
Abstract
In addition to mRNAs whose primary function is transmission of genetic information from DNA to proteins, numerous other classes of RNA molecules exist, which are involved in a variety of functions, such as catalyzing biochemical reactions or performing regulatory roles. In analogy to proteins, the function of RNAs depends on their structure and dynamics, which are largely determined by the ribonucleotide sequence. Experimental determination of high-resolution RNA structures is both laborious and difficult, and therefore, the majority of known RNAs remain structurally uncharacterized. To address this problem, computational structure prediction methods were developed that simulate either the physical process of RNA structure formation (“Greek science” approach) or utilize information derived from known structures of other RNA molecules (“Babylonian science” approach). All computational methods suffer from various limitations that make them generally unreliable for structure prediction of long RNA sequences. However, in many cases, the limitations of computational and experimental methods can be overcome by combining these two complementary approaches with each other. In this work, we review computational approaches for RNA structure prediction, with emphasis on implementations (particular programs) that can utilize restraints derived from experimental analyses. We also list experimental approaches, whose results can be relatively easily used by computational methods. Finally, we describe case studies where computational and experimental analyses were successfully combined to determine RNA structures that would remain out of reach for each of these approaches applied separately.
Introduction
RNA is a polymer composed mainly of only four basic building blocks. This complexity appears relatively low compared with proteins that are composed of far more diverse functional groups on the side chains five times the diversity of residues. Nevertheless, RNA is capable of performing a wide range of biological functions that resemble those of proteins, and some unique functions because of the RNA’s coding potential. RNA molecules have been long known to carry genetic information and to synthesize proteins. They may detect the presence of ions or small molecules in the environment, regulate gene expression on various levels (from DNA, to RNA, to proteins), and catalyze chemical reactions (reviewed comprehensively in ref. Citation1). While the role of protein-coding RNA in transmission of genetic information encoded in triplets of residues depends essentially just on the ribonucleotide sequence, most of the other roles depend also (or mostly) on the spatial structure of the ribonucleotide chain. There exist non-coding RNA molecules such as tRNAs or certain ribozymes that form very stable tertiary structures that define their function in a very similar manner to how the function of a protein enzyme is defined by the stable structures that are formed. Many functions depend not on one structure, but on the RNA ability to form alternative structures or to undergo transformations between the structured and unstructured state or even both. For example, riboswitches, regulatory elements located within mRNA that switch protein production on and off, function owing to the ability to undergo conformational changes depending on binding of specific ligands or on sensing other environmental changes (review ref. Citation2). Thus, the understanding of manifold mechanisms of RNA function beyond protein coding requires detailed knowledge of the RNA tertiary structure in the same way as complete understanding of protein function is heavily dependent on the knowledge of protein structure (reviewed comprehensively in ref. Citation3).
Unfortunately, the experimental determination of RNA structures at atomic level of precision is very difficult and rather expensive. Currently, it is significantly more difficult than protein structure determination.Citation4 As of February 2014, only 632 unique RNA crystal structures determined at high or medium resolution (up to 3 Å) have been deposited in the Protein Data Bank, which is dwarfed by the number of representative protein structures of equivalent quality determined thus far (about 28 000 structures with resolution up to 3 Å and up to 90% identity). On the other hand, experimental determination of sequence and structural features of the transcriptome (e.g., by using second generation sequencing methods that rely on reverse transcription of RNA into DNA followed by DNA sequencing) is much cheaper and faster. Given the rapid growth of RNA sequence information and very slow growth of information about RNA structures, it seems unlikely that structures will be determined experimentally for all RNA molecules in the foreseeable future. This situation parallels a similar problem concerning protein sequences and structures, and both these problems have been approached by the development of computational methods for prediction of three-dimensional structures from the sequence information. Although we are still far from solving these problems completely, thus far, a few successful approaches have been proposed, which allow for reasonably accurate and practically useful prediction of RNA and protein tertiary structures. It is worth emphasizing that many methods for RNA structure prediction have been strongly inspired by computational methods developed earlier for protein structure prediction.Citation5
In opposition to high-resolution experimental structure determination, low-resolution studies on RNA structure that focus on the level of residues rather than atoms have been methodologically more advanced than the corresponding low-resolution studies on protein structure. This is to large extent because direct sequencing of RNA molecules is easier than protein sequencing and because nucleic acid molecules can be easily amplified (by the polymerase chain reaction; PCR), while protein molecules essentially cannot. As a consequence, studies on RNA structure have frequently involved computational predictions aided by the use of additional information derived from low-resolution experiments. This synergy has been further reinforced by the recent development of novel computational approaches methods for RNA modeling as well as by the popularization of experimental methods used for low-resolution protein shape determination to analyze complex RNA structures.
In this article, we first describe contemporary methods for computational modeling of RNA structure; we indicate conceptual differences between the dominant approaches, with emphasis on those that can most easily use data resulting from experiments (examples are listed in ). Second, we briefly introduce experimental techniques that provide the types of structural information that can be relatively easily utilized by RNA modeling methods (examples are listed in ). Finally, we discuss the utility of various methods for RNA structure prediction depending on the source of data, and provide a few examples of RNA structure modeling with the use of experimental restraints.
Table 1. Examples of computational methods for RNA 3D structure modeling that are capable of using experimental restraints
Table 2. Low-resolution experimental methods that generate particularly useful data for computational prediction of RNA 3D structure
Classification of RNA 3D Structure Modeling Methods
There exist a wide variety of methods for macromolecular 3D structure prediction that are applicable to nucleic acids, proteins, and their complexes. They can be classified in various ways. One classification divides structure modeling methods into those based on the fundamental laws of physics that govern the process of folding (i.e., the “Greek science” approach, in reference to their work on first principles that do not make assumptions such as empirical model and fitting parameters), and all others, which typically extensively use information about other structures, available in various databases (i.e., the “Babylonian science” approach, in reference to the original contribution of their massive libraries). Another classification divides methods based on whether they use some of the available structures as templates. One more way to divide methods is into those that are fully automated and those that require the manipulation of spatial coordinates by human experts. Finally, a number of methods may perform model building with the use of restraints derived from biochemical or biophysical experiments (i.e., to bias the modeling process to fit the experimental data), while others have been designed to perform unrestrained simulations and only the resulting models can be compared with the experimental results.
Modeling Based on Fundamental Laws of Physics is Accurate (for Small Molecules) but Very Slow
The “Greek science” approach that employs the fundamental laws of physics without information from databases is often referred to as “ab initio.” It is based on the thermodynamic hypothesis formulated by Anfinsen, according to which the native structure of a macromolecule corresponds to the global minimum of the free energy of the system under consideration, which includes the solvent.Citation6 Accordingly, physics-based methods model the process of folding by simulating the conformational changes of a macromolecule, while it searches for the state of minimal free energy (review ref. Citation7). Each conformation is “scored” by calculating the physical energy based on the interactions within the macromolecule and between the macromolecule and the solvent.Citation8 The function with which to calculate the energy should be ideally based on quantum-mechanical (QM) description of the system; however, such calculations are extremely complex and slow, therefore they are applicable only to very small molecules. Therefore, various simplifications are used and typically the system is described in terms of Newtonian dynamics.
Molecular dynamics (MD) is an example of such simplifications, where the moves of particles are determined by numerically solving Newton's equations of motion. The functional form and parameter sets used to describe the forces between the particles and the potential energy of a system is called a force field. There exist a number of software packages for simulation of macromolecular folding by MD that employ force fields such as AMBER,Citation9 CHARMM,Citation10 or GROMOSCitation11 to calculate the energy. Even with various simplifications used in these methods, both conformational sampling and energy calculations are very costly in terms of computational power required. Typically, the free energy landscape is extremely rugged (i.e., it possesses multiple local minima), so for RNA molecules comprising hundreds or thousands of atoms, it is essentially impossible to perform an exhaustive evaluation of all these minima to identify the one with the globally lowest value. Further, some of the components of the free energy function (e.g., the entropy) are very difficult to calculate, and may not be inferred accurately for large molecules. For these reasons, ab initio methods can only be used to model very small molecules or systems, where the search is limited to only a small part of the conformational space (e.g., optimization of models generated by other methods). An alternative approach is to limit the conformational space by user-defined restraints. For instance, molecular dynamics simulations initiated with experimentally determined structures and heavily restrained by experimental data have been used to model the conformational transitions of RNA-containing macromolecular complexes as large as entire ribosome (review ref. Citation12).
Simplifications Make the Modeling Faster, but There is a Cost Associated with Reduced Accuracy
Full-atomic resolution models represented in Cartesian coordinates utilize three degrees of freedom (x, y, z) for each atom; therefore, even for small molecules the conformational space becomes enormous and equals 3 * number of atoms - 6 (we typically ignore six degrees of freedom required to describe translation and rotation of the entire molecule). Such a huge space to search for correct conformations is very impractical in terms of computational requirements to perform a simulation. One way to reduce the number of degrees of freedom is to restrict bond lengths and/or angles to idealized values to reduce approximately 10-fold the number of adjustable parameters that characterize a model. For instance, in torsion angle dynamics, torsion angles are used instead of Cartesian coordinates as degrees of freedom, and the only degrees of freedom are rotations about single bonds. The molecule may be also transformed into an internal coordinate system. RNA can be thus represented as a tree structure consisting of n+1 atoms connected by n rotatable bonds of fixed length. This approach has been implemented e.g., in program DYANA,Citation13 used for RNA structure determination based on nucleic magnetic resonance (NMR) data.
