
Abstract
The establishment of an immunosuppressive tumor microenvironment is a hallmark feature driving cancer cell evasion of immunosurveillance. In a murine melanoma model, we recently demonstrated that decreased intratumoral CD4+ T-cell expression of CD40L and interferon γ (IFNγ) is critical to maintain this immunosuppressive microenvironment. Altered effector functions of tumor-associated CD4+ T cells is essential for B-RafV600E inhibitor-mediated restoration of antitumor immunity.
Oncogenic mutations in the gene encoding BRAF kinase are driver mutations found in roughly half of human melanoma patients. Remarkably a single lesion, the V600E mutation (BRAFV600E), is the predominant mutation found in these patients. Oncogenic BRAFV600E constitutively activates MEK-MAPK signaling cascade to promote proliferation and survival as well as protect transformed melanocytes from apoptosis.Citation1 Selective BRAFV600E inhibitors, vemurafenib and dabrafenib, are effective at halting disease progression or even inducing tumor regression in a high frequency of melanoma patients harboring BRAFV600E mutation. However, melanomas resistant to BRAFV600E inhibitor inevitably arise after a few months of treatment, after which there are no further therapeutic options. The lack of treatment for this aggressive form of melanoma demands a better understanding of tumor progression and regression to prevent resistance and identify novel therapies.Citation2
The tumor microenvironment consists of tumor cells, stromal cells and a variety of immune cells. Clinically, the increase of tumor-infiltrating T lymphocytes has been shown to correlate with prolonged survival of patients with colon cancer as well as other types of cancers.Citation3 Increasing the frequency of intratumoral T cells by transferring tumor-specific T cells can also diminish tumor progression in small portion of patients. These findings reveal that immune cells may be able to eliminate tumor cells by recognizing tumor antigens or moieties. However, recent studies have suggested that tumor-infiltrating immune cells may provide both pro-tumorigenic and anti-tumorigenic effects in the tumor microenvironment. The tumor microenvironment is generally immunosuppressive and the accumulation of different immunomodulatory cells, including regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), within the tumor microenvironment can compromise antitumor immunity.Citation4 Immunotherapies that target checkpoints of immune responses (including PD-1 and CTLA-4 blockade) or the immunomodulatory cells themselves have been shown partially restore antitumor immunity.Citation5 Host immunity has also been suggested to inhibit melanoma progression in melanoma-bearing mice receiving B-RafV600E inhibitor treatment, but the underlying immune responses are not well defined.Citation6 Therefore, understanding how cancer cells promote an immunosuppressive microenvironment in tumors, and, whether the underlying mechanisms can be targeted to activate antitumor responses, are critical, clinically relevant aspects of tumor immunology. The knowledge attained from targeted therapies and chemotherapies may facilitate the development of new treatment regimens that harness the power of the immune system in combating malignant disease.
In our recent study, we first determined the kinetics of both adaptive and innate immune responses during tumor growth using a genetically modified melanoma mouse model (referred as Braf/Pten mice).Citation7 In this mouse model, melanomas were induced by the oncogenic B-RafV600E mutation and simultaneous depletion of Pten in melanocytes.Citation8 Since Braf/Pten mice harbor a common driver mutation and exhibit similar pathophysiological features as those observed in melanoma patients, we took the advantage of this model to examine the immune responses in an immunocompetent setting. We first characterized whether specific immune responses are diminished during tumor progression and, if so, whether these immune responses could be restored after B-RafV600E inhibitor treatment. We found that Braf/Pten melanomas accumulated multiple immunosuppressive cells during tumor growth, including increased frequencies of intratumoral Tregs, MDSCs, immature tumor-associated macrophages (TAMs), and tumor-infiltrating dendritic cells (TIDCs). Additionally, intratumoral effector CD4+ T cells lost their effector cytokine production and CD40L expression in advanced stage melanomas. Surprisingly, treating tumor-bearing mice with PLX4720, an analog of the B-RafV600E inhibitor vemurafenib, reduced tumor growth and diminished the frequencies of intratumoral Tregs and MDSCs while increasing the populations of tumor-infiltrating CD4+ and CD8+ T cells. PLX4720 also promoted the maturation of both TAMs and TIDCs. Importantly, more intratumoral CD4+ T cells produced interferon γ (IFNγ) and expressed CD40L in mice receiving B-RafV600E inhibitor therapy than in mice receiving control treatment. Although the increase in tumor-infiltrating CD8+ T cells has been suggested to impede tumor progression by elimination of cancer cells, the role of intratumoral CD4+ T cells in shaping antitumor immunity has been underappreciated. To our surprise, our results showed that PLX4720 treatment suppresses melanoma growth in a CD4+, but not CD8+, T cell-dependent manner. Our study also revealed that the effector (IFNγ) and helper (CD40L) functions of intratumoral CD4+ T cells might be involved in promoting the maturation of TAMs and TIDCs and preventing the accumulation of MDSCs and Tregs differentially (). Critically, we demonstrated that treating mice with agonistic anti-CD40 antibody to mimic CD40L could provide significant antitumor effect and promote maturation of TAMs.
