
Abstract
Pancreatic cancer remains largely an incurable disease necessitating the development of novel therapeutic approaches. Adoptive immunotherapy using chimeric antigen receptor (CAR)-transduced T cells represents an alternative treatment with curative potential. We present an overview of the engineering of novel CARs targeting prostate stem cell antigen (PSCA), implications for the development of immunotherapies, and potential strategies to circumvent on-target/off-tumor toxicities.
Recent clinical success in the treatment of B-cell cancers using chimeric antigen receptor (CAR)-transduced T cellsCitation1-Citation3 has sparked great interest. Genetic manipulation to “re-educate” the patient’s own immune cells to detect and destroy tumors opens a plethora of new possibilities for the clinical management of incurable diseases. Among these, pancreatic ductal adenocarcinoma, the most common type of pancreatic cancer, represents an ideal candidate due to the lack of treatment options for patients with advanced disease.
Limited clinical success using CAR-transduced cells for the treatment of solid tumors has been reported so far, primarily an anti-GD2 CAR treatment in neuroblastoma patients.Citation4 In the case of pancreatic cancer, clinical trials based on CARs targeting carcinoembryonic antigen have been initiated in the US (NCT01723306, NCT00004178) and Europe (NCT01212887), but with no results reported so far. Two parallel trials targeting mesothelin (MSLN) with a mouse antibody-based CAR are underway at the National Cancer Institute (NCT01583686) and the Abramson Cancer Center (NCT01897415). In the Abramson Cancer Center study, multiple doses of RNA-electroporated T cells were administered to patients, with evidence suggestive of potential clinical responses.Citation5 However, repeated murine CAR dosing stimulated the development of human anti-mouse antibodies, and was associated with cardiac arrest resulting from anaphylactic shock in one of the patients so treated.Citation6
Among many parameters that must be optimized for the development of CAR-based therapies, selection of the target antigen and design of an appropriate immune receptor are crucial. In a recently published study,Citation7 we generated and characterized a CAR directed against prostate stem cell antigen (PSCA), a small, extracellular glycoprotein of unknown function that is overexpressed in pancreatic cancer cells from early stages of malignant transformation. As prior CARs targeting PSCA were derived from mouse antibodies, we sought to generate a receptor derived entirely from human proteins hoping to prevent human anti-mouse toxicities, and further, determine whether these human sequences would stabilize CAR expression in transduced human T cells.
We found that the human CAR does have a greater surface expression and induces greater reactivity in vitro than both the mouse counterpart and the anti-MSLN CAR used in the NCI clinical trial, suggesting that it may induce more potent anticancer immune responses. We next tested the antitumor potential of two anti-PSCA CAR variants (differing in the number of costimulatory moieties) in vivo using a mouse model permissive to tumor and primary human T-cell engraftment.
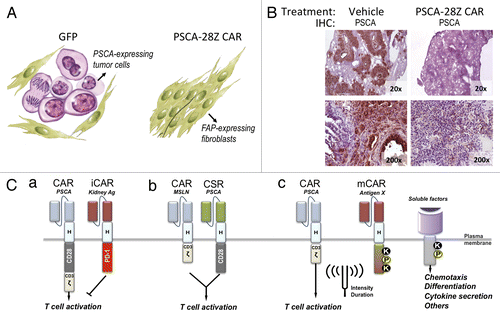
Adoptive transfer of CAR-transduced human CD8+ cells resulted in reduction of tumor volume and/or replacement of tumor cells by fibroblasts (). After treatment, tumors from control mice contained abundant tumor cells with intense expression of PSCA, interspersed with fibroblasts expressing fibroblast-activating protein (FAP). In contrast, residual masses resected from PSCA CAR-treated mice contained minimal amounts of PSCA-positive cells and large number of FAP-expressing cells, indicating that the tumor tissue had been replaced by scar tissue. Interestingly, the antitumor effect observed in this animal model was not restricted to subcutaneous xenografts. Control mice receiving saline solution presented macroscopic lung nodules comprised of PSCA-positive tumor cell deposits (i.e., metastases) in addition to subcutaneous lesions at the site of implantation. In contrast, mice treated with the second generation PSCA-CAR did not have nodules or PSCA staining in their lungs (). Although further analysis will be required to determine whether this phenomenon resulted from the elimination of established lung metastases or prevention of metastatic spread, our findings show that CAR-transduced T cells have the potential to mediate a potent and systemic antitumor effect. Of note, although a third-generation CAR containing CD28 and 4–1BB costimulatory domains induced greater persistence of T cells in vivo, the second-generation CAR containing only a CD28 costimulatory domain induced a superior antitumor effect, indicating that CAR-expressing T cell persistence does not necessarily equate to enhanced tumor cell killing.