Further simplification can be achieved by coarse-graining. The coarse-grained representation replaces an atomistic description of a molecular system with a low-level model. Groups of atoms may be treated as single interaction centers or “pseudoatoms,” so that a smaller number of elements and interactions need to be considered (review ref. Citation14). The simplification can range by defining interaction centers at different levels of detail—from several interaction centers per nucleotide to a single pseudoatom per helix; such approach has been used e.g., for the refinement of low-resolution structures of rRNAs with restraints from experimental data.Citation15 Examples of modern methods for RNA 3D structure prediction that utilize coarse-graining include NASTCitation16 that represents RNA by just one pseudoatom per nucleotide residue, VfoldCitation17 and DMDCitation18 that represent RNA by three pseudoatoms per residue, and SimRNA that uses five interactions centers per residue.Citation19
Force fields derived for coarse-grained systems typically yield a much smoother energy surface than those used for all-atom systems. As a result, many local minima are removed, thus reducing the probability that a molecule is trapped in a suboptimal energy state during the simulation. However, it must be emphasized that simplifications of the model representation and the energy function enhances the modeling speed usually at the cost of accuracy of the structures obtained. Thus, it is not practical to expect that a folding simulation with a coarse-grained representation would confidently predict a native-like RNA structure with a precisely estimated energy. On the other hand, the use of such simplified methods may be the only practical way to computationally fold a structure that is too complex for methods utilizing a full-atom representation and a physical potential that is more expensive to calculate.
Knowledge-Based Modeling Can Utilize the Principles of Statistical Thermodynamics
The increasing pace of macromolecular structure determination by X-ray crystallography and NMR has led to the accumulation of sizeable data sets describing protein, and more recently, also RNA structures. The availability of these data has in turn enabled the development of methods for macromolecular structure prediction that are not based on first principles, but extensively use the knowledge of “what the structures should look like.” The “Babylonian science” approach that exploits databases to derive force fields for structure prediction has a long tradition. The derivation of statistical potentials was reported for proteins as early as in the 1970s,Citation20 and more recently, also for nucleic acids. The so-called mean force potentials are compiled by using the Boltzmann's principle to approximate the distribution of different energy states by extracting relative frequencies of these states from a database. The definition of a database-derived mean force potentials introduced by SipplCitation21 is E(r) = -kT ln[p(r)], where r is a particular parameter describing a feature of a molecular system (such as the distance between two atoms of type A and B, a value of a dihedral angle etc.), E(r) is the energy at r, p(r) is the probability at r, k is Boltzmann's constant, and T is the absolute temperature. Mean force potentials can take into account all forces acting between atoms of the molecule under study as well as the influence of the environment, without the need of defining each type of interactions separately.
Knowledge-Based Modeling Can Also Rely on Principles of Molecular Evolution
A completely different approach to molecular modeling, developed initially for protein structure prediction, attempts to model not the physical process of macromolecular structure formation, but a different one: how do the structures of macromolecules change in the course of the evolution, in response to mutations accumulated in the sequence of genes. This type of structure prediction relies on empirical observation, made initially for proteins, that evolutionarily related (homologous) molecules usually retain the same three-dimensional structure despite the accumulation of divergent mutations.Citation22 It was also found that structural divergence is much slower than sequence divergence, although these two features are strongly correlated. Comparative analyzes of evolutionarily related structured RNAs (e.g., ref. Citation23), revealed patterns of conservation that are analogous to those observed in proteins.
Thus, methods have been developed to build models for molecules with unknown structure based on experimentally determined structures of molecules that are expected to be related by homology. For this, the sequence of a “target” molecule to be modeled must be aligned to the sequence of a “template” molecule with known structure (to define the correspondence between homologous residues), and then the sequence of the template is replaced with the sequence of the target in the context of the target structure, while attempting to reduce any physical conflicts caused by replacement, removal, and addition of atoms and residues (review ref. Citation5). This approach is often referred to as “comparative modeling,” “homology modeling,” or “template-based modeling.”
Two major approaches have been developed for template-based modeling, initially for proteins and then for RNA (review ref. Citation5). One is to model the structure by copying the coordinates of the template, followed by introduction of insertions/deletions using structural fragments from a database, and finally by “mutating” the residues that differ between the template and the target. This approach has been implemented in a comparative modeling method ModeRNACitation24 developed in the authors’ group. A highlight of ModeRNA is that it can model not only RNAs composed of the four canonical residues, but can also handle post-transcriptional modifications. Another approach utilizes distance and torsion angles and interatomic distances from the aligned regions of the template structure as restraints for the modeling of the target sequence. This strategy has been implemented in a program initially named RNABuilder and renamed as MacroMoleculeBuilder (MMB) as it also enables modeling of proteins.Citation25 A highlight of this approach is that it can more easily incorporate additional restraints defined by a user.
Modeling by Fragment Assembly
Another “Babylonian science” approach that is intermediate between template-based and coarse-grained modeling involves the assembly of macromolecular structures from fragments. The building blocks may comprise short sequence stretches, local structural motifs, isolated elements of secondary structure (e.g., helices, loops, junctions), or even entire domains, and may be taken from experimentally determined structures or modeled with any other approach. The fragment selection and assembly can be either fully automated or left to the user. The scoring functions used can utilize physical or statistical potentials or the combinations of both. For RNA, the paucity of RNA structures hampered the development of statistics-based approaches. Consequently, the RNA modeling field has been initially driven by manual (user-guided) modeling methods that could utilize fragments from the few structures available, as well as custom-made idealized structural elements, with the assembly assisted by the use of simplified physics-based energy functions. Examples of methods that allow the expert user to rearrange and recombine multiple RNA structures include S2S/Assemble,Citation26 ERNA-3D,Citation27 and RNA2D3D.Citation28
Only recently, the increased size of the RNA structure database prompted the development of fully automated fragment-assembly methods that have the power to explore much larger space of conformations than possible for any expert structural biologist working with a graphical user interface. These include FARNA/FARFAR,Citation29,Citation30 which is essentially an adaptation of the ROSETTA method, initially developed for protein folding, so it could be used also for RNA. The FARNA procedure assembles an RNA 3D structure from short linear fragments, using a knowledge-based energy function derived from experimentally determined RNA structures. FARFAR uses a full-atom refinement with a hybrid physics- and statistics-based scoring function to optimize the models generated by FARNA. MC-Fold|MC-SymCitation31 is another method, developed specifically for RNA, which assembles RNA structures from a library of “nucleotide cyclic motifs,” i.e., fragments in which all nucleotides are circularly connected by covalent, pairing, or stacking interactions. MC-Fold|MC-Sym implements a hybrid scoring function that includes physical terms as well as statistics derived from experimentally determined structures. RNA ComposerCitation32 is a recently developed method that assembles structures from tertiary motifs taken from the RNA FRABASE database; it has a hybrid scoring function that can be supplemented by user-defined restraints.
Template-based and fragment-based methods share a major limitation in that they can generate conformations only within the space defined by a database of templates/motifs. Hence, they are not appropriate for the modeling of novel 3D motifs. Moreover, they cannot be used in a straightforward manner for the simulation and analysis of RNA folding, only for the determination of final, defined structures.
Modeling by Base Pairing Prediction
One of the most coarse-grained approaches is to predict the pattern of base-paired nucleotide residues of an RNA molecule. This was first attempted in the early 1960s.Citation33,Citation34 There are a variety of computational techniques that have been introduced over the last five decades to predict the base-pairing pattern computationally. One general approach relies on ranking the base pairing using thermodynamic weights or some form of stochastic context-free grammar (SCFG).Citation35 The earliest approaches use the laws of thermodynamics and aim to compute a structure of minimum free-energy;Citation36 examples of well-known and commonly used programs are MFold,Citation37 Vienna RNA,Citation38 and RNAstructure.Citation39 More recent attempts have explored SCFG methods such as SfoldCitation40 and base pairing probability strategies like Centroidfold.Citation41 Another general approach relies on molecular evolutionary analysis wherein the method looks for correlated substitutions in sets of aligned homologous sequences and weights these with the above thermodynamic method or a SCFG method. Examples of thermodynamic weighting can be found in DYNALIGNCitation42 and PETfold.Citation43 Examples of SCFG methods include pfold.Citation44 Examples of base pairing probability methods include CentroidAlifoldCitation45 and MXScarna.Citation46 It is worth emphasizing that base-pairing may define not only secondary structure in the simplest sense, but also tertiary interactions such as pseudoknots and kissing loops. While many of the above-mentioned programs ignore such interactions for the sake of computational speed, there are algorithms that specialize in predictions for potentially pseudoknotted structures, such as IPknot.Citation47
Our group has recently published a large-scale test of base-pairing prediction methods that compares their accuracy for different types of RNAs, when used with sequence information alone, i.e., without experimental data.Citation48 According to our benchmarks, the combination of sequence alignment with structure prediction is the most reliable approach, while methods based on unguided thermodynamic search only work well for short sequences. A weakness in all these strategies is that they only predict the base pairing of the sequence and provide little information about the 3D structural arrangement. They are however comparatively very fast in generating the base pairing information, and such information can be employed to aid 3D structure prediction programs.Citation49
Restraints Used for RNA Secondary and Tertiary Structure Prediction
Computational methods for RNA 3D structure modeling can yield accurate and reliable models for small RNA molecules that are topologically simple, and for RNAs that have closely related homologs of known structure. However, it is still not feasible to predict theoretically structures of large RNAs with complex topologies (e.g., with long-range tertiary interactions and pseudoknots) that do not have related structures in the database. Modeling of conformational changes and interactions of RNA with other molecules (e.g., with proteins) is also extremely difficult. The major bottlenecks are the vast conformational space, which grows exponentially with the RNA size, and inaccuracies of the force fields. These problems can be partially overcome by incorporation of experimental restraints, which can be used either explicitly in the modeling process or for scoring and ranking of alternative models generated by the program used. In the simplest form, modeling assisted by the use of experimental data involves selection of a model that agrees with additional information that is not used in the very modeling process. In the more advanced form, information derived from experimental studies can be included as additional terms of the scoring function and/or by modifications of the conformational sampling scheme. The conditions concerning the satisfaction of restraints can be expressed in a binary (e.g., Boolean) or probabilistic manner.