Figure 1. Intratumoral CD4+ T cells functional alterations during melanoma progression and PLX4720 therapy. Tumor-infiltrating CD4+ T cells lose the expression of interferon γ (IFNγ) and CD40L during tumor growth accompanied by accumulation of Tregs, MDSCs and immature APCs (TAMs and TIDCs). Treating tumor-bearing mice with PLX4720 stimulated the expression of IFNγ and CD40L on intratumoral CD4+ T cells. Thus, PLX4720 treatment promotes the immunostimulatory properties of the tumor microenvironment and suppress the accumulation of Tregs and MDSCs. Tregs, regulatory T cells; MDSCs, myeloid-derived suppressor cells; APCs, antigen-presenting cells; TAMs, tumor-associated macrophages; TIDCs, tumor-infiltrating dendritic cells.
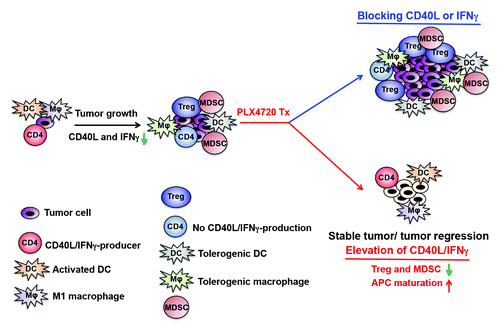
Since depletion of CD4+ T cells and inhibition of IFNγ or CD40L significantly blunted PLX4720-mediated antitumor responses, our results suggested that altering the functions of intratumoral CD4+ T cells could be the driving force to restore antitumor immunity in the tumor microenvironment. Of note, blocking IFNγ and CD40L also efficiently inhibited maturation of TAMs and TIDCs in response to PLX4720 therapy. Therefore, it is less likely that the maturation of these intratumoral APCs was induced by elevation of damage-associated molecular patterns (DAMPs) subsequent to B-RafV600E inhibitor therapy.Citation9 Given that B-RafV600E inhibitors targets mutant B-Raf kinase and affect melanoma cells’ proliferation and metabolism, we hypothesize that B-RafV600E inhibitors may alter certain activities of melanoma cells that normally allow them to suppress T helper cell type 1 (TH1) effector and helper functions.
In summary, these findings inspire conceptual advances on the roles of immune signaling in BRAFV600E inhibitor-mediated antitumor responses. Most importantly, treating Braf/Pten mice with agonistic anti-CD40 mAb is a proof-of-concept that re-introduction of CD40L:CD40 signal could impede tumor growth, suggesting that diminished CD40L expression on intratumoral CD4+ T cells may be involved in immune escape. Of note, we showed that the therapeutic benefit of agonistic anti-CD40 mAb is T cell-independent. Therefore, an important future direction would be exploring whether combining agonistic anti-CD40 mAb with immunotherapies targeting inhibitory checkpoints can elicit stronger antitumor response in both BRAFV600E inhibitor-sensitive and –resistant melanomas. Additionally, determining whether the CD40L expression level after BRAFV600E inhibitor therapy correlates with the antitumor responses and the emergence of resistant melanomas will be of great interest for clinical applications.
| Abbreviations: | ||
| DAMP | = | damage-associated molecular pattern |
| MDSC | = | myeloid-derived suppressor cell |
| TAM | = | tumor-associated macrophage |
| TIDC | = | tumor-infiltrating dendritic cell |
| Treg | = | regulatory T cell |
Disclosure of Potential Conflicts of Interst
No potential conflicts of interest were disclosed. P.-C. H. is supported by National Cancer Center Postdoctoral Fellowship. S.M.K. is suppoted by Howard Hughes Medical Institute. The study was financially supported in part by Yale Cancer Center, Melanoma Research Alliance Development Award and NIH R01AI074699 (to SMK), NCI Specialized Programs of Research Excellence in Skin Cancer (5 P50 CA121974, PI: Ruth Halaban).
References
- Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 2012; 367:1694 - 703; http://dx.doi.org/10.1056/NEJMoa1210093; PMID: 23020132
- Bollag G, Tsai J, Zhang J, Zhang C, Ibrahim P, Nolop K, Hirth P. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat Rev Drug Discov 2012; 11:873 - 86; http://dx.doi.org/10.1038/nrd3847; PMID: 23060265
- Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, Tosolini M, Camus M, Berger A, Wind P, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006; 313:1960 - 4; http://dx.doi.org/10.1126/science.1129139; PMID: 17008531
- Shiao SL, Ganesan AP, Rugo HS, Coussens LM. Immune microenvironments in solid tumors: new targets for therapy. Genes Dev 2011; 25:2559 - 72; http://dx.doi.org/10.1101/gad.169029.111; PMID: 22190457
- Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer 2012; 12:237 - 51; http://dx.doi.org/10.1038/nrc3237; PMID: 22437869
- Knight DA, Ngiow SF, Li M, Parmenter T, Mok S, Cass A, Haynes NM, Kinross K, Yagita H, Koya RC, et al. Host immunity contributes to the anti-melanoma activity of BRAF inhibitors. J Clin Invest 2013; 123:1371 - 81; http://dx.doi.org/10.1172/JCI66236; PMID: 23454771
- Ho PC, Meeth KM, Tsui YC, Srivastava B, Bosenberg MW, Kaech SM. Immune-based antitumor effects of BRAF Inhibitors rely on signaling by CD40L and IFNγ. Cancer Res 2014; ; In press http://dx.doi.org/10.1158/0008-5472.CAN-13-3461; PMID: 24736544
- Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE Jr., You MJ, DePinho RA, McMahon M, Bosenberg M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet 2009; 41:544 - 52; http://dx.doi.org/10.1038/ng.356; PMID: 19282848
- De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 2013; 23:277 - 86; http://dx.doi.org/10.1016/j.ccr.2013.02.013; PMID: 23518347