Figure 1. Treatment with anti-PSCA CAR and potential safety strategies to prevent on target/off tumor toxicity. (A) Schematic representation of the results previously obtained in a humanized mouse model of pancreatic cancer. Nod/SCID gamma (NSG) mice bearing human pancreatic adenocarcinoma (HPAC) subcutaneous xenografts were treated with human CD8+ T cells transduced with GFP (control) or with an anti-prostate stem cell antigen (PSCA) chimeric antigen receptor (CAR). Forty days after treatment, residual tumors were excised and analyzed for PSCA expression. (B) Immunohistochemical staining of PSCA in lung sections from mice at the end of the treatment. Magnification is indicated at the bottom right corner of each micrograph. (C) Potential strategies to restrict cytotoxicity of CAR-transduced cells to tumor tissue. (a) Coexpression of anti-PSCA CAR, together with an inhibitory CAR (iCAR) directed to an antigen expressed in critical normal tissues that express PSCA. (b) The T cell activation (CD3-zeta) and costimulatory (CD28) domains can be split into two different receptors, each targeting a different antigen, such that ligation of both receptors is necessary to unleash the cytotoxic potential of T cells. CSR: Costimulatory receptor. (c) Combination of CARs with more sophisticated modulatory receptors (mCAR) to ‘fine tune’ the biological activity of CAR-transduced lymphocytes in terms of intensity and/or duration. Synthetic signaling domains can be generated that contain docking sites for kinases (K), phosphates (P) or other signal transducers allowing for the activation of signaling cascades in response to virtually any antigen.
A major difference between the CD19- and the PSCA- targeting therapeutic scenarios is the pattern of antigen expression. Whereas CD19 is expressed exclusively in malignant cells and non-essential tissues, PSCA shows basal expression in the collecting tubules of the kidney, as well as in gastric and esophageal epithelia. Thus, treatment with a potent PSCA-targeting CAR may be too risky for immediate clinical application as a single agent due to the potential of life-threating toxicities.
Although we succeeded in improving the in vivo antitumor efficacy and potentially preventing any anti-mouse responses, many hurdles remain before this treatment can be applied to patients. Combination of a PSCA CAR with safety modules, such as inducible suicide systems, would potentially protect patients from life-treating toxicities but would not prevent them Citation9. shows three potential strategies to restrict the cytotoxicity of CAR-transduced cells to tumor tissue: (1) Combination of anti-PSCA CAR with an inhibitory CAR (iCARCitation8) directed to an antigen expressed in critical normal tissues that express PSCA (e.g., kidney collecting tubules), may result in inhibition of T cell activation (). (2) The T-cell activation (CD3-zeta) and costimulatory (CD28) domains can be split into two different receptors, each targeting a different antigen, such that ligation of both receptors is necessary to unleash the cytotoxic potential of T cells. Because of their simultaneous expression in pancreatic cancer cells and non-overlapping expression in normal tissues, mesothelin and PSCA would be an appropriate pair of target antigens for this approach (). (3) Combination of CARs with more sophisticated modulatory receptors (mCARs) may constitute the next generation of immune receptors, designed to ‘fine tune’ the biological activity of CAR-transduced lymphocytes in terms of intensity and/or duration (). Synthetic signaling domains can be generated that contain docking sites for kinases, phosphates or other signal transducers allowing the activation of signaling cascades in response to virtually any antigen. Integration of multiple artificial receptors to detect antigens or soluble factors from the microenvironment, coupled to the control of migration, metabolism, differentiation or other processes, may ultimately turn T cells into intelligent biological agents armed with real-time sensing and decision-making capabilities.
Ultimately, as the field of systems biology progresses toward a better understanding of the complex signaling networks governing cellular functions, we envisage the development of more sophisticated information-sensing modules. As a result, CARs will not be limited to transducing T-cell activation signals, but may integrate other functions, such as chemotactic control, T-cell differentiation and polarization, and modulation of the intensity and duration of cytotoxicity.Citation10
| Abbreviations: | ||
| CAR | = | chimeric antigen receptor |
| FAP | = | fibroblast-activating protein |
| PSCA | = | prostate stem cell antigen |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, Maric I, Raffeld M, Nathan DA, Lanier BJ, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010; 116:4099 - 102; http://dx.doi.org/10.1182/blood-2010-04-281931; PMID: 20668228
- Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 2011; 365:725 - 33; http://dx.doi.org/10.1056/NEJMoa1103849; PMID: 21830940
- Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med 2013; 5:77ra38; http://dx.doi.org/10.1126/scitranslmed.3005930; PMID: 23515080
- Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, Huls MH, Liu E, Gee AP, Mei Z, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med 2008; 14:1264 - 70; http://dx.doi.org/10.1038/nm.1882; PMID: 18978797
- Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, Chew A, Zhao Y, Levine BL, Albelda SM, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res 2014; 2:112 - 20; http://dx.doi.org/10.1158/2326-6066.CIR-13-0170; PMID: 24579088
- Maus MV, Haas AR, Beatty GL, et al. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res 2013; 1:26 - 31; http://dx.doi.org/10.1158/2326-6066.CIR-13-0006
- Abate-Daga D, Lagisetty KH, Tran E, et al. A novel chimeric antigen receptor against PSCA mediates tumor destruction in a humanized mouse model of pancreatic cancer. Hum Gene Ther 2014; Forthcoming http://dx.doi.org/10.1089/hum.2013.209
- Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med 2013; 5:ra172; http://dx.doi.org/10.1126/scitranslmed.3006597; PMID: 24337479
- Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med 2011; 365:1673 - 83; http://dx.doi.org/10.1056/NEJMoa1106152; PMID: 22047558
- Fischbach MA, Bluestone JA, Lim WA. Cell-based therapeutics: the next pillar of medicine. Sci Transl Med 2013; 5:ps7; http://dx.doi.org/10.1126/scitranslmed.3005568; PMID: 23552369