The restraints can describe various structural parameters of an RNA molecule (), such as pairing of nucleotide residues (secondary structure), solvent accessibility, interatomic distances (e.g., tertiary contacts), and a 3D shape of entire molecule. In each of these categories restraints can be obtained based on the results of various biophysical and biochemical experiments, and in many cases they can also be predicted from additional computational analyzes, or both. For the single nucleotide resolution experiments, a terminology of techniques used is captured in an ontology framework.Citation50 Here, we will discuss examples of data that have been successfully used to aid computational modeling of RNA structures.
Secondary Structure and Local Flexibility
Experimental techniques for RNA secondary structure determination typically utilize chemical or enzymatic probing, and can be used either in vitro or in vivo. A whole palette of chemicals and enzymes with an established relationship between RNA structural feature and cleavage/modification activity exist and can be used to obtain a broad picture of structural features and their precise localization in selected RNA molecules (review ref. Citation51). The main principle is that chemical reagents and nucleases used for this type of analysis interact differentially with paired and unpaired nucleotide residues, e.g., nuclease V1 is reactive toward residues in double-stranded RNA, and RNase S1 is reactive toward single-stranded regions. The use of base-selective chemical reagents (DMS, kethoxal, CMCT) provides structural information about the base stacking, hydrogen bonding, and electrostatic environment adjacent to the base. Local nucleotide flexibility and dynamics can be inferred from experiments that interrogate all four RNA nucleotides. For instance, selective 2'-hydroxyl acylation analyzed by primer extension (SHAPE) technique uses hydroxyl-selective electrophiles that react with the 2′-hydroxyl group at flexible or disordered nucleotides.Citation52 The in-line probing method does not require the use of any chemicals but exploits the natural instability of RNA molecules. The RNA is incubated at slightly alkaline pH, and the spontaneous cleavage of the sugar backbone by adjacent 2′-hydroxyl groups, which reflects the local nucleotide flexibility, is monitored.Citation53
Although there is a clear correlation between the local reactivities of RNA molecules and base pairing probabilities, the problem of how to incorporate the probing data into computational modeling procedure is not straightforward. The difficulty originates from the fact that reactivities depend on the structural context and are influenced by tertiary contacts.Citation54 Thus, computational methods have been adapted to allow transforming the reactivities to discrete states (paired or unpaired), or calibrating the interaction energy term proportionally to the reactivities.Citation55,Citation56
Solvent Accessibility
Hydroxyl radical probing (HRP) is yet another chemical probing method useful for RNA structure determination. Hydroxyl radicals attack the ribose ring of RNA, which results in breaking of the sugar-phosphate backbone. Nucleotides protected against breaking are considered solvent inaccessible and are buried within the core of a RNA molecule. After an RT-PCR experiment, the cleavage pattern observed in gel electrophoresis indicates exposed nucleotides, and moreover, the strength of protection can be also devised. Solvent exposed nucleotides have high HRP reactivity, and respectively, buried nucleotides show low HRP reactivity. Importantly, the method does not identify nucleotides that are base-paired, instead, it detects nucleotides that hydroxyl radicals cannot access, and in fact, HRP reactivity is correlated with backbone solvent accessibility.Citation57 Data derived from HRP experiments can be used to evaluate or filter models;Citation16 however, recently it has been shown that the method can be applied even to drive in a systematic and quantitative way, molecular dynamics simulations within the DMD program.Citation58
Non-Local Interactions
An interesting addition to the panel of methods for experimental probing of RNA structure is a recently developed “mutate-and-map” strategy.Citation59 It is based on the observation that when a paired nucleotide is mutated, its partner becomes more accessible to reagent, which can be readily detected by subsequent chemical probing (e.g., by SHAPE). Importantly, this strategy can reveal not only pairings in secondary structure, but also tertiary contacts between sequentially distant fragments of the molecule.
Multiplexed hydroxyl radical cleavage analysis (MOHCA) is another technique that provides information about long-range contacts. There, RNAs are created with randomly incorporated nucleotides tethered to a Fe(II)–EDTA moiety, which can be used to induce through-space cleavage of nearby residues in the RNA. Sites of that cleavage and the location of the probe nucleotide can be identified by two-dimensional gel electrophoresis.Citation60
Long-range restraints are important for the modeling process, as even a small number of them are sufficient to reduce the conformational space sufficiently to allow accurate prediction of native RNA structures.Citation61 Experimental methods that are used to probe long-range contacts include UV- or chemically induced cross-linking, site-directed cleavage, fluorescence resonance energy transfer (FRET), electron spin resonance (ESR/EPR), nuclear Overhauser effect spectroscopy (NOESY-NMR). Some of these methods (FRET, ESR, NMR) cannot only give information about residue–residue contacts, but also high-quality distance information. NMR provides large number of distances between hydrogen atoms, together with large amount of other structural data, which for small RNA structures is enough to solve the 3D structure, and as such, NMR is beyond the scope of this review.
Through use of bifunctional cross-linking reagents or a group that is tethered to one part of an RNA molecule and able to react with a second region, it is possible to use chemical mapping methods to obtain very useful long-range restraints for RNA 3D structure modeling. The residues that are involved in chemical cross-links can be detected by mass spectrometry. This approach is known as MS3D, and involves the MS analysis of cross-linked oligonucleotides obtained from fragmentation (e.g., by RNases) of a chemically cross-linked RNA molecule. A major challenge of this approach is the identification of the cross-linked oligonucleotides in a composite mixture, as well as the identification of the residues involved in the cross-link. MS3D has been applied to elucidate e.g., the structure of HIV-1 Psi-RNA.Citation62 There exist different classes of bifunctional chemical reagents that are reactive toward nucleic acid substrates.Citation63
FRET is one of the most used and most versatile techniques of fluorescence spectroscopy.Citation64 For RNA structure determination it allows for measurement of inter-residue distances in the range of 20–100Å. FRET is based on the observation that if two fluorophores are in proximity the excitation can be non-radiatively transferred from the initially excited one (donor dye) to the one which initially was in its electronic ground state (acceptor dye). The radiation emitted from donor and acceptor fluorophores can be distinguished based on the wavelength if these dyes have non-overlapping emission spectra. In contrast to most methods of RNA structural probing of RNA that return values averaged over ensembles of molecules, FRET can also be measured for single molecules. In order to be used for RNA structure determination, FRET requires chemical modification of RNA molecules by the introduction of fluorescent residues.Citation65 These residues can be connected to the RNA molecules using flexible linkers, or can be themselves analogs of nucleotide bases.
The structure reconstruction from the FRET data obtained using freely moving dyes tethered to the RNA is quite complex computationally, because of different influence of environment on the dynamics of the dye. This procedure can be simplified if the accessible volumes for the dyes are constrained by RNA structural motifs of known structure (e.g., RNA B-helices). In such case, the accessible volumes can be precomputed and FRET observables can be used during modeling as distance restraints. Such procedure has been used to study the structure of e.g., RNA three-way junction.Citation65 An alternative strategy in using FRET measurements for RNA structure determination is to use fluorescent dyes which are analogs of the nucleotide bases. Several such fluorophores are even commercially available in the form or nucleotide precursors for phosphoramidite oligonucleotide synthesis. Theoretically, for a sufficiently large number of RNA variants, labeled at various helical regions of the RNA structure, the mutual positions and orientations of helices can be determined based on FRET data alone.Citation66 However, the disadvantage of the currently available fluorescent nucleotide base analogs is their low quantum yield, which makes single molecule measurements relatively impractical.
