorspheres derived from patient tumor specimens revealed a similar increase in STAT3 phosphorylation following in vitro infection with HCMV. This finding correlated with increased tumorsphere size and cellularity, in comparison to mock-infected control tumorspheres. In this setting, STAT3 inhibition successfully abolished the proliferative advantage caused by HCMV infection, indicating that the activation of STAT3 is involved in the oncomodulatory effects of CMV on both murine and human GBM.
This study contains the first in vivo data demonstrating the effects of MCMV on murine GBM-related death in the setting of oncosuppressor gene mutations. Genomics analyses of human GBM samples revealed that the murine models that we employed bear mutations that affect the core signal transduction pathways most commonly mutated in the course of GBM. The genes coding for phosphatase and tensin homolog (PTEN) and neurofibromin 1 (NF1) are mutated or deleted in 36% and 18% of GBM cases, respectively, and play an essential role as negative regulators of the signal transduction pathway involving receptor tyrosine kinases (RTKs), RAS, and phosphoinositide-3-kinase (PI3K), which is hyperactivated (hence delivering mitogenic and antiapoptotic signals) in 88% of GBM patients. The TP53 oncosuppressive pathway is disrupted in 87% of GBM cases and is essential for cell cycle arrest and the initiation of apoptotic cell death in response to both intracellular and extracellular stress conditions.Citation7 It is therefore likely that the molecular environment generated by genetic manipulations employed in this study is representative of the conditions encountered during the infection of human GBMs by HCMV.
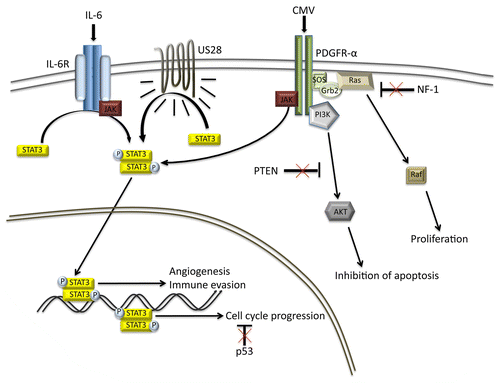
The role of STAT3 in the pathogenesis of GBM has gained significant attention over last several years.Citation6 In particular, the aberrant activation of STAT3 has been shown to induce cell cycle progression, angiogenesis, and immune evasion. CMV can activate STAT3 by several mechanisms (). Thus, US28, a CMV-encoded chemokine receptor, has been shown to convey proliferative signals via the interleukin-6 (IL-6)-STAT3 pathway.Citation8 HCMV-infected GBM cells are known to secrete increased levels of IL-6 and express high levels of US-28.Citation9 The activation of the platelet-derived growth factor receptor α chain (PDGFRα) is a required for productive CMV infection.Citation10 The gene coding for PDFGRα is commonly amplified in GBM patients, which may ultimately explain both the tropism of HCMV for GBM cells and the pronounced HCMV-mediated STAT3 hyperactivation.
Figure 1. Mechanisms of STAT3 activation by CMV. Cytomegalovirus (CMV) can activate signal transducer and activator of transcription 3 (STAT3) in glioblastoma multiforme (GBM) cells by 3 different mechanisms. CMV-infected cells secrete increased levels of interleukin-6 (IL-6), stimulating signal transduction via the IL-6 receptor (IL6R)-STAT3 axis.Citation8 US28 is a CMV-encoded constitutively active chemokine receptor that is capable of activating STAT3.Citation9 The CMV virion can activate STAT3 via platelet-derived growth factor receptor α chain (PDGFRα), presumably by means of the viral surface glycoprotein B.Citation10 The combination of STAT3 activation and oncosuppressor gene loss significantly exacerbates the malignancy of GBM cells.
The results of Price et al. lend support to the notion that the cellular and molecular alterations induced by CMV in combination with pathogenetic defects in core signal transduction pathways increase the malignancy of GBM cells much more than either of these processes alone. Whether HCMV constitutes an effective target for the treatment of GBM patients remains an open question. The hope is that a deeper understanding of the oncomodulatory effects of CMV on GBM will allow for the delineation of novel therapeutic strategies against this aggressive malignancy.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Cobbs CS, Harkins L, Samanta M, Gillespie GY, Bharara S, King PH, et al. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res 2002; 62:3347 - 50; PMID: 12067971
- Sampson JH, Mitchell DA. Is cytomegalovirus a therapeutic target in glioblastoma?. Clin Cancer Res 2011; 17:4619 - 21; http://dx.doi.org/10.1158/1078-0432.CCR-11-0992; PMID: 21632859
- Michaelis M, Doerr HW, Cinatl J. The story of human cytomegalovirus and cancer: increasing evidence and open questions. Neoplasia 2009; 11:1 - 9; PMID: 19107226
- Price RL, Song J, Bingmer K, Kim TH, Yi JY, Nowicki MO, et al. Cytomegalovirus contributes to glioblastoma in the context of tumor suppressor mutations. Cancer Res 2013; 73:3441 - 50; http://dx.doi.org/10.1158/0008-5472.CAN-12-3846; PMID: 23729642
- Price RL, Bingmer K, Harkins L, Iwenofu OH, Kwon CH, Cook C, et al. Cytomegalovirus infection leads to pleomorphic rhabdomyosarcomas in Trp53+/- mice. Cancer Res 2012; 72:5669 - 74; http://dx.doi.org/10.1158/0008-5472.CAN-12-2425; PMID: 23002204
- Brantley EC, Benveniste EN. Signal transducer and activator of transcription-3: a molecular hub for signaling pathways in gliomas. Mol Cancer Res 2008; 6:675 - 84; http://dx.doi.org/10.1158/1541-7786.MCR-07-2180; PMID: 18505913
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008; 455:1061 - 8; http://dx.doi.org/10.1038/nature07385; PMID: 18772890
- Slinger E, Maussang D, Schreiber A, Siderius M, Rahbar A, Fraile-Ramos A, et al. HCMV-encoded chemokine receptor US28 mediates proliferative signaling through the IL-6-STAT3 axis. Sci Signal 2010; 3:ra58; http://dx.doi.org/10.1126/scisignal.2001180; PMID: 20682912
- Soroceanu L, Matlaf L, Bezrookove V, Harkins L, Martinez R, Greene M, et al. Human cytomegalovirus US28 found in glioblastoma promotes an invasive and angiogenic phenotype. Cancer Res 2011; 71:6643 - 53; http://dx.doi.org/10.1158/0008-5472.CAN-11-0744; PMID: 21900396
- Soroceanu L, Akhavan A, Cobbs CS. Platelet-derived growth factor-alpha receptor activation is required for human cytomegalovirus infection. Nature 2008; 455:391 - 5; http://dx.doi.org/10.1038/nature07209; PMID: 18701889