ESR spectroscopy is another method based on different physical mechanism than FRET, which nonetheless provides similar data and suffers from similar difficulties. It is based on measurement of direct spin–spin couplings between localized unpaired electrons within the molecule or molecular complex. The experimental setup for ESR is similar to one used for NMR, with most differences resulting from magnetic moment of the electron being thousands times greater than nuclear magnetic moments. To be studied with this technique selected residues within RNA need to be chemically labeled using stable free-radicals (most commonly nitroxide radicals).Citation67 The spin–spin coupling does depend on both the distance between unpaired electrons and on the angle between the position vector and the external magnetic field. This angle can be determined from the ESR experiment in a model independent way. In contrast to FRET measurements, the spins of the electrons are aligned to the magnetic field, unlike the transition moments of the dyes, which are freely rotating. Also, the paramagnetic spin-labels used in ESR are typically much smaller than the fluorescent dyes used in FRET, which does reduce the error introduced by the finite size of these probes.Citation68 For RNA modeling, ESR is able to provide reliably measured distances of 5–20 Å (5–20 Å with continuous wave setup and 20–80 Å with pulsed ESR) with uncertainty of a few Ångströms.Citation69 In contrast to most other methods of RNA structure determination, with ESR it is possible to elucidate the positions of metal-ions within RNA molecules (for paramagnetic ions like Mn2+).
RNA Molecule Shape
Another source of restraints for the RNA structure modeling is small angle scattering (SAS) that can utilize either X-ray (SAXS) or neutron (SANS) radiation. Classically, SAS has been used to determine the radius of gyration and maximum dimension of the scattering particles. Currently, there are many algorithms available that enable ab initio reconstruction of the scattering particle envelope at low-resolution, e.g., DAMMIN/FCitation70). The envelope may be consequently used to aid building a full-atom model of the macromolecule by docking, e.g., as it was done for the determination of a structure of T box RNA complexed with tRNA.Citation71 It must be borne in mind, however, that SAS is a low information content technique and special care must be taken to avoid over-parameterization.Citation72
The global shape of large RNA molecules and RNA–protein complexes can also be directly observed using the electron microscopy (EM). The measurement is done in cryogenic conditions (cryo-EM) in order to reduce the radiation damage of macromolecules under study.Citation73 The three-dimensional structure reconstruction is done based on large number of collected two-dimensional projections. While cryo-EM data has rarely been used to restrain the modeling of RNA structures in isolation, it has been instrumental for the modeling of RNA structures in the context of many important RNA–protein complexes, e.g., the modeling of tmRNA structure bound to the ribosome with the use of MMB and ModeRNA methods mentioned earlier in this article.Citation74
RNA 3D Structure Modeling with Restraints: Case Studies
Since 1969, when the first manually predicted tertiary structure of tRNA was published,Citation75 the RNA structure modeling procedures have evolved considerably and a variety of programs that enable systematic and quantitative restraints-guided predictions became available. In this paragraph, we provide a subjective selection of a few examples from the history of RNA modeling.
Manual building of RNA models was an excellent and intuitive approach for putting together different types of restraints, and allowed for expressing the researcher’s “feel” about a given RNA fold. Levitt manually assembled from space-filling components the first detailed model of tRNA in 1969.Citation75 Similar to the contemporary models, this model had to satisfy many constraints: stereochemical (sufficiently large distances between atoms, realistic dihedral angles about single bonds), topological (proximity between the loops deduced from chemical reactions), energetic (energy minimization with a force field), regarding shape (radius of gyration), and local stability (base stacking). Compiled with the data on the base-paired nucleotides, and revealed by the analysis of correlated substitutions in homologous sequences, these constraints appeared to be sufficient to obtain the correct layout of the helices in tRNA molecule. Models resulting from subsequent studies, including topologically complex 16S rRNA,Citation76 established the concept of using the experimental restraints in the RNA modeling process.
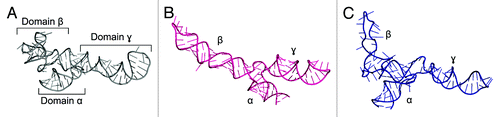
The growing repository of X-ray structures triggered the development of methods based on fragment assembly. In the technique introduced by Westhof and co-workers, structural motifs are either taken from the database or created based on established stereochemical rules, bases are substituted according to the desired sequence, fragments are joined together, and finally the obtained model is subjected to stereochemical and energetic refinement.Citation77 This procedure was integrated with experimental data during the modeling of 5S rRNA from Escherichia coliCitation78 (). Moreover, the secondary structure of 5S rRNA was probed in solution using various chemical and enzymatic probes, which identified the presence of three branches. The model was predicted not to contain any long-range interactions between the different domains, and it displays a different fold from the one seen in the crystal structure of an E. coli ribosome (PDB ID: 3OAS). Another atomic model of 5S rRNA structure was modeled interactively with ERNA-3D, based on a low-resolution cryo-EM reconstruction of the 50S ribosomal subunit and cross-linking data.Citation79 Despite the correct arrangement of the three domains, the model did not predict a correct fold of the second domain. However, much can be learned by comparing the results of these first efforts to model 5S rRNA with the crystal structure solved at atomic resolution.Citation80
Figure 1. Crystal structure of Escherichia coli 5S rRNA (PDB ID: 3OAS) (A) and computational models predicted with the fragment assembly approach based on structural probingCitation78 (B) and manual modeling based on cryo-EM data of the 50S subunitCitation79 (C).
Even though the approaches described above had a considerable history of success, an introduction of physics- and statistics-based potential functions enabled studying RNA folding in a systematic and physics-consistent way. As a “proof-of-concept” for molecular mechanics of RNA, correct tRNA architecture was predicted using a simplified potential function with consciously parametrized terms.Citation81 The potential comprised only harmonic-type functions, which are easy to minimize and have a unique minimum. In the experiment cross-linking and probing data were represented as distance restraints with force constants reflecting the precision of the data. The simple framework, designed specifically for large RNA molecules, opened up a new generation of programs based on potential functions.
Despite the undeniable progress in experimental techniques for RNA 3D structure determination, they are still not well suited for fully automated determination of large RNA molecules that are often highly flexible and dynamically change the structure in physiological conditions. Recent modeling of the 3D structure of the 240 nt long HIV-1 5′UTR—packaging signal (Psi) regionCitation82—provides a good example how these difficulties can be overcome by performing additional experiments, which explore secondary structure and long-distance contacts, and thus, yield geometric restraints for computer simulations. Previously, only a secondary structure model was available, composed of four hairpins connected by short linkers.Citation83 The model building was divided into five steps. In the first stage, secondary structure was predicted with the RNAstructure program supplied with SHAPE reactivities as pseudo-free-energy restraints. Then, a crude 3D representation of the secondary structure model was generated with the software RNA2D3D.Citation28 However, the model was not assumed to be close to the optimum in terms of conformational space. To increase the searchable structure space, 10 different models were generated after applying randomized sets of pseudo-experimental FRET restraints. Each of the models was subsequently subjected to global energy minimization using a simulated annealing procedure in the CNS program.Citation84 During the simulations, experimentally determined FRET distances and SHAPE-derived base pairing data were expressed as distance restraints represented by harmonic-type function terms added to the total energy function. Because CNS was not specifically designed to deal with RNA structures, additional restraints had to be incorporated on angles and distances of the RNA backbone to maintain regular double helical conformations. Next, the resulting models were scored with the RMSD function with FRET distances (FRET-RMSD). Finally, room-temperature MD simulations were performed to relax the structures and the best model in terms of FRET-RMSD and total minimized energy was selected. Importantly, the model was validated by comparison to the previously solved structures of three individual hairpins of the Psi region, which were not used in the modeling process.
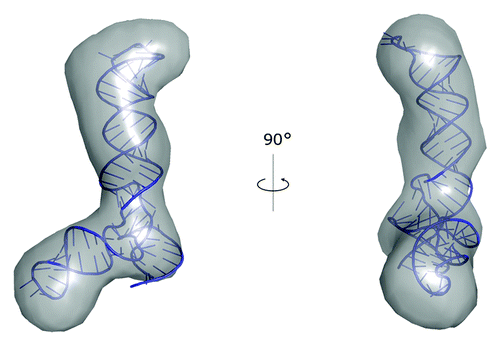
Recently, we used our software PyRy3D (http://genesilico.pl/pyry3d/) in the study of the adenovirus virus-associated RNA lacking the terminal stem (VAIΔTS).Citation85 A 3D structure model was built based on a set of ab initio SAXS reconstructions determined using DAMMIN and averaged using DAMAVER.Citation86 The averaged ab initio reconstruction was converted into a pseudo EM-map with sfall and fft programs from the CCP4 suite.Citation87 The initial atomic model of RNA was obtained using our coarse-grained method SimRNA, with restraints on experimentally determined secondary structure, but without the use of SAXS data. Subsequently, the RNA model was divided into hairpin-loop segments and a three-way junction, and fitted into the pseudo-EM map using PyRy3D, with restraints at the junction to maintain the continuity of the nucleotide chain. The final model was obtained following a refinement with phenix.refine,Citation88 with restrains on the pseudo-density map (). The radius of gyration (Rg) and maximum spatial dimension (Dmax) values obtained from the model of VAI∆TS are 3.2 nm and 11.2 nm, respectively. Both values are in agreement with those obtained directly from the SAXS analysis. Furthermore, the chi-square value calculated with CRYSOLCitation89 was found to be 1.03, indicating that the model explains well the experimental data.
Figure 2. A model of VAI∆TS RNA structure obtained with SimRNA and built into the SAXS reconstruction using PyRy3D.Citation85
Benchmarking of RNA Modeling Programs Used with Experimental Restraints
Since the construction of the popular methods for automatic 3D structure modeling is mainly a heuristic procedure, the best way to prove their usefulness is to test how the predicted models are similar to the native structures. However, there are no systematic studies comparing performance of all these methods used with various combinations of experimental restraints, although several of them have been used extensively in modeling of various targets.
In 2010, RNA Puzzles was launched as the first blind experiment for RNA structure prediction assessment.Citation90 In most of the puzzles, additional information was provided that could be encoded in various restraints. The level of automation in RNA modeling vs. human intervention varied from group to group, thus RNA Puzzles serves as a benchmark for comparison of entire workflows rather than individual computational programs. Results published thus far for three puzzles suggest that predictions are improved owing to careful investigation of local structural motifs, inclusion of as much experimental restraints as possible, and global fold refinement, but they also inform us that it is important to avoid over-constraining the simulations.
Relative performance of several programs for 3D structure prediction in making use of secondary structure information was also assessed in two benchmarks.Citation91,Citation92 These studies were focused on a comparison of four programs for automatic RNA 3D structure modeling used with restraints on the secondary structure. FARNA and MC-Fold|MC-Sym were selected as the most established fragment-based approaches, while NAST and DMD—as the most popular programs with knowledge-based energy functions to sample coarse grained RNA structures. In the most comprehensive benchmarks published to date,Citation91 the test set consisted of 43 different RNA structures of various lengths and motifs, including hairpins, junctions, and pseudoknots. The prediction performance was evaluated with different measures, taking into account global geometry (i.e., RMSD over backbone atoms) and correctly predicted base pair and base stacking interactions (i.e., as sensitivity and specificity). Although the accuracy of each program varied from structure to structure, certain trends were clearly visible. Most importantly, secondary structure information improved most of the predictions done with FARNA and MC-Fold|MC-Sym. Although MC-Fold|MC-Sym generated the most accurate structures in most cases, it did not generate any folded structure for some sequences, especially for larger molecules, and was rarely able to predict non-canonical base pairs. The best RMSD for hairpins and junctions were 9.5 Å and 18.5 Å, respectively. However, topologically complex structures were not correctly predicted by any program even with restraints on the secondary structure.
Selection of Computational Programs for RNA Structure Modeling with Experimental Data
Confronted with a plethora of computational approaches and a variety of experimental techniques as explained above, it is understandably confusing and difficult for a researcher to decide which techniques to use to address any specific biological problem. In , we present a flowchart to help aid in deciding when (or where) to apply a given computational method and how to integrate the experimental data in such way as to make effective use of these tools.
Figure 3. A flowchart describing relations between different types of data, computer programs, and RNA 3D structure modeling strategies.
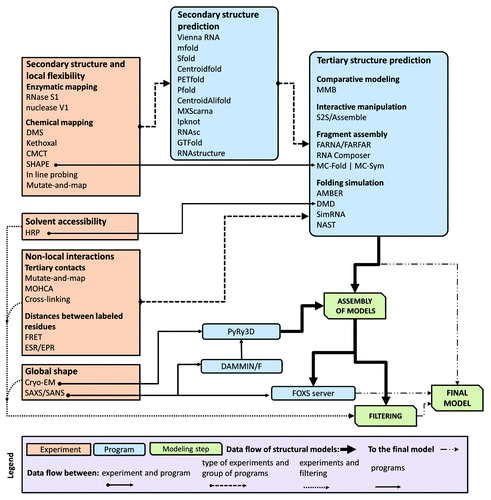
RNA secondary structure is the most common type of restraints used in modeling of RNA 3D structures and can be utilized by all computational methods for the tertiary structure prediction described in this work. The simplest way to obtain these restraints is to use computational methods for secondary structure prediction. However, for long RNAs these computational prediction strategies may yield rather low accuracy predictions. Therefore, we highly recommend the incorporation of experimental data into these methods to improve the reliability of the predictions. For example, secondary structure prediction can be restrained using SHAPE mapping data to improve the accuracy of structure prediction.Citation56 Data from SHAPE experiments can be incorporated directly into the prediction using RNAstructure,Citation93 GTFold,Citation94 RNAsc,Citation95 and MC-Fold|MC-Sym. Chemical probing with RNase S1, Nuclease V1, DMS, etc. can often be incorporated into RNA secondary structure prediction programs and these results then incorporated into 3D structure prediction ().
Hydroxyl radical probing (HRP) is another technique that can provide valuable restraints to describe the approximate solvent accessibility for ribonucleotide residues. Thus far, only the DMD method has been adapted to utilize HRP data directly in RNA folding simulations.Citation58 However, results of HRP experiments can be transformed into information about solvent accessibility and used to identify “good” structures among a set of models generated by essentially any modeling method. For discrimination of models that agree with experimental data we can recommend e.g., the FILTREST3D method developed in our group and available as a standalone program as well as a web server.Citation96
The second most common group of restraints used for RNA 3D modeling are ones encoded as pairwise distance ranges. These types of restraints can be obtained from experimental techniques for detecting long-range interactions between different residues or structural segments, and can be divided into two groups. The first group includes experimental techniques, such as cross-linking, mutate-and-map, and MOHCA. They detect spatial proximity of ribonucleotide residues and depending on the method they may identify residues that are in direct contact or up to 20 Å from each other. Experimental techniques such as FRET and EPR belong to the second group. They provide important information about distance range between specific labeled residues. The distances can range between 20 and 100 Å for FRET and between 5 and 80 Å for EPR. A list of various residues and pairwise distance ranges between them can be utilized by most of computational methods for RNA 3D structure prediction listed in . It is important to mention that a distance range between just one pair of residues is a relatively weak restraint, and usually a large set of such restraints is required to guide the prediction, in the order of a number of ribonucleotide residues in the modeled RNA molecule divided by 5. Pairwise distance restraints can be also easily combined with secondary structure restraints.
The last type of restraints involves probing the actual shape of the RNA molecule. The physical shape of RNA molecules can be inferred based on results from small angle scattering (SAS) or cryo-EM experiments. SAS data can be used for ranking the model structures within large data sets to identify structures that best explain the observed dataCitation97 using for example the FOXS server.Citation98 However, the direct comparison of the SAS and model-derived scattering curves is computationally expensive. Alternatively, the models may be also ranked against SAS data with respect to the distributions of pairwise distances.Citation16 Data from cryo-EM can be utilized to obtain a low-resolution shape envelope. SAS data can be also used with DAMMIN/F to obtain ab initio shape reconstructions. Various modeling programs can be then used to perform fitting of pre-modeled RNA 3D structures into these shapes, and best-fitting structures may be selected as the most promising models.
One of computational problems that plagues RNA 3D structure prediction is the large number of local minima of the scoring function, which need to be sampled. This problem is even more pronounced when restraints are used. Many of the new minima are formed in this case, and some of them are genuine topological traps. These conflicts cannot be resolved without the unfolding of large parts of the structure or allowing for unphysical tunneling of RNA chains through themselves. This problem can be addressed by using different weight of structural restraints during different stages of the modeling. Initially, low weights should be used, so that the RNA does not get trapped in any of any spurious minima. Once enough models have been generated that approximately satisfy the imposed restraints, these models can be selected and used as starting structures for the next stage of the modeling procedure with increased weight on the restraints. An alternative approach, implemented in PyRy3D (http://genesilico.pl/pyry3d/), is by gradually switching various restraints on during the simulation. A similar technique is used in the MMB (Macro Molecule Builder)Citation25 program, which allows for alternating periods of simulation with restraints switched completely on or completely off, but this approach has not been used for de-novo modeling of large RNA structures. The danger of being trapped in local minima of incorrect topology can be avoided by using whatever available prior knowledge exists about the RNA structure in the construction of a suitable initial structure for the modeling.
Future Prospects
It this review, we described main advancements in computational modeling of RNA, with the emphasis on methods that can take into account experimental data. Currently existing computational methods are successful in “purely theoretical” structure prediction only for relatively small molecules. For example, methods such as DMD, FARNA, SimRNA, and V-fold can be used with relatively high confidence to obtain models of RNA molecules shorter than ~40 nt, and with moderate confidence for molecules up to ~80 nt. Folding of larger molecules is possible, but average reliability of such predictions drops down dramatically with increasing length. The upper limit of RNA sequence size foldable by computational methods is not only related to the increased simulation time, but also to accuracy of the scoring functions used. Prediction of compact conformations of very large molecules requires detection of stable long-range contacts that knit together complete domains. These, however, may be difficult to recognize for standard scoring functions that rely on the mutual positions of individual nucleotides; in particular, when sampling of the conformational space is very rough like in case of fragment assembly methods. Therefore, detection of the stabilizing tertiary interactions may require the mutual docking of super-secondary structure motifs or even complete domains. The studies on the tertiary interactions in RNA on the level of complete structural motifs are still relatively scarce,Citation99,Citation100 and we believe that further exploration of this problem may lead to significant improvement in RNA structure prediction.
Given the current limitations of computational prediction of RNA 3D structure, the inclusion of experimental restraints in the structure modeling process greatly increases the maximal length of RNA that can be modeled with confidence as well as increases the precision of the models obtained. Data from structurally informative experimental analyses can be used to reduce the conformational space that has to be sampled or to post-filter the obtained candidate models and select only those that agree with the data. Currently, the “Babylonian science” methods for RNA 3D structure determination outperform “Greek science” methods in most applications that are practically useful for molecular biologists. We are convinced that further accumulation of experimental restraints will bring a database of “bricks” for the libraries of Babel and a deeper understanding will yield the mortar of wisdom that binds them. On the other hand, the advances in computer hardware and software will hopefully improve the speed and accuracy of calculations required for the “Greek science” methods, thereby increasing the size of RNA molecules and the accuracy of predictions that can be obtained from first principles alone.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The research on the development of methods for RNA 3D structure modeling in the Bujnicki laboratory has been funded from several sources. Boniecki M and in general the development and application of RNA folding methods that use distance restraints from FRET experiments have been supported by the German Science Foundation (DFG) within SPP 1258 (grant no. SE 1195/12-2 coordinated by Claus Seidel). Chojnowski G, Łach G, Magnus M, Matelska D, and Purta E were supported by the Foundation for Polish Science (FNP, grant TEAM/2009-4/2 to Bujncki JM). Bujncki JM and Dunin-Horkawicz S were supported by the European Research Council (ERC, StG grant RNA+P = 123D to Bujncki JM). Dawson W was supported by the European Commission (EC; FP7 grant FishMed, 316125). The development of our bioinformatics servers was funded mainly by the Polish Ministry of Science and Higher Education (MNiSW, grant POIG.02.03.00-00-003/09 to Bujncki JM). Bujncki JM was additionally supported by the “Ideas for Poland” fellowship from the FNP. Dunin-Horkawicz S and Chojnowski G were additionally supported by the National Science Centre (NCN, grants 2011/03/D/NZ8/03011 to Dunin-Horkawicz S and 2011/01/D/NZ1/00212 to Chojnowski G). Dunin-Horkawicz S also acknowledges support from the Polish Ministry of Science and Higher Education (MNiSW, fellowship for outstanding young scientists).
We thank Juliusz Stasiewicz, Tomasz Waleń, Irina Tuszyńska, and Anna Philips for their participation in the development of RNA modeling methods in the Bujnicki laboratory. We also thank Eric Westhof, Jan Gorodkin, Claus Seidel, and Stanislav Kalinin for stimulating discussions and helpful advice on various occasions. Bujncki JM thanks Andrzej Sokalski for an inspirational reference to “Greek science” and “Babylonian science” in the context of chemical structures and interactions.
References
- Gesteland RF, Cech TR, Atkins JF. The RNA World. New York: Cold Spring Harbor Press 2005:768
- Serganov A, Patel DJ. Molecular recognition and function of riboswitches. Curr Opin Struct Biol 2012; 22:279 - 86; http://dx.doi.org/10.1016/j.sbi.2012.04.005; PMID: 22579413
- Leontis N, Westhof E. RNA 3D structure analysis and prediction. Berlin Heidelberg: Springer-Verlag, 2012.
- Doudna JA. Structural genomics of RNA. Nat Struct Biol 2000; 7:Suppl 954 - 6; http://dx.doi.org/10.1038/80729; PMID: 11103998
- Rother K, Rother M, Boniecki M, Puton T, Bujnicki JM. RNA and protein 3D structure modeling: similarities and differences. J Mol Model 2011; 17:2325 - 36; http://dx.doi.org/10.1007/s00894-010-0951-x; PMID: 21258831
- Anfinsen CB. Principles that govern the folding of protein chains. Science 1973; 181:223 - 30; http://dx.doi.org/10.1126/science.181.4096.223; PMID: 4124164
- Hardin C, Pogorelov TV, Luthey-Schulten Z. Ab initio protein structure prediction. Curr Opin Struct Biol 2002; 12:176 - 81; http://dx.doi.org/10.1016/S0959-440X(02)00306-8; PMID: 11959494
- Scheraga HA. Recent developments in the theory of protein folding: searching for the global energy minimum. Biophys Chem 1996; 59:329 - 39; http://dx.doi.org/10.1016/0301-4622(95)00126-3; PMID: 8672720
- Case DA, Cheatham TE 3rd, Darden T, Gohlke H, Luo R, Merz KM Jr., Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem 2005; 26:1668 - 88; http://dx.doi.org/10.1002/jcc.20290; PMID: 16200636
- Brooks BR, Brooks CL 3rd, Mackerell AD Jr., Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, et al. CHARMM: the biomolecular simulation program. J Comput Chem 2009; 30:1545 - 614; http://dx.doi.org/10.1002/jcc.21287; PMID: 19444816
- Christen M, Hünenberger PH, Bakowies D, Baron R, Bürgi R, Geerke DP, Heinz TN, Kastenholz MA, Kräutler V, Oostenbrink C, et al. The GROMOS software for biomolecular simulation: GROMOS05. J Comput Chem 2005; 26:1719 - 51; http://dx.doi.org/10.1002/jcc.20303; PMID: 16211540
- Sanbonmatsu KY, Tung CS. High performance computing in biology: multimillion atom simulations of nanoscale systems. J Struct Biol 2007; 157:470 - 80; http://dx.doi.org/10.1016/j.jsb.2006.10.023; PMID: 17187988
- Stein EG, Rice LM, Brünger AT. Torsion-angle molecular dynamics as a new efficient tool for NMR structure calculation. J Magn Reson 1997; 124:154 - 64; http://dx.doi.org/10.1006/jmre.1996.1027; PMID: 9424305
- Tozzini V. Multiscale modeling of proteins. Acc Chem Res 2009; 43:220 - 30; PMID: 19785400
- Malhotra A, Tan RK, Harvey SC. Prediction of the three-dimensional structure of Escherichia coli 30S ribosomal subunit: a molecular mechanics approach. Proc Natl Acad Sci U S A 1990; 87:1950 - 4; http://dx.doi.org/10.1073/pnas.87.5.1950; PMID: 2408047
- Jonikas MA, Radmer RJ, Laederach A, Das R, Pearlman S, Herschlag D, Altman RB. Coarse-grained modeling of large RNA molecules with knowledge-based potentials and structural filters. RNA 2009; 15:189 - 99; http://dx.doi.org/10.1261/rna.1270809; PMID: 19144906
- Cao S, Chen SJ. A new computational approach for mechanical folding kinetics of RNA hairpins. Biophys J 2009; 96:4024 - 34; http://dx.doi.org/10.1016/j.bpj.2009.02.044; PMID: 19450474
- Ding F, Sharma S, Chalasani P, Demidov VV, Broude NE, Dokholyan NV. Ab initio RNA folding by discrete molecular dynamics: from structure prediction to folding mechanisms. RNA 2008; 14:1164 - 73; http://dx.doi.org/10.1261/rna.894608; PMID: 18456842
- Rother K, Rother M, Boniecki M, Puton T, Tomala K, Lukasz P, Bujnicki JM. Template-based and template-free modeling of RNA 3D structure: Inspirations from protein structure modeling. In: Leontis NB, Westhof E, eds. RNA 3D structure analysis and prediction. Berlin: Springer-Verlag, 2012.
- Tanaka S, Scheraga HA. Medium- and long-range interaction parameters between amino acids for predicting three-dimensional structures of proteins. Macromolecules 1976; 9:945 - 50; http://dx.doi.org/10.1021/ma60054a013; PMID: 1004017
- Sippl MJ. Boltzmann’s principle, knowledge-based mean fields and protein folding. An approach to the computational determination of protein structures. J Comput Aided Mol Des 1993; 7:473 - 501; http://dx.doi.org/10.1007/BF02337562; PMID: 8229096
- Chothia C, Lesk AM. The relation between the divergence of sequence and structure in proteins. EMBO J 1986; 5:823 - 6; PMID: 3709526
- Dror O, Nussinov R, Wolfson H. ARTS: alignment of RNA tertiary structures. Bioinformatics 2005; 21:Suppl 2 ii47 - 53; http://dx.doi.org/10.1093/bioinformatics/bti1108; PMID: 16204124
- Rother M, Rother K, Puton T, Bujnicki JM. ModeRNA: a tool for comparative modeling of RNA 3D structure. Nucleic Acids Res 2011; 39:4007 - 22; http://dx.doi.org/10.1093/nar/gkq1320; PMID: 21300639
- Flores SC, Wan Y, Russell R, Altman RB. Predicting RNA structure by multiple template homology modeling. Pac Symp Biocomput 2010; 216 - 27; PMID: 19908374
- Jossinet F, Westhof E. Sequence to Structure (S2S): display, manipulate and interconnect RNA data from sequence to structure. Bioinformatics 2005; 21:3320 - 1; http://dx.doi.org/10.1093/bioinformatics/bti504; PMID: 15905274
- Zwieb C, Müller F. Three-dimensional comparative modeling of RNA. Nucleic Acids Symp Ser 1997; 69 - 71; PMID: 9478210
- Martinez HM, Maizel JV Jr., Shapiro BA. RNA2D3D: a program for generating, viewing, and comparing 3-dimensional models of RNA. J Biomol Struct Dyn 2008; 25:669 - 83; http://dx.doi.org/10.1080/07391102.2008.10531240; PMID: 18399701
- Das R, Baker D. Automated de novo prediction of native-like RNA tertiary structures. Proc Natl Acad Sci U S A 2007; 104:14664 - 9; http://dx.doi.org/10.1073/pnas.0703836104; PMID: 17726102
- Das R, Karanicolas J, Baker D. Atomic accuracy in predicting and designing noncanonical RNA structure. Nat Methods 2010; 7:291 - 4; http://dx.doi.org/10.1038/nmeth.1433; PMID: 20190761
- Parisien M, Major F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature 2008; 452:51 - 5; http://dx.doi.org/10.1038/nature06684; PMID: 18322526
- Popenda M, Szachniuk M, Antczak M, Purzycka KJ, Lukasiak P, Bartol N, Blazewicz J, Adamiak RW. Automated 3D structure composition for large RNAs. Nucleic Acids Res 2012; 40:e112; http://dx.doi.org/10.1093/nar/gks339; PMID: 22539264
- Fresco JR, Alberts BM, Doty P. Some molecular details of the secondary structure of ribonucleic acid. Nature 1960; 188:98 - 101; http://dx.doi.org/10.1038/188098a0; PMID: 13701785
- Devoe H, Tinoco I Jr.. The stability of helical polynucleotides: base contributions. J Mol Biol 1962; 4:500 - 17; http://dx.doi.org/10.1016/S0022-2836(62)80105-3; PMID: 13885894
- Rivas E, Eddy SR. The language of RNA: a formal grammar that includes pseudoknots. Bioinformatics 2000; 16:334 - 40; http://dx.doi.org/10.1093/bioinformatics/16.4.334; PMID: 10869031
- Tinoco I Jr., Uhlenbeck OC, Levine MD. Estimation of secondary structure in ribonucleic acids. Nature 1971; 230:362 - 7; http://dx.doi.org/10.1038/230362a0; PMID: 4927725
- Zuker M, Stiegler P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res 1981; 9:133 - 48; http://dx.doi.org/10.1093/nar/9.1.133; PMID: 6163133
- Lorenz R, Bernhart SH, Höner Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL. ViennaRNA Package 2.0. Algorithms Mol Biol 2011; 6:26; http://dx.doi.org/10.1186/1748-7188-6-26; PMID: 22115189
- Mathews DH. RNA secondary structure analysis using RNAstructure. Curr Protoc Bioinformatics 2006; Chapter 12:Unit 12 6.
- Ding Y, Lawrence CE. A statistical sampling algorithm for RNA secondary structure prediction. Nucleic Acids Res 2003; 31:7280 - 301; http://dx.doi.org/10.1093/nar/gkg938; PMID: 14654704
- Hamada M, Kiryu H, Sato K, Mituyama T, Asai K. Prediction of RNA secondary structure using generalized centroid estimators. Bioinformatics 2009; 25:465 - 73; http://dx.doi.org/10.1093/bioinformatics/btn601; PMID: 19095700
- Mathews DH, Turner DH. Dynalign: an algorithm for finding the secondary structure common to two RNA sequences. J Mol Biol 2002; 317:191 - 203; http://dx.doi.org/10.1006/jmbi.2001.5351; PMID: 11902836
- Seemann SE, Gorodkin J, Backofen R. Unifying evolutionary and thermodynamic information for RNA folding of multiple alignments. Nucleic Acids Res 2008; 36:6355 - 62; http://dx.doi.org/10.1093/nar/gkn544; PMID: 18836192
- Knudsen B, Hein J. RNA secondary structure prediction using stochastic context-free grammars and evolutionary history. Bioinformatics 1999; 15:446 - 54; http://dx.doi.org/10.1093/bioinformatics/15.6.446; PMID: 10383470
- Hamada M, Sato K, Asai K. Improving the accuracy of predicting secondary structure for aligned RNA sequences. Nucleic Acids Res 2011; 39:393 - 402; http://dx.doi.org/10.1093/nar/gkq792; PMID: 20843778
- Tabei Y, Kiryu H, Kin T, Asai K. A fast structural multiple alignment method for long RNA sequences. BMC Bioinformatics 2008; 9:33; http://dx.doi.org/10.1186/1471-2105-9-33; PMID: 18215258
- Sato K, Kato Y, Hamada M, Akutsu T, Asai K. IPknot: fast and accurate prediction of RNA secondary structures with pseudoknots using integer programming. Bioinformatics 2011; 27:i85 - 93; http://dx.doi.org/10.1093/bioinformatics/btr215; PMID: 21685106
- Puton T, Kozlowski LP, Rother KM, Bujnicki JM. CompaRNA: a server for continuous benchmarking of automated methods for RNA secondary structure prediction. Nucleic Acids Res 2013; 41:4307 - 23; http://dx.doi.org/10.1093/nar/gkt101; PMID: 23435231
- Shapiro BA, Yingling YG, Kasprzak W, Bindewald E. Bridging the gap in RNA structure prediction. Curr Opin Struct Biol 2007; 17:157 - 65; http://dx.doi.org/10.1016/j.sbi.2007.03.001; PMID: 17383172
- Rocca-Serra P, Bellaousov S, Birmingham A, Chen C, Cordero P, Das R, Davis-Neulander L, Duncan CD, Halvorsen M, Knight R, et al. Sharing and archiving nucleic acid structure mapping data. RNA 2011; 17:1204 - 12; http://dx.doi.org/10.1261/rna.2753211; PMID: 21610212
- Weeks KM. Advances in RNA structure analysis by chemical probing. Curr Opin Struct Biol 2010; 20:295 - 304; http://dx.doi.org/10.1016/j.sbi.2010.04.001; PMID: 20447823
- Merino EJ, Wilkinson KA, Coughlan JL, Weeks KM. RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE). J Am Chem Soc 2005; 127:4223 - 31; http://dx.doi.org/10.1021/ja043822v; PMID: 15783204
- Nahvi A, Green R. Structural analysis of RNA backbone using in-line probing. Methods Enzymol 2013; 530:381 - 97; http://dx.doi.org/10.1016/B978-0-12-420037-1.00022-1; PMID: 24034334
- Washietl S, Hofacker IL, Stadler PF, Kellis M. RNA folding with soft constraints: reconciliation of probing data and thermodynamic secondary structure prediction. Nucleic Acids Res 2012; 40:4261 - 72; http://dx.doi.org/10.1093/nar/gks009; PMID: 22287623
- Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc Natl Acad Sci U S A 2004; 101:7287 - 92; http://dx.doi.org/10.1073/pnas.0401799101; PMID: 15123812
- Hajdin CE, Bellaousov S, Huggins W, Leonard CW, Mathews DH, Weeks KM. Accurate SHAPE-directed RNA secondary structure modeling, including pseudoknots. Proc Natl Acad Sci U S A 2013; 110:5498 - 503; http://dx.doi.org/10.1073/pnas.1219988110; PMID: 23503844
- Cate JH, Gooding AR, Podell E, Zhou K, Golden BL, Kundrot CE, Cech TR, Doudna JA. Crystal structure of a group I ribozyme domain: principles of RNA packing. Science 1996; 273:1678 - 85; http://dx.doi.org/10.1126/science.273.5282.1678; PMID: 8781224
- Ding F, Lavender CA, Weeks KM, Dokholyan NV. Three-dimensional RNA structure refinement by hydroxyl radical probing. Nat Methods 2012; 9:603 - 8; http://dx.doi.org/10.1038/nmeth.1976; PMID: 22504587
- Kladwang W, VanLang CC, Cordero P, Das R. A two-dimensional mutate-and-map strategy for non-coding RNA structure. Nat Chem 2011; 3:954 - 62; http://dx.doi.org/10.1038/nchem.1176; PMID: 22109276
- Das R, Kudaravalli M, Jonikas M, Laederach A, Fong R, Schwans JP, Baker D, Piccirilli JA, Altman RB, Herschlag D. Structural inference of native and partially folded RNA by high-throughput contact mapping. Proc Natl Acad Sci U S A 2008; 105:4144 - 9; http://dx.doi.org/10.1073/pnas.0709032105; PMID: 18322008
- Lavender CA, Ding F, Dokholyan NV, Weeks KM. Robust and generic RNA modeling using inferred constraints: a structure for the hepatitis C virus IRES pseudoknot domain. Biochemistry 2010; 49:4931 - 3; http://dx.doi.org/10.1021/bi100142y; PMID: 20545364
- Yu ET, Hawkins A, Eaton J, Fabris D. MS3D structural elucidation of the HIV-1 packaging signal. Proc Natl Acad Sci U S A 2008; 105:12248 - 53; http://dx.doi.org/10.1073/pnas.0800509105; PMID: 18713870
- Zhang Q, Yu ET, Kellersberger KA, Crosland E, Fabris D. Toward building a database of bifunctional probes for the MS3D investigation of nucleic acids structures. J Am Soc Mass Spectrom 2006; 17:1570 - 81; http://dx.doi.org/10.1016/j.jasms.2006.06.002; PMID: 16875836
- Klostermeier D, Millar DP. Time-resolved fluorescence resonance energy transfer: a versatile tool for the analysis of nucleic acids. Biopolymers 2001-2002; 61:159 - 79; http://dx.doi.org/10.1002/bip.10146; PMID: 11987179
- Kalinin S, Peulen T, Sindbert S, Rothwell PJ, Berger S, Restle T, Goody RS, Gohlke H, Seidel CA. A toolkit and benchmark study for FRET-restrained high-precision structural modeling. Nat Methods 2012; 9:1218 - 25; http://dx.doi.org/10.1038/nmeth.2222; PMID: 23142871
- Preus S, Kilsa K, Miannay FA, Albinsson B, Wilhelmsson LM. FRETmatrix: a general methodology for the simulation and analysis of FRET in nucleic acids. Nucleic Acids Res 2013; 41:e18; PMID: 22977181
- Qin PZ, Dieckmann T. Application of NMR and EPR methods to the study of RNA. Curr Opin Struct Biol 2004; 14:350 - 9; http://dx.doi.org/10.1016/j.sbi.2004.04.002; PMID: 15193316
- Schiemann O, Weber A, Edwards TE, Prisner TF, Sigurdsson ST. Nanometer distance measurements on RNA using PELDOR. J Am Chem Soc 2003; 125:3434 - 5; http://dx.doi.org/10.1021/ja0274610; PMID: 12643697
- Cai Q, Kusnetzow AK, Hideg K, Price EA, Haworth IS, Qin PZ. Nanometer distance measurements in RNA using site-directed spin labeling. Biophys J 2007; 93:2110 - 7; http://dx.doi.org/10.1529/biophysj.107.109439; PMID: 17526583
- Svergun DI. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J 1999; 76:2879 - 86; http://dx.doi.org/10.1016/S0006-3495(99)77443-6; PMID: 10354416
- Grigg JC, Chen Y, Grundy FJ, Henkin TM, Pollack L, Ke A. T box RNA decodes both the information content and geometry of tRNA to affect gene expression. Proc Natl Acad Sci U S A 2013; 110:7240 - 5; http://dx.doi.org/10.1073/pnas.1222214110; PMID: 23589841
- Jacques DA, Guss JM, Svergun DI, Trewhella J. Publication guidelines for structural modelling of small-angle scattering data from biomolecules in solution. Acta Crystallogr D Biol Crystallogr 2012; 68:620 - 6; http://dx.doi.org/10.1107/S0907444912012073; PMID: 22683784
- Gopal A, Zhou ZH, Knobler CM, Gelbart WM. Visualizing large RNA molecules in solution. RNA 2012; 18:284 - 99; http://dx.doi.org/10.1261/rna.027557.111; PMID: 22190747
- Ramrath DJ, Yamamoto H, Rother K, Wittek D, Pech M, Mielke T, Loerke J, Scheerer P, Ivanov P, Teraoka Y, et al. The complex of tmRNA-SmpB and EF-G on translocating ribosomes. Nature 2012; 485:526 - 9; PMID: 22622583
- Levitt M. Detailed molecular model for transfer ribonucleic acid. Nature 1969; 224:759 - 63; http://dx.doi.org/10.1038/224759a0; PMID: 5361649
- Expert-Bezançon A, Wollenzien PL. Three-dimensional arrangement of the Escherichia coli 16 S ribosomal RNA. J Mol Biol 1985; 184:53 - 66; http://dx.doi.org/10.1016/0022-2836(85)90043-9; PMID: 2411936
- Westhof E, Romby P, Romaniuk PJ, Ebel JP, Ehresmann C, Ehresmann B. Computer modeling from solution data of spinach chloroplast and of Xenopus laevis somatic and oocyte 5 S rRNAs. J Mol Biol 1989; 207:417 - 31; http://dx.doi.org/10.1016/0022-2836(89)90264-7; PMID: 2754730
- Brunel C, Romby P, Westhof E, Ehresmann C, Ehresmann B. Three-dimensional model of Escherichia coli ribosomal 5 S RNA as deduced from structure probing in solution and computer modeling. J Mol Biol 1991; 221:293 - 308; http://dx.doi.org/10.1016/0022-2836(91)80220-O; PMID: 1717695
- Mueller F, Sommer I, Baranov P, Matadeen R, Stoldt M, Wöhnert J, Görlach M, van Heel M, Brimacombe R. The 3D arrangement of the 23 S and 5 S rRNA in the Escherichia coli 50 S ribosomal subunit based on a cryo-electron microscopic reconstruction at 7.5 A resolution. J Mol Biol 2000; 298:35 - 59; http://dx.doi.org/10.1006/jmbi.2000.3635; PMID: 10756104
- Leontis NB, Westhof E. The 5S rRNA loop E: chemical probing and phylogenetic data versus crystal structure. RNA 1998; 4:1134 - 53; http://dx.doi.org/10.1017/S1355838298980566; PMID: 9740131
- Malhotra A, Tan RK, Harvey SC. Modeling large RNAs and ribonucleoprotein particles using molecular mechanics techniques. Biophys J 1994; 66:1777 - 95; http://dx.doi.org/10.1016/S0006-3495(94)80972-5; PMID: 7521223
- Stephenson JD, Li H, Kenyon JC, Symmons M, Klenerman D, Lever AM. Three-dimensional RNA structure of the major HIV-1 packaging signal region. Structure 2013; 21:951 - 62; http://dx.doi.org/10.1016/j.str.2013.04.008; PMID: 23685210
- Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW Jr., Swanstrom R, Burch CL, Weeks KM. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 2009; 460:711 - 6; http://dx.doi.org/10.1038/nature08237; PMID: 19661910
- Brunger AT. Version 1.2 of the Crystallography and NMR system. Nat Protoc 2007; 2:2728 - 33; http://dx.doi.org/10.1038/nprot.2007.406; PMID: 18007608
- Dzananovic E, Patel TR, Chojnowski G, Boniecki MJ, Deo S, McEleney K, Harding SE, Bujnicki JM, McKenna SA. Solution conformation of adenovirus virus associated RNA-I and its interaction with PKR. J Struct Biol 2014; 185:48 - 57; http://dx.doi.org/10.1016/j.jsb.2013.11.007; PMID: 24291322
- Volkov VV, Svergun DI. Uniqueness of ab-initio shape determination in small-angle scattering. J Appl Cryst 2003; 36:860 - 4; http://dx.doi.org/10.1107/S0021889803000268
- Cowtan K, Emsley P, Wilson KS. From crystal to structure with CCP4. Acta Crystallogr D Biol Crystallogr 2011; 67:233 - 4; http://dx.doi.org/10.1107/S0907444911007578; PMID: 21460440
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr 2012; 68:352 - 67; http://dx.doi.org/10.1107/S0907444912001308; PMID: 22505256
- Svergun D, Barberato C, Koch MHJ. CRYSOL – a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Cryst 1995; 28:768 - 73; http://dx.doi.org/10.1107/S0021889895007047
- Cruz JA, Blanchet MF, Boniecki M, Bujnicki JM, Chen SJ, Cao S, Das R, Ding F, Dokholyan NV, Flores SC, et al. RNA-Puzzles: a CASP-like evaluation of RNA three-dimensional structure prediction. RNA 2012; 18:610 - 25; http://dx.doi.org/10.1261/rna.031054.111; PMID: 22361291
- Laing C, Schlick T. Computational approaches to 3D modeling of RNA. J Phys Condens Matter 2010; 22:283101; http://dx.doi.org/10.1088/0953-8984/22/28/283101; PMID: 21399271
- Sripakdeevong P, Beauchamp K, Das R. Why can't we predict RNA structure at atomic resolution? In: Leontis N, Westhof E, eds. RNA 3D Structure Analysis and Prediction, 2012.
- Reuter JS, Mathews DH. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics 2010; 11:129; http://dx.doi.org/10.1186/1471-2105-11-129; PMID: 20230624
- Swenson MS, Anderson J, Ash A, Gaurav P, Sükösd Z, Bader DA, Harvey SC, Heitsch CE. GTfold: enabling parallel RNA secondary structure prediction on multi-core desktops. BMC Res Notes 2012; 5:341; http://dx.doi.org/10.1186/1756-0500-5-341; PMID: 22747589
- Zarringhalam K, Meyer MM, Dotu I, Chuang JH, Clote P. Integrating chemical footprinting data into RNA secondary structure prediction. PLoS One 2012; 7:e45160; http://dx.doi.org/10.1371/journal.pone.0045160; PMID: 23091593
- Gajda MJ, Tuszynska I, Kaczor M, Bakulina AY, Bujnicki JM. FILTREST3D: discrimination of structural models using restraints from experimental data. Bioinformatics 2010; 26:2986 - 7; http://dx.doi.org/10.1093/bioinformatics/btq582; PMID: 20956242
- Yang S, Parisien M, Major F, Roux B. RNA structure determination using SAXS data. J Phys Chem B 2010; 114:10039 - 48; http://dx.doi.org/10.1021/jp1057308; PMID: 20684627
- Schneidman-Duhovny D, Hammel M, Sali A. FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res 2010; 38:W540 - 4; http://dx.doi.org/10.1093/nar/gkq461; PMID: 20507903
- Chojnowski G, Walen T, Bujnicki JM. RNA Bricks--a database of RNA 3D motifs and their interactions. Nucleic Acids Res 2014; 42:D123 - 31; http://dx.doi.org/10.1093/nar/gkt1084; PMID: 24220091
- Xin Y, Laing C, Leontis NB, Schlick T. Annotation of tertiary interactions in RNA structures reveals variations and correlations. RNA 2008; 14:2465 - 77; http://dx.doi.org/10.1261/rna.1249208; PMID: 